
Abstract
A Mo(VI) complex, [MoO2HL(HOCH3)], HL = 2-[(2-hydroxy-benzylidene)-amino]-3-(4-hydroxy-phenyl)-propionic acid, was reacted with 3-(chloropropyl)-trimethoxysilane-functionalized silica that had been fused with magnetite to yield a magnetically separable catalyst in which the Mo complex was covalently linked to the silica matrix through a silane linkage. Both the free [MoO2HL(HOCH3)] and the immobilized complex function as efficient catalysts for the oxidation of olefins in the presence of tert-butylhydroperoxide as primary oxidant. The Fe3O4@SiO2/[MoO2L(HOCH3)] showed lower catalytic activity and turnover numbers compared to its homogeneous counterpart. However, the immobilized catalyst could be readily recovered from the reaction mixture by using a magnet and could be reused up to seven times without any loss of activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Molybdenum possesses several oxidation states and variable coordination numbers from four to eight. Together with its roles in many molybdoenzymes, this has led to much interest in Mo-based catalysts [1]. Epoxidation and hydroxylation of olefins [2] are important enzymatic roles of molybdenum.
In the last decades, homogeneous catalysts have been used for the oxidation of alkenes [3, 4]. Despite this attention, such materials have disadvantages such as difficulty in separation from the reaction mixture, deactivation of the catalyst via self-aggregation of active sites, high price and difficulty of recycling, which make them inappropriate for large-scale applications [5]. Hence, new oxidative processes based on the activation of tert-butylhydroperoxide (TBHP) by robust, efficient and recyclable heterogeneous catalysts have been developed. In this regard, the immobilization of homogeneous catalysts onto inorganic solid supports constitutes a potential strategy [6].
In continuation of our research into the design and synthesis of organic–inorganic hybrid materials and their applications as catalysts for oxidation reactions [5, 7], we now report the synthesis, characterization and application of a magnetic nanoparticle (MNP)-supported molybdenum catalyst for the oxidation of various olefins to the corresponding epoxides.
Experimental
Materials and characterization
Chemicals were purchased from Merck. Elemental analyses were determined on a CHN PerkinElmer 2400 analyzer. FTIR spectra were recorded as KBr pellets on a PerkinElmer model 597 FTIR spectrometer. Magnetic measurements were taken on an MPMS XL7 magnetometer. The 57Fe Mossbauer spectra were recorded in transmission mode at room temperature using a conventional constant acceleration spectrometer and a 50-mCi 57Co source in a Rh matrix. The velocity scale was calibrated using α-Fe foil. The spectra were fitted to Lorentzian lines using the WinNormos software program [8], and the isomer shifts are reported relative to metallic α-Fe at room temperature. Metal contents were determined by atomic absorption spectroscopy. Scanning electron micrographs were taken on a MIRA3FE6 SEM instrument. XRD patterns were recorded on a Philips PW1130 X-ray diffractometer with Cu Kα target (λ = 1.54 Å). The products of the oxidation reactions were analyzed with an HP Agilent 6890 gas chromatograph, equipped with an HP-5 capillary column (phenyl methyl siloxane 30 m × 320 μm × 0.25 μm) and a flame ionization detector.
Synthesis
The Schiff base ligand H3L was synthesized by the condensation of salicylaldehyde with l-tyrosine in 1:1 molar ratio in methanol [9].
FTIR (KBr, cm −1 )
3208 (w) (O–H), 3026 (w), 2963 (w), 2930 (w), 1612 (vs) (C=N), 1609 (s), 1514 (m), 1455 (m), 1364 (m), 1332 (s), 1245 (s), 1100 (m), 842 (s), 740 (m), 650 (s), 576 (s), 530 (s), 493 (w), 434 (w), cm−1.
Its molybdenum complex, [MoO2HL(HOCH3)] was synthesized according to a published procedure [10], and its identity was confirmed by CHN elemental analysis. Calc.: C 46.1, H 3.9, N 3.2, Mo 21.7 %. Found: C 45.9, H 3.6, N 3.0, Mo 21.2 %.
FTIR (KBr, cm−1)
3467 (br) ν(o–H), 3195 (m), 2924 (w), 2843 (w), 1634 (s) (C=N), 1590 (vs) [ν as(COO)], 1514 (s), 1446, 1368 (s) [ν s(COO)], 1337 (s), 1248 (m), 1115 (m), 1033 (m), 943 (m), 884 (m), 747 (w), 652 (m), cm−1.
Magnetite Fe3O4 nanoparticles were prepared according to the procedure of Wang et al. [11] and characterized by the FTIR band at 586 (vs) cm−1. Silica-coated magnetite nanoparticles, denoted as Fe3O4@SiO2, were prepared according to the literature method [12] and characterized by their FTIR bands: 3437 (br, O–H), 1110 (vs), 963 (w), 809 (w), 591 (s), 482 (m), 434 (w) cm−1. The Fe3O4@SiO2 nanoparticles were functionalized with 3-(chloropropyl)trimethoxysilane (CPTMS) to give a material, denoted by Fe3O4@SiO2/CPTMS, according to a reported procedure [13]. CPTMS loading was 0.65 mmol g−1 MNP silica gel.
To prepare the immobilized catalyst, Fe3O4@SiO2/CPTMS (0.25 g) and [MoO2HL(HOCH3)] (0.04 g) were refluxed in CH2Cl2 for 24 h followed by washing with CH2Cl2 for 4 h in order to eliminate all the adsorbed [MoO2HL(HOCH3)] (Scheme 1). The resulting black material, Fe3O4@SiO2/[MoO2L(HOCH3)], was dried at room temperature and characterized by CHN, SEM, A.A, IR spectroscopy, VSM and Mossbauer measurement. The Mo loading and the complex loading were 0.30 mmol g−1 MNP silica gel and 0.3 mmol g−1 MNP silica gel, respectively.

Schematic representation of the formation of Fe3O4@SiO2/[MoO2(L)(HOCH3)]
FTIR (KBr, cm−1)
3437 (br, O–H), 1631 (s), 1592 (s), 1515 (m), 1440 (w), 1416 (m), 1367 (m), 1327 (w), 1293 (w), 1124 (vs), 949 (m), 843 (w), 742 (w), 708 (w), 577 (vs), 482 (vs).
General oxidation procedure
Oxidation reactions were carried out under air at 80 °C, with 1,2-dichloroethane as solvent and aqueous TBHP (80 %) as oxidant. In a typical experiment, a mixture of 0.013 mmol of [MoO2HL(HOCH3)], 1.0 mL solvent and 1.0 mmol of cyclooctene was placed in a 25-mL round-bottomed glass flask. The mixture was heated to 80 °C, and after addition of TBHP, the solution was placed in an oil bath at 80 °C. At appropriate intervals, aliquots were removed and analyzed immediately by GC. The oxidation products were identified by the comparison of their retention times with those of authentic samples. Yields are based on cyclooctene.
Results and discussion
Characterization of the catalyst
The MoO2(HL)(CH3OH) complex was prepared in good yield by the condensation of 2-[(2-hydroxy-benzylidene)-amino]-3-(4-hydroxy-phenyl)-propionic acid (H3L) with MoO2(acac)2 in methanol. The FTIR spectrum of the free Schiff base (Fig. 1a) showed an intense band at 1614 associated with the C=N stretching frequency, which was shifted to 1634 cm−1 for the corresponding dioxo complex (Fig. 1b), consistent with coordination of the azomethine nitrogen. In the spectrum of the complex (Fig. 1b), an extremely broad band at about 3467 cm−1 is assigned to –OH, possibly involved in intermolecular hydrogen bonding. The ν as(COO) band is observed at 1590 cm−1, while ν s(COO) is identified as a medium to strong bond at 1368 cm−1. Hence, Δν [ν as(COO)–ν s(COO)] is 191–225 cm−1, characteristic of monodentate coordination of the carboxylate group [14].

FTIR spectra of a H3L, b [MoO2(HL)(HOCH3)] (from 500–4000 and 500–1750 cm−1)
Finally, two characteristic bands assigned to ν(Mo=O) of the cis-MoO2 moiety at 884 and 943 cm−1 confirm the presence of this unit in MoO2(HL)(CH3OH) [15] (Fig. 1b).
The FTIR spectrum of Fe3O4 (Fig. 2a) exhibited the expected Fe–O stretching absorption at 586 cm−1. This peak also appears in all the FTIR spectra of the heterogeneous compounds. In the FTIR spectra of both Fe3O4/SiO2 and Fe3O4@SiO2/[MoO2L(HOCH3)], the absorption intensity of the Fe–O group decreases with the addition of silica [16] and a strong absorption arising from the Si–O–Si group was observed. In Fig. 2b, bands at 482, 963 and 1110 cm−1 are assigned to δ(Si–O–Si), ν(Si–OH) and ν as(Si–O–Si), respectively.

FTIR spectra of a Fe3O4, b Fe3O4@SiO2, c Fe3O4@SiO2/[MoO2L(HOCH3] (fresh), d Fe3O4@SiO2/[MoO2L(HOCH3] (recycled)
The FTIR spectrum of the supported catalyst, Fe3O4@SiO2/[MoO2L(HOCH3)] (Fig. 2c), exhibits characteristic new bands assigned to the complex units at 1631[ν(C=N)], 1592 [ν as(COO)], 1368 [ν s(COO)] and 949 cm−1 [ν(Mo=O)], together with silica and magnetite matrix bands. Hence, the FTIR characterization confirmed the successful immobilization of the Mo(VI) complex on the modified Fe3O4@SiO2 nanoparticles.
57Fe Mossbauer spectroscopy measurements were taken at room temperature for samples of Fe3O4, Fe3O4@SiO2 and the catalyst, Fe3O4@SiO2/[MoO2L(HOCH3)], in order to verify the stability of the magnetic nanoparticles after all stages of the synthesis. The room temperature 57Fe Mossbauer spectra of these compounds are shown in Fig. 3.

Room-temperature 57Fe Mossbauer spectra of a Fe3O4, b Fe3O4@SiO2, c Fe3O4@SiO2/[MoO2L(HOCH3)]
Experimental and fitted Mossbauer spectra for Fe3O4, Fe3O4@SiO2 and Fe3O4@SiO2/[MoO2L(HOCH3)] collected at room temperature are illustrated in Fig. 3, and the hyperfine parameters are shown in Table 1. The best fits to the spectra were obtained with two strong sextets and a weak doublet, indicating dominant ferromagnetic behavior. For Fe3O4, the sextet with hyperfine field 481.71 kOe and isomer shift (I. S.) 0.20 mm s−1 indicates the presence of tetrahedral Fe3+ sites, while the other sextet with hyperfine field 450.30 kOe and isomer shift 0.50 mm s−1 is assigned to the presence of both octahedral Fe3+ and Fe2+ [17]. The extra doublet with isomer shift −0.01 mm s−1 and quadrupole shift (Q. S.) 0.49 mm s−1 may be due to the finite size effect which can cause some of the Fe3O4 display superparamagnetic behavior.
Magnetic hysteresis curves obtained by vibrating sample magnetometry at room temperature are shown in Fig. 4.

Hysteresis loops of a Fe3O4, b Fe3O4@SiO2, c Fe3O4@SiO2/[MoO2L(HOCH3)]
Although the starting magnetite powder shows saturation magnetization behavior around 0.017 Nβg−1, as expected for Fe3O4, it is observed that coating of the magnetic nanoparticles with the silica layer and subsequent functionalization with the Mo complex results in a reduction in the magnetization of about ~0.012 Nβg−1 for Fe3O4@SiO2 and 0.0085 at Fe3O4@SiO2/[MoO2L(HOCH3)]. Although the mass saturation magnetization in the last two composites is decreased, due to the contribution of the non-magnetic silica shell and functionalized groups [18], they still could be efficiently separated from solution media with a permanent magnet.
The crystalline structures of the nanoparticles were identified by XRD (Fig. 5). Figure 5 shows the XRD patterns of the nanoparticles at different steps: specifically, (a) Fe3O4 nanoparticles, (b) Fe3O4@SiO2 nanoparticles and (c) Fe3O4@SiO2/[MoO2L(HOCH3)] nanoparticles. As shown in Fig. 5a, the X-ray powder diffractogram of the iron oxide prepared in this study matches that of magnetite (JCPDS No. 72-2303) [18]; however, when coated with silica, the intensity of the Fe3O4 phase decreases but some new low-intensity peaks appear. As peaks corresponding to silica are absent, the material must be amorphous [19], with the Fe3O4 phase retaining its crystallinity [20]. For Fe3O4@SiO2/[MoO2L(HOCH3)], peak characteristic of [MoO2HL(HOCH3)] is absent, which is indicative of an amorphous nature [21].

XRD pattern of a Fe3O4, b Fe3O4@SiO2, c Fe3O4@SiO2/[MoO2L(HOCH3)]
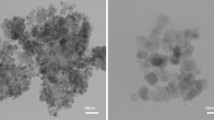
The morphologies of Fe3O4@SiO2 and Fe3O4@SiO2/[MoO2L(HOCH3)] were further investigated via SEM, as revealed in Fig. 6. The nanoparticle sizes of Fe3O4@SiO2 and Fe3O4@SiO2/[MoO2L(HOCH3)] were about 39 and 52 nm, respectively, both with spherical shape.

SEM image of a Fe3O4@SiO2, b Fe3O4@SiO2/[MoO2HL(HOCH3)]
Catalytic activity
Using [MoO2HL(HOCH3)] as a homogeneous catalyst for the oxidation of cyclooctene to the corresponding epoxide, oxidation did not take place in the absence of tert-butylhydroperoxide. Without the catalyst, the oxidation was only 5 % complete after 24 h [22]. The conversion increased with increasing amounts of tert-butylhydroperoxide (Table 2, entries 1–3).
The oxidation was also affected by the nature of the solvent (Table 3, entries 1–6).
High epoxide yields were obtained in a range of chlorinated solvents such as CCl4, CHCl3, 1,1,2,2-tetrachloroethane and 1,2-dichloroethane [23]. This can be explained by hard–soft acid–base theory. Solvents such as methanol, ethanol and acetonitrile, containing hard donor atoms (O and N), tend to coordinate strongly to hard metal centers such as Mo(VI). On the other hand, chlorinated solvents coordinate only very weakly to the metal [molybdenum(VI)]. As the coordination ability of the solvent increases, the solvent competes with TBHP for coordination to the molybdenum(VI), resulting in a decrease in the conversion.
When the temperature was increased (from 40 to 80 °C), the conversion correspondingly increased (from 2 to 98 %). Also the amount of catalyst had a significant effect on the oxidation of cyclooctene. A relatively low conversion (89 %) was obtained with 6.5 × 10−3 mmol catalyst, but a higher conversion (98 %) with 13 × 10−3 mmol catalyst. A still larger quantity resulted in lower conversion, 84 % with 19.5 × 10−3 mmol catalyst. This may be due to increased degradation of the oxidant at higher concentration of catalyst [24].
To establish the scope for the activity of [MoO2L(HOCH3), this study was further extended to the catalytic epoxidation of several linear and cyclic olefins, namely styrene, α-methylstyrene, 1-heptene, 1-hexene, cyclohexene, norbornene and indene, using [MoO2L(HOCH3)] (Table 4).
Generally, excellent epoxide selectivity was observed for the tested aliphatic substrates and cyclooctene (Table 4, entries 6–8). A similar result for cyclooctene was also reported for Schiff base molybdenum complexes immobilized on MCM-41 [25]. Moreover, the present catalytic system was completely selective for the epoxide for all substrates except for styrene and α-methyl styrene, with benzaldehyde and acetophenone, respectively, as the side products. The production of benzaldehyde in the oxidation of styrene is due to the overoxidation of styrene oxide with TBHP. The conversion of styrene and selectivity for styrene oxide were also affected by the reaction time. The selectivity for styrene oxide dramatically increased from 65 to 87 % upon decreasing the reaction time from 7 to 2 h, which is mainly caused by overoxidation at higher reaction times and temperatures (Table 4, entry 1) [26].
The lower catalytic activity of α-methyl styrene compared to styrene seems to be due to the steric hindrance of the methyl substituent of the former [27]. It should be noted that if electronic effects due to the methyl substituent were important, then α-methyl styrene as a more electron-rich olefin would be expected to show higher reactivity than styrene.
The relatively low catalytic activity for the epoxidation of indene does not seem to be caused by catalyst decomposition, since an increase in the reaction time indene resulted in a significant increase in the yield of oxidation, as shown in Table 4, entry 4.
In the present system, the oxidizing agent TBHP is transformed to tert-butyl alcohol during the reaction, which can coordinate to the Mo(VI) center and consequently retard the reaction, as shown in Table 2, entry 4 [28].
To confirm that these oxidation reactions are catalyzed under heterogeneous conditions with Fe3O4@SiO2/[MoL(HOCH3)], we tested the oxidation reaction in the presence of Fe3O4 and Fe3O4@SiO2 instead of the catalyst. Under these conditions, the oxidations were only about 12 and 10 % complete after 7 h, respectively [29, 30]. The epoxide yields for the homogeneous and heterogeneous systems under the same experimental conditions are compared in Table 5.
The heterogeneous catalyst is less active than its homogenous analogue (Tables 4, 5); however, some species show enhanced selectivity and longer lifetime due to the site isolation that prevents catalyst deactivation in the case of heterogeneous catalyst. Also, since the catalyst is intimately combined with magnetite, it can be easily separated on the completion of the reaction by magnetic decantation. In this way, the heterogeneous catalyst could be reused at least seven times without noticeable loss of activity (Table 6, entries 1–8).
Other important features of the system include the high formation of benzaldehyde over styrene oxide for the heterogeneous catalyst. In the homogeneous phase, the formation of epoxide was favored. The reason for benzaldehyde formation may be linked to the high acidity of the host materials, which according to a proposed mechanism leads to preferential formation of that product [31].
Table 7 compares the efficiency of our catalyst with some found in the literature.
Overall, all reported systems showed high activity and recyclability. For instance, Grivani and coworkers have also performed epoxidation of cyclooctene, styrene, α-methylstyrene, indene, 1-octene with TBHP promoted by a polymer-bound molybdenum carbonyl Schiff base catalyst. The performance of their catalyst was better than the present example. Similarly, oxidation of styrene also yielded benzaldehyde as major product [27].
Compared with the previously reported catalysts, both [MoO2HL(HOCH3)] and [Fe3O4@SiO2/[MoO2L(HOCH3)] exhibited superior activity for the oxidation of hydrocarbons. The previously reported glypy-Mo(VI) [32] showed lower catalytic activity for the oxidation of cyclooctene. Moreover, our catalysts showed higher catalytic activity than the glypy-Mo(VI), PMO-Mo(VI), MCM-glypy-Mo(VI) and [MoO2L(CH3CN)] for the oxidation of styrene. Our homogeneous and heterogeneous catalysts are also very efficient compared to ZPS-PVPA-MoO2(HOEt)(Schiff base) [23] and cis-MoO2{salnptn(3-OMe)2}, cis-MoO2{hnaphnptn} for the oxidation of α-methylstyrene [33]. However, for the oxidation of indene, our catalytic system is less effective than the cis-MoO2{salnptn(3-OMe)2} and cis-MoO2{hnaphnptn} systems. In addition, the Mo-oxodiperoxo core was previously used by Thiel’s group in the oxidation of olefins using TBHP as oxidant. Their catalysts showed high activity even for terminal unbranched olefins such as 1-octene [34].
Catalyst recycling
The present catalyst, Fe3O4@SiO2/[MoO2L(HOCH3)], could be reused seven times without any perceptible loss of activity, and leaching was not observed in the subsequent runs by ICP-AES analysis, so that no regeneration was required at the end of the reaction [35]. The stability of the catalyst is also indicated by the similarity of the FTIR spectra (Fig. 1d).
Possible reaction mechanism
In recent years, many researchers have investigated the mechanistic aspects of the molybdenum(VI)-catalyzed epoxidation reactions in the presence of TBHP. The mechanism for the oxidation of various olefins to epoxides using the catalyst [MoO2HL(HOCH3)] has been proposed by comparison with the literature report [23]. According to the suggested mechanism (Scheme 2), the second stage of the process is the interaction between the olefin and the TBHP molecule, activated in the coordination sphere of the molybdenum complex. It is well known that early transition metal ions in their highest oxidation states such as Mo(VI) tend to be stable toward changes in their oxidation state. Thus, in epoxidation reactions with alkyl hydroperoxides, they form adducts (MOOR) which are the key intermediates in the epoxidation and the role of the metal ion is that of a Lewis acid site. The metal center acts as a Lewis acid by removing charge from the O–O bond, facilitating its dissociation and activating the proximal O atom for insertion into the olefin double bond, whereas the distal oxygen constitutes a good leaving group in the form of –OtBu (see Scheme 2).

Probable mechanism of epoxidation of cyclooctene with TBHP in the presence of [MoO2(HL)(HOCH3)]
Conclusion
In summary, in the present study, we demonstrated that [MoO2HL(HOCH3)] as homogeneous catalyst and Fe3O4@SiO2/[MoO2L(HOCH3)] as heterogeneous catalyst both promote oxidation reactions efficiently in association with tert-butylhydroperoxide as an oxidant. The oxide yields show that the efficiency of the catalytic system is strongly dependent on the temperature and nature of solvent, and the best yields were achieved in 1,2-dichloroethane and at 80 °C. The supported catalyst Fe3O4@SiO2/[MoO2L(HOCH3)] gave lower activity and turnover number compared to its homogeneous counterpart. However, the supported catalyst was easily separated from the reaction mixture by the use of a magnet and could be recycled for several catalytic runs, without significant loss of activity. This system could prove useful for many other base-catalyzed oxidations.
References
Maurya MR (2012) Curr Org Chem 16:73–88
Bagherzadeh M, Tahsini L, Latifi R, Woo LK (2009) Inorg Chim Acta 362:3698–3702
Javadi MM, Moghadam M, Mohammadpoor-Baltork I, Tangestaninejad S, Mirkhani V, Kargar H, Nawaz Tahir M (2014) Polyhedron 72:19–26
Kuhn FE, Santos AM, Abrantes M (2006) Chem Rev 106:2455–2475
Ghorbanloo M, Ghamari S, Shahbakhsh N, Ng SW (2014) J Braz Chem Soc 25:2073–2079
Clark JH (1994) Supported reagents in organic reactions. VCH, Weinheim
Amini M, Mehdi Haghdoost M, Bagherzadeh M (2013) Coord. Chem Rev 257:1093–1121
Klencsar Z (2013) Hyperfine Interact 217:117–126
Heinert D, Martell AE (1962) J Am Chem Soc 84:3257–3263
Bagherzadeh M, Zare M, Amani V, Ellern A, Keith Woo L (2013) Polyhedron 53:223–229
Wang J, Zheng S, Shao Y, Liu J, Xu Z, Zhu D (2010) J Colloid Interface Sci 349:293–299
Deng YH, Wang CC, Hu JH, Yang WL, Fu SK (2005) Colloid Surf A 262:87–93
Saeedi MS, Tangestaninejad S, Moghadam M, Mirkhani V, Mohammadpoor-Baltork I, Khosropour AR (2013) Polyhedron 49:158–166
Nakamoto K (1986) Infrared and raman spectra of inorganic and coordination compounds. Wiley, New York
Topich J (1981) Inorg Chem 20:3704–3707
Zhang X, Niu H, Pan Y, Shi Y, Cai Y (2011) J Colloid Interface Sci 362:107–112
De Grave E, Persoons RM, Vanderberghe RE, Bakker PMA (1993) Phys Rev B 47:5881
Goya GF, Berquo TS, Fonseca FC, Morales MP (2003) J Appl Phys 94:3520–3527
Parida KM, Sahoo M, Singha S (2010) J Catal 276:161–169
Maiti SK, Dinda S, Gharah N, Bhattacharyya R (2006) New J Chem 30:479–489
Zhang Y, Zhen L, Sun W, Xia C (2008) Catal Commun 10:237–242
Fernandes CI, Carvalho MD, Ferreira LP, Nunes CD, Vaz PD (2014) J Organomet Chem 760:2–10
Li Y, Fu X, Gong B, Zou X, Tu X, Chen J (2010) J Mol Catal A: Chem 322:55–62
Sheldon RA (1973) Vav Doorn JA. J Catal 31:427–437
Masteri-Farahani M, Farzaneh F, Ghandi M (2006) J Mol Catal A: Chem 248:53–60
Deng Y, Cai Y, Sun Z, Liu J, Liu C, Wei J, Li W, Liu C, Wang Y, Zhao D (2010) J Am Chem Soc 132:8466–8473
Grivani G, Tangestaninejad S, Halili A (2007) Inorg Chem Commun 10:914–917
Kuhn FE, Groarke M, Bencze E, Herdtweck E, Prazeres A, Santos AM, Calhorda MJ, Romao CC, Lopes AD, Pillinger M, Goncalves IS (2002) Chem Eur J 8:2370–2383
Choudhary VR, Jha R, Jana P (2008) Catal Commun 10:205–207
Mohammadikish M, Masteri-Farahani M, Mahdavi S (2014) J Magn Magn Mater 354:317–323
Silva NU, Fernandes CI, Nunes TG, Saraivaa MS, Nunes Cd, Vaz PD (2011) Appl Catal A 408:105–116
Moradi-Shoeili Z, Boghaei DM, Amini M, Bagherzadeh M, Notash B (2013) Inorg Chem Commun 27:26–30
Rayati S, Rafiee N, Wojtczak A (2012) Inorg Chim Acta 386:27–35
Shylesh S, Schweizer J, Demeshko S, Schunemann V, Ernst S, Thiel WR (2009) Adv Synth Catal 351:1789–1795
Nakayama N, Tsuchiya S, Ogawa S (2007) J Mol Catal A 277:61–71
Acknowledgments
Authors are thankful to University of Zanjan for financial support of this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ghorbanloo, M., Mohamadi, A., Amini, M. et al. Use of a molybdenum(VI) dioxide complex as a homogeneous and heterogeneous magnetically recoverable epoxidation catalyst. Transition Met Chem 40, 321–331 (2015). https://doi.org/10.1007/s11243-015-9920-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-015-9920-7