
Abstract
The oxidation of glycerol by molecular oxygen in the aqueous phase over Au/TiO2 was investigated in both a batch reactor and a continuous upflow fixed bed reactor. The effects of catalyst particle size, gas flow rate, liquid flow rate, reaction temperature, dioxygen pressure, and solution pH were examined in the fixed bed system. The unique hydrodynamics of the fixed bed system allowed for secondary oxidation products such as tartronic acid and oxalic acid to form in substantial amounts, which contrasts the product distribution observed in a batch system. These results suggest that reactor configuration can play an important role in the observed product selectivity from oxidation reactions over highly active gold catalysts.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Glycerol is a co-product of the transesterification of triglycerides with methanol to form biodiesel fuel (methyl esters of fatty acids). Glycerol is obtained in 1:3 mole ratio (approximately 10 wt%) of the total biodiesel produced. Biodiesel production is expected to rise significantly over next few years since governments in the US and EU have introduced legislation to replace a percentage of conventional fossil fuels with biofuels [1]. Glycerol can also be produced through hydrogenolysis of sugar derivatives obtained from biomass [2, 3]. Thus, glycerol is a viable renewable feedstock for the production of value added chemicals.
Prior works on glycerol oxidation have reported high activities for supported gold catalysts, but the product distribution can vary widely. Although some research groups have observed very high selectivity (>90%) to glyceric acid, others have found substantially lower selectivity (50–70%) to glyceric acid because of significant production of other oxidation products [4–7]. One can easily envision a possible role of support, gold particle size, solution pH, dissolved O2 concentration, and temperature on the selectivity of glycerol oxidation. The mechanism of glycerol oxidation under basic conditions over supported gold catalysts has been well supported in the literature whereby high pH is required for the initial deprotonation of glycerol and subsequent dehydrogenation to glyceraldehyde, which undergoes further oxidation to glyceric acid [4–8]. Glyceric acid results from oxidation of a terminal carbon in glycerol without rupturing any C–C bonds. Further oxidation of glyceric leads to tartronic acid. The formation of glycolic acid necessarily requires rupture of a C–C bond in glycerol [4]. The Prati group has suggested a path to glycolic acid through oxidation of dihydroxyacetone forming hydroxypyruvic acid as an intermediate whereas the Claus group proposes formation of glycolic acid by further oxidation of tartronic acid obtained through oxidation of glyceric acid [5, 6]. More recently, Ketchie et al. reported that peroxide formed during the glycerol oxidation over gold under basic conditions is correlated to the formation of glycolic acid [4]. These previous studies generally point toward an oxidation mechanism that involves both solution phase and metal-catalyzed reactions [4, 6]. Thus, glycerol oxidation over supported gold catalysts under basic conditions is a complex reaction sequence in which subtle changes can affect the ultimate product distribution. The major products observed during glycerol oxidation are presented in Scheme 1.

Main products of glycerol oxidation over Au/TiO2 under basic conditions
The very high activity reported by different research groups for glycerol oxidation over supported gold catalysts suggests that transport limitations may influence some of the results [4, 6, 7]. In a three phase system such as glycerol oxidation with molecular oxygen over supported Au, mass transfer artifacts can be a strong function of reactor configuration. In all of the studies cited above, the reaction was carried out in a semibatch system in which powdered Au catalyst was slurried with glycerol solution and contacted with a static pressure of dioxygen. However, large scale processes that would be anticipated for a future biorefinery would likely utilize a continuous reaction system instead of a batch process.
Thus, in this work, we compare activity and selectivity of glycerol oxidation over a Au/TiO2 catalyst provided by World Gold Council (WGC) in both a batch autoclave reactor and a fixed bed continuous upflow reactor.
2 Experimental Methods
2.1 Materials
The Au/TiO2 catalyst (1.6 wt% Au, average Au particle size 3.5 nm, prepared by deposition–precipitation method) provided by the World Gold Council (WGC) was used as received [9]. Although the powdered catalyst was used in its original form for the batch reactor study, it was pelletized before loading into the continuous fixed bed reactor to avoid a large pressure drop across the catalyst bed. Three particle sizes were size separated using sieve trays: −40/+140 mesh (106–420 μm), −80/+170 mesh (90–180 μm) and −170/+270 mesh (53–90 μm). The particles with −80/+170 mesh size gave a reasonable pressure drop and were used for most of the study. Glycerol [Acros, 99.6% purity] and NaOH [Mallinckrodt, 99%] were used to prepare the liquid feed for the reaction. All gases used in the study (dioxygen, dinitrogen, and helium) were of ultra high purity grade [GT&S].
2.2 Glycerol Oxidation
Batch studies of glycerol oxidation were performed in a 50 cm3 Parr Instrument Company 4592 batch autoclave reactor. The experimental protocol as described by Ketchie et al. [4] was followed. The feed solution (30 mL, 0.3 M glycerol, 0.1–0.6 M NaOH, vacuum degassed) and a proper amount of catalyst were added to the reactor that was then sealed, purged with He and heated to 333 K before pressurizing with O2. Dioxygen was continually fed to maintain constant head pressure (10 bar) in the reactor. The system was adjusted to 8000:1 glycerol:Au mole ratio to avoid dioxygen mass transfer limitations from the gas to the liquid in the batch reactor, as determined by Ketchie et al. [4].
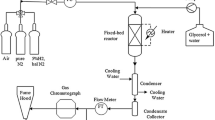
Glycerol oxidation was also studied in a continuous upflow fixed bed reactor system represented schematically in Fig. 1. This system is similar to the aqueous phase reforming (APR) reactor system used by the Dumesic group [10]. A stainless steel tubular reactor with ¼ inch (6.35 mm) OD and 4 mm ID was used with packed catalyst bed at the end of the tube supported by a stainless steel frit (20 μm, pressed) on one side and by glass wool on the other. A packed bed of 3 mm height was formed using pelletized Au/TiO2 catalyst (0.050 g). The liquid feed solution consisting of appropriate quantities of glycerol and NaOH was vacuum degassed and then pumped into the reactor using a high pressure Teledyne ISCO series D (500D) type syringe pump. Dioxygen flow was regulated by a mass flow controller. A filter (0.5 μm) was placed downstream of the reactor to trap any solid catalyst particles that may have eluted. A gas–liquid separator was used to collect liquid product between sampling intervals. Dinitrogen was used to purge the gas–liquid separator and to maintain the pressure in the system, which was controlled by a back pressure regulator. The dinitrogen purge gas along with unreacted dioxygen were fed to a gas chromatograph (GC) equipped with a thermal conductivity detector (TCD). A CTR I column was used to separate dioxygen and dinitrogen which allowed the exit flow rate of dioxygen to be quantified. No CO2 was observed in the gas phase. Any CO2 that was formed was subsequently converted to Na2CO3 in the basic solution and was quantified with the other liquid products. The liquid product collected in the gas–liquid separator was analyzed intermittently during the course of reaction. Three to four samples were collected over the course of each run and reasonable consistency of the results indicated steady state operation of the fixed bed reactor. The liquid products were analyzed using a high pressure liquid chromatograph (HPLC) equipped with a refractive index detector. Product separation in the HPLC was carried out using an Aminex HPX-87H column (Bio-Rad) with 5 mM H2SO4 as eluent.

Continuous upflow fixed bed reactor system used for glycerol oxidation
The reactor temperature was controlled by heating tape wrapped around its external surface. A thermocouple was inserted into the reactor to measure the temperature of the bed. Pressure gauges were placed on each side of the reactor to measure the pressure drop across the bed. Less than 0.5 bar pressure drop was observed for −80/+170 mesh catalyst particles under typical reaction conditions. Peroxide has been detected as a side product during glycerol oxidation over gold catalysts [4]. The colorimetric method described by Satterfield and Bonnell was used to quantify the peroxide formed in this work [11]. A Varian Cary 3E UV–VIS spectrometer was used to measure the absorbance at 405 nm of the complex formed by peroxide with TiO(SO4). The detailed procedure for peroxide analysis is explained by Ketchie et al. [4]. The lower detection limit for peroxide was 0.005 mM.
3 Results
3.1 Batch Reactor Studies
To provide a baseline comparison for the fixed bed reactor studies, we first performed glycerol oxidation in a batch autoclave reactor. The reaction conditions used were 0.3 M glycerol, 333 K, pO2 = 10 bar, 8000:1 glycerol:Au mole ratio and concentrations of NaOH ranging from 0.1 to 0.6 M. Table 1 presents the initial turnover frequency (TOF) based on glycerol conversion and the product selectivity based on C2 and C3 products. The initial TOFs were calculated from initial global reaction rates [mol glycerol converted (mol Ausurface)−1 s−1] measured at t = 30 min, unless otherwise noted. The dispersion of Au (0.29) was estimated from the average particle size reported by the World Gold Council. The TOF was observed to increase with base concentration as has been reported in many of the previous batch studies. Indeed, our results match reasonably well those of Ketchie et al. [4]. An increase in activity of glycerol oxidation with increase in base concentration was also observed by the Prati group and the Claus group [5, 6]. The selectivity to glyceric acid was nearly double that of glycolic acid, with both tartronic and oxalic acid forming in small quantities (<5%) regardless of the conversion.
Glyceric, glycolic, tartronic and oxalic acid were the major oxidation products observed during the HPLC analysis. In addition to these primary and secondary oxidation products, formic acid and Na2CO3 were also formed during the reaction. Acetic acid is formed in small amounts and is therefore not presented as a product in the tables and figures. The C1 products (formic acid and Na2CO3) were formed in 1:1 or lower mole ratio to C2 products. The overall carbon balance for the glycerol oxidation typically closes to within 5%. Selectivity is defined as moles of product formed divided by moles of C2 and C3 products formed.
3.2 Continuous Fixed Bed Reactor Studies
3.2.1 Influence of Particle Size
To examine the effects of transport limitations in the fixed bed reactor, three different catalyst particle sizes, −40/+140 mesh (106–420 μm), −80/+170 mesh (90–180 μm) and −170/+270 mesh (53–90 μm), were studied. For particles with diameter <180 μm, the observed turnover frequencies (TOFs) based on glycerol conversion were nearly constant, which suggests the influence of internal mass transfer limitations were minimal (Fig. 2). The slight decrease in TOF for 90 μm particles as compared to 180 μm particles was likely due to the excessive pressure drop observed across the bed for the smallest particles. Therefore, we decided to use 180 μm particles for further analysis.

Influence of particle size on activity of Au/TiO2 for glycerol oxidation in the fixed bed reactor. Reaction conditions: 0.3 M glycerol, 0.3 M NaOH, 333 K, total pressure (P tot) 10 bar, gas flow rate (Q G) = 30 cm3 (STP) min−1, liquid flow rate (Q L) = 1 cm3 min−1, amount of catalyst = 50 mg
3.2.2 Influence of Dioxygen Flow Rate
The effect of dioxygen flow rate on TOF and conversion of glycerol in our upflow fixed bed reactor is summarized in Fig. 3. The TOF (s−1) and conversion increased from 0.25 and 5% to 0.8 and 16%, respectively, as the dioxygen flow rate (Q G) increased from 5 to 30 cm3 (STP) min−1. Above 30 cm3 (STP) min−1, only slight increases in TOF and conversion were observed. Thus, a dioxygen flow rate of 30 cm3 (STP) min−1 was used for further study to avoid the higher pressure drop and possible liquid entrainment associated with higher gas flow rates.

Influence of dioxygen flow rate on rate of glycerol oxidation in the fixed bed reactor. Reaction conditions: 0.3 M glycerol, 0.3 M NaOH, 333 K, P tot = 10 bar, Q L = 1 cm3 min−1, catalyst particle size = 180 μm, amount of catalyst = 50 mg
3.2.3 Influence of Liquid Flow Rate
The fixed bed reactor was also operated at different liquid flow rates (Q L) to examine its effect on reaction rate and product distribution during glycerol oxidation. For the typical reaction conditions of 0.3 M glycerol, 0.3 M NaOH, 333 K, 10 bar total pressure, the liquid flow rate was varied from 0.2 to 4 cm3 min−1 and the results are given in Table 2. The TOF depended on Q L for flow rates less than 1 cm3 min−1, but was fairly constant at higher flow rates. The lowest flow rates gave conversions exceeding 20%, which indicates the reactor may have no longer been operating differentially. Moreover, artifacts from mass transfer limitations may have influenced the reaction rates at low liquid flow rates. Therefore, we decided to operate at or above 1 cm3 min−1 liquid flow rate. As seen in Table 2, selectivity to glyceric acid increased and that for oxalic and tartronic acid decreased with increasing liquid flow rate. Evidently, the primary oxidation products (glyceric acid and glycolic acid) undergo further oxidation with increasing contact with the metal catalyst under reacting conditions.
3.2.4 Influence of Temperature and Pressure
Glycerol oxidation was studied under typical reaction conditions over the range of temperatures from 308 to 348 K in the fixed bed reactor. Figure 4 represents an Arrhenius type plot of the reaction rate versus temperature. The reaction rate at the highest temperature falls below the trendline in Fig. 4, which suggests mass transfer might be limiting the rate. These transport limitations could possibly result from increased reaction rates and lower dioxygen solubility at higher temperatures. The apparent activation energy (E A) was calculated from the Arrhenius type plot and was found to be about 5.3 kJ mol−1 for the oxidation of glycerol, which is quite low. The selectivity to glyceric, oxalic and tartronic acid increased with temperature whereas selectivity to glycolic acid decreased significantly, as illustrated in Fig. 5.

Arrhenius type plot illustrating the influence of temperature on reaction rate. Reaction conditions: 0.3 M glycerol, 0.3 M NaOH, P tot = 10 bar, Q L = 1 cm3 min−1, Q G = 30 cm3 (STP) min−1, catalyst particle size = 180 μm, rate = mol glycerol converted (mol Au)−1 min−1, amount of catalyst = 50 mg

Influence of temperature on product selectivity during glycerol oxidation in the fixed bed reactor. Reaction conditions: 0.3 M glycerol, 0.3 M NaOH, P tot = 10 bar, Q L = 1 cm3 min−1, Q G = 30 cm3 (STP) min−1, catalyst particle size = 180 μm, amount of catalyst = 50 mg
The influence of dioxygen pressure on glycerol oxidation was also examined. The TOF increased with O2 pressure as expected for an oxidation reaction. However, the order in dioxygen was significantly less than first order since the TOF only approximately doubled for tripling of the O2 pressure, as seen in Fig. 6. Interestingly, O2 pressure has been reported to have no effect on the rate of glycerol oxidation in batch studies [5]. The product distribution was unaffected by the variation in pressure for the fixed bed reactor in this study.

Influence of pressure on the turnover frequency of glycerol oxidation in the fixed bed reactor. Reaction conditions: 0.3 M glycerol, 0.3 M NaOH, 333 K, Q L = 1 cm3 min−1, Q G = 30 cm3 (STP) min−1, catalyst particle size = 180 μm, amount of catalyst = 50 mg
3.2.5 Influence of Base Concentration
The influence of the NaOH:glycerol ratio on the TOF and product distribution was studied for glycerol oxidation in the fixed bed reactor. As evident in Fig. 7, very little activity was observed in the absence of base. In contrast to the batch results (Table 1), the turnover frequency was not a strong function of base above a NaOH:glycerol ratio of 0.33. However, at very low NaOH concentration (0.015 M, NaOH:glycerol = 0.05) the TOF decreased significantly indicating that below a threshold pH, activity of glycerol oxidation over gold decreases. Moreover, the increase in base concentration increased production of tartronic and oxalic acid whereas glycolic acid formation decreased significantly over the same range. The selectivity to glyceric acid was fairly constant.

Influence of NaOH:glycerol mole ratio on product selectivity and rate of glycerol oxidation in the fixed bed reactor. Reaction conditions: 0.3 M glycerol, 333 K, P tot = 10 bar, Q L = 1 cm3 min−1, Q G = 30 cm3 (STP) min−1, catalyst particle size = 180 μm, amount of catalyst = 50 mg
Figure 8 shows the concentration of peroxide collected from the fixed bed reactor system as a function of NaOH concentration. As previously reported by our lab, higher amounts of peroxide are detected at elevated pH [4].

Influence of NaOH:glycerol mole ratio on peroxide formation during glycerol oxidation in the fixed bed reactor. Reaction conditions: 0.3 M glycerol, 333 K, P tot = 10 bar, Q L = 1 cm3 min−1, Q G = 30 cm3 (STP) min−1, catalyst particle size = 180 μm, amount of catalyst = 50 mg
4 Discussion
4.1 Possible Influence of Mass Transfer on Glycerol Oxidation
Gold catalysts are reported to be highly active for glycerol oxidation under basic conditions. For example earlier work in our lab found turnover frequencies (TOFs) as high as 17.5 s−1 for Au/C and 4.1 s−1 for Au/TiO2 [4]. We also observed high TOFs in our batch and fixed bed reactors. Mass transfer limitations may influence reactions in a three phase gas–liquid–solid system, especially when the intrinsic rate is so high. A few previous studies on glycerol oxidation have attempted to account for mass transfer limitations in their results. For example, Claus et al. used results from empirical relations to suggest their reactor system was free from any mass transfer limitations [5, 12, 13]. Ketchie et al. [4] used a sulfite oxidation method to find the maximum dioxygen mass transfer rate from the gas to the liquid and operated the batch autoclave reactor accordingly to avoid gas–liquid transfer limitations.
The following sections describe how empirical relations were used to estimate the importance of mass transfer limitations on experimental observations for the different reactor configurations used in this work. The criteria, proposed by Mills and Chaudhari, were derived for gas–liquid–solid (G-L-S) three phase reactions such as \( A(l) + B(g)\xrightarrow{catalyst(s)}products, \) where the reaction is first order in both reactants [14]. However, the criteria are used here more as general indicators of possible mass transfer problems instead of quantitative correction factors of the rate data because of the unknown reaction orders for glycerol and dioxygen and the unknown flow pattern in the fixed bed reactor.
4.1.1 Gas–Liquid Mass Transfer
Mills and Chaudhari [14] proposed a criterion to determine the significance of gas–liquid mass transfer in a three phase gas–liquid–solid catalyzed reactions:
where R A is the intrinsic rate of reaction, k L a is gas–liquid mass transfer coefficient, and \( C_{A}^{*} \) concentration of gas in solution.
Since dioxygen is assumed to be the limiting reactant under our reacting conditions, R A in this transport analysis is the rate of oxygen consumption. The reaction rate for the fixed bed reactor (R A = 10.6 mol O2 consumed m−3 s−1) was calculated for typical reaction conditions used: 0.3 M glycerol, 0.3 M NaOH, 333 K, P tot = 10 bar, liquid flow rate (Q L) = 1 cm3 min−1, gas flow rate (Q G) = 30 cm3 (STP) min−1, catalyst particle size = 180 μm.
The gas–liquid mass transfer coefficient for cocurrent flow in a fixed bed reactor with pulsing flow regime, k L a, is calculated from the correlation proposed by Blok et al. [15].
where S is specific packing area (estimated) = \( \frac{1}{2} \cdot \frac{3}{{{\text{R}}_{\text{p}} }} = 16.7 \times 10^{3} \left( {{\text{m}}^{2} /{\text{m}}^{3} {\text{column}}} \right), \) ε is packing porosity = 0.34 (m3 void/m3 column), u L is superficial liquid velocity = 1.33 × 10−3 (m/s), u g is superficial gas velocity = 4.45 × 10−3 (m/s).
Also, \( C_{A}^{*} = \frac{P}{{H_{i} }} = 10.03\frac{mol}{{m^{3} }}, \) where P is pressure of the system = 10 bar, H i = 99.7 kPa m3 mol−1 is the Henry’s law constant for dioxygen in water [13].
For our fixed bed reactor system, the calculated \( \alpha_{gl} = 1.73, \) which is higher than the limit of the proposed criterion \( \left( {\alpha_{gl} < 0.1} \right), \) and indicates there may be some influence of gas–liquid mass transfer on observed rates.
4.1.2 Liquid–Solid Mass Transfer
The criterion to assess the significance of liquid–solid mass transfer is [14]:
where
where w is catalyst mass per unit volume of fixed bed = 1326 kg m−3, ρ p is catalyst particle density = 2000 kg m−3, d p is particle diameter = 180 × 10−6 m.
The liquid–solid mass transfer coefficient for the fixed bed reactor, k s , is calculated from the correlation for laboratory reactors proposed by Hidajat et al. [16]:
where \( \text{Re} = \frac{{u_{L} \cdot \rho \cdot d_{p} }}{\mu } = 0.51 \) and \( Sc = \frac{\mu }{{\rho .D_{{O_{2} - H_{2} O}} }} = 98.3 \)
The calculated value of k s from this relation is 1.1 × 10−4 m/s. The α ls criterion for the fixed bed reactor was found to be 0.43, which is close to the proposed criterion (α ls < 0.1), indicating slight influence of liquid–solid mass transfer on measured kinetics.
4.1.3 Internal Diffusion
The Thiele modulus is used to determine the influence of internal diffusion on the measured rates [14]:
where the effective diffusivity = De = 4.78 × 10−9 . \( \frac{\varepsilon }{\tau } = 4.0 \times 10^{ - 10} {\text{m}}^{ 2} {\text{s}}^{ - 1} \) (Porosity ɛ = 0.5 and tortuosity τ = 6 were assumed)
The Thiele modulus for the fixed bed reactor was found to be greater than the criterion (ϕ exp = 1.9), but was not excessively large. Evidently, some influence of mass transfer within the catalyst particle likely affects the measured rates.
Thiele modulus calculation for the batch reactor using Eq. 6 gave ϕ exp = 0.90, indicating that the internal mass transfer limitations could also be present to some extent in the batch reactor. The reaction rate (R A = 0.0201 mol O2 consumed m−3 s−1) was calculated for typical reaction conditions of 0.3 M glycerol, 0.3 M NaOH, 333 K, pO2 = 10 bar, glycerol:Au = 8000:1. The w, catalyst mass per unit volume of the batch reactor, is 0.5 kg m−3. To determine the particle size for the powdered catalyst provided by the World Gold Council, we sieved the catalyst using a 400 mesh (38 μm) sieve and found that although most of the catalyst passed through, some was retained. Nevertheless, 38 μm was used as an approximation for the particle diameter for above calculation.
4.2 Batch Versus Continuous Flow Reactor Studies
Because of the high dioxygen flow rates used, gas–liquid–solid contacting in our fixed bed reactor is considered to be quite different than in an idealized fixed bed reactor. Under conditions typically used here, regions of direct gas–solid contact are possible in the reactor, which would allow for exposure of the catalyst particles to a significant concentration of dioxygen without the solubility limit of a liquid film. Moreover, the high gas flow rates could also reduce the liquid film thickness surrounding wetted solid particles in the bed thereby reducing the resistance to dioxygen transfer to the catalyst surface. This type of G–L–S contacting is very different than that observed in typical laboratory batch reactors where gas dissolves into the liquid through the gas pressure applied in the head space of the reactor and solid catalyst particles are completely wetted by liquid. It appears that it is possible to enhance dioxygen at the catalyst surface by using a fixed bed reactor operating at high gas flow as compared to a batch reactor in which dioxygen is supplied to the catalyst solely through the bulk solvent. The empirical relations discussed above indicate diffusion limitations might be present in our system, but are not severe. Moreover, commonly used experimental tests for mass transfer limitations such as effect of liquid flow rate on observed activity, effect of particle size on activity and influence of temperature on activity suggest that transport artifacts did not severely affect our observations.
Therefore, we must address how different gas–liquid–solid contacting schemes for the different reactor configurations can influence the product distribution during glycerol oxidation over supported gold catalysts. Both tartronic acid and oxalic acid were formed in very small quantities over the course of reaction in our batch study (<5% selectivity). Previous batch studies have also found similar low selectivity to tartronic and oxalic acid using both Au/TiO2 and Au/C catalysts [4, 6, 7]. Since tartronic and oxalic acid are subsequent oxidation products of glyceric and/or glycolic acid, the low selectivity to the diacids in a batch reactor, even at high conversions of glycerol, is rather unexpected. In the continuous upflow fixed bed reactor, under similar conditions of concentration, pressure, temperature and pH, a higher selectivity towards secondary oxidation products like tartronic and oxalic acid was observed, even at low conversions. Thus, selectivity of gold catalysts for primary oxidation is a strong function of reactor configuration. Apparently the high dioxygen availability made possible by the direct gas–solid contact associated with the fixed bed reactor enhances the subsequent oxidation of glyceric acid and glycolic acid to the diacids compared to that observed in a standard slurry-type batch reactor.
The turnover frequency of glycerol oxidation was observed to increase with increasing initial base concentration in the batch reactor whereas the TOF was nearly constant with pH in the fixed bed reactor. It should be noted, however, that the pH drops as the reaction proceeds by converting the acids to salts. In a fixed bed system, we are continuously feeding reactants and removing products, thus stabilizing the pH in the system. Apparently, above a certain threshold pH, the rate in the fixed bed reactor remained constant. Therefore, the fixed bed reactor can be used for glycerol oxidation with lower initial base concentration than that typically used in batch reactors.
The generally accepted reaction path for the aqueous-phase, gold-catalyzed oxidation of glycerol under basic conditions involves the intermediate formation of glyceraldehyde and dihydroxyacetone in equilibrium with each other, with glyceraldehyde oxidation to glyceric acid being favored [4, 6, 7]. However, the subsequent formation of glycolic, tartronic, and oxalic acid is not as clear as different routes are proposed in literature [4, 6, 7, 13]. Therefore, oxidation of glyceric acid (0.015 M) and glycolic acid (0.05 M) was carried out under typical reaction conditions (0.3 M NaOH, 333 K, 10 bar total pressure, liquid flow rate (Q L) = 1 cm3 min−1, gas flow rate (Q G) = 30 cm3 (STP) min−1, catalyst particle size = 180 μm) in our fixed bed reactor system. Approximately, 19% of the glyceric acid converted under these conditions to give primarily tartronic acid (72%) with some formation of oxalic acid (28%). Glycolic acid was oxidized to oxalic acid at a level of conversion equal to 16%. Clearly, tartronic acid is formed from subsequent oxidation of glyceric acid under reaction conditions, which is consistent with the literature [4, 6, 7, 13]. Tartronic acid (0.01 M) was fed to the fixed bed reactor under typical oxidation conditions of 0.3 M NaOH, 333 K, 10 bar total pressure, liquid flow rate (Q L) = 1 mL min−1, gas flow rate (QG) = 30 mL (STP) min−1, catalyst particle size = 180 μm, and no primary or secondary oxidation products were observed during HPLC analysis. Oxalic acid can be formed by further oxidation of either glycolic acid or glyceric acid. The formation of glycolic acid apparently does not involve a glyceric acid reaction intermediate. The C–C bond cleavage needed to form glycolic acid has been attributed to the formation of peroxide intermediate [4]. In our fixed bed reactor studies, peroxide was found to increase with increasing pH, as seen previously [4]. However, the selectivity to glycolic acid was actually lower at higher pH in the continuous flow system. The selectivity pattern observed in the fixed bed system is significantly affected by subsequent oxidation of diacids which is not observed in batch studies.
5 Conclusions
The product selectivity during glycerol oxidation was affected by the reactor system used to perform the reaction which can be attributed to the difference in gas–liquid–solid contacting between a batch reactor and a fixed bed reactor. The higher selectivity to the secondary oxidation products, oxalic and tartronic acid, in the fixed bed system was likely the result of direct gas–solid contact which allowed for a higher inventory of dioxygen on the catalyst compared to a batch system. Tartronic acid was formed from glyceric acid. Oxalic acid could be produced from both glyceric and glycolic acid. In agreement with earlier works in our lab, peroxide was also formed during glycerol oxidation and its concentration was correlated with increasing base concentration. Conditions were selected to minimize the effects of mass transfer on the reaction rates since highly active gold catalysts can be very easily operated in a transport limited regime.
References
Ragauskas A, Williams C, Davison B, Britovsek G, Cairney J, Eckert C, Frederick W, Hallett J, Leak D, Liotta C, Mielenz J, Murphy R, Templer R, Tschaplinski T (2006) Science 311:484–489
Huber GW, Iborra S, Corma A (2006) Chem Rev 106:4044–4098
Clark IT (1958) J Ind Eng Chem 50:1125–1126
Ketchie W, Murayama M, Davis R (2007) Top Catal 44:307–317
Demirel-Gulen S, Lucas M, Claus P (2005) Catal Today 102–103:166–172
Porta F, Prati L (2004) J Catal 224:397–403
Carrettin S, McMorn P, Johnston P, Griffin K, Hutchings GJ (2002) Chem Commun 696–697
Carrettin S, McMorn P, Johnston P, Griffin K, Kiely CJ, Attard GA, Hutchings GJ (2004) Top Catal 27:131–136
Holliday R Data sheets accompanying gold reference catalysts
Shabaker JW, Davda RR, Huber GW, Cortright RD, Dumesic JA (2003) J Catal 215:344–352
Satterfield CN, Bonnell AH (1955) Anal Chem 27:1174–1175
Demirel S, Lehnert K, Lucas M, Claus P (2007) Appl Catal B 70:637–643
Demirel S, Lucas M, Wärnå J, Salmi T, Murzin D, Claus P (2007) Top Catal 44:299–305
Mills PL, Chaudhari RV (1997) Catal Today 37:367–404
Blok JR, Koning CE, Drinkenburg AAH (1984) AIChE J 30:393–401
Hidajat K, Aracil FJ, Carberry JJ, Kenney CN (1987) J Catal 105:245–248
Acknowledgement
This work was supported by a grant from the National Science Foundation (CTS-0624608). Partial support by the NSF PIRE program is also acknowledged (OISE-0730277).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zope, B.N., Davis, R.J. Influence of Reactor Configuration on the Selective Oxidation of Glycerol over Au/TiO2 . Top Catal 52, 269–277 (2009). https://doi.org/10.1007/s11244-008-9163-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11244-008-9163-3