
Abstract
This study reports the usage of molybdovanadophosphoric acid catalyst on amino-functionalized SBA-15(PMoV2/SBA-15-NH2) for the removal of sulfur from model oil (dibenzothiophene dissolved in n-hexane). To increase the tendency for adsorption of heteropoly acids, mesoporous SBA-15 silica was functionalized with amino groups by postsynthesis grafting, using 3-aminopropyltrimethoxy silane as the coupling agent. Immobilization of molybdovanadophosphoric acid on pure SBA-15 (PMoV2/SBA-15) was also studied for comparison and the catalysts were characterized by physicochemical and spectroscopic methods. It was found that the catalysts exhibit high catalytic activities and PMoV2/SBA-15-NH2 is more durable than PMoV2 impregnated on unmodified mesoporous SBA-15 silica. The results may bring about improvement for oxidative desulfurization of transportation fuels.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Sulfur removal from fuels has become a very important and active research subject worldwide in the past decade since the production and use of environmentally friendly fuels have been attracting increasing attention in many countries. Furthermore, the regulations for sulfur contents in the liquid transportation fuels are becoming more and more stringent [1]. Therefore, deep desulfurization has become an urgent issue for the petroleum refining industry [2]. At present, hydrodesulfurization (HDS) is a very useful conventional method for the removal of aliphatic and alicyclic sulfur-containing compounds in the petroleum refining industry. However, it has its limit on the desulfurization of dibenzothiophene (DBT), benzothiophene (BT) and 4,6-dimethyldibenzothiophene (4,6-DBT) due to the steric hindrance effect of these compounds [3, 4]. To achieve ultra-low sulfur fuels, more severe conditions are required, such as higher temperature, higher hydrogen pressure and catalysts with higher activity, inevitably leading to higher operating cost. Therefore, it is desirable to develop some alternatives, such as oxidative desulfurization (ODS) [5, 6], adsorptive desulfurization [7], extractive desulfurization [8] and bio-desulfurization [9], to obtain ultra clean fuels. Because of moderate reaction conditions, high reactivity and no hydrogen requirement, oxidative desulfurization turned out to be one of the most promising desulfurization processes [10, 11]. In the ODS process, DBT and its derivatives can be selectively oxidized to their corresponding sulfoxides and sulfones, which can be then removed by extraction using water-soluble polar solvents as the extractants [12].
Various oxidants, including H2O2 [13], organic peroxide [14], potassium superoxide [15], molecular oxygen [10], ozone [16], and nitric acid/NO2 [17], were investigated in ODS processes. Among them, H2O2 has been widely used due to its high reactivity and affordable cost. Moreover, the only product is water in the ODS process. Heteropoly acid (HPA) catalysts in H2O2 oxidation systems have also exhibited high catalytic activity for the oxidation of BTs and DBTs. HPA has been widely used as a catalyst in the oxidative desulfurization process due to its high catalytic activity for the oxidation of benzothiophene and its derivatives [18, 19]. However, the challenges for these catalysts are the difficulties in separation and recovery. Therefore, supported catalysts have been used in the ODS process to overcome these disadvantages. Yazu et al. [11] immobilized phosphotungstic acid on an anion-exchange resin to catalyze the oxidation of DBT with H2O2 as the oxidant. Results showed that the supported catalyst exhibited higher catalytic activities and could be easily recovered. Yan et al. [20] investigated the oxidation of DBT with H2O2 using phosphotungstic acid/TiO2 as the catalyst. The selective desulfurization ratio reaches to 95.2 % under optimal conditions. Yang et al. [21] studied the supported phosphotungstic acid for the oxidative desulfurization and the catalyst turned out to be of high activity and easily recovered. Although immobilization of heteropolyacids on a suitable support can make them easily separated from the reaction mixture, weak bonding interaction of heteropolyanion with the silanol group of these silicate materials leads to partial leaching especially with polar solvent media [22, 23].
Herein, in the present work, we report on the molybdovanadophosphoric acid (PMoV2) supported onto mesoporous silica both by adsorption to SBA-15 and by electrostatic binding to SBA-15 modified with amino groups (SBA-15-NH2). Influences of reaction conditions, such as reaction time and temperature, catalyst amount, the molar ratio of H2O2 to DBT (n(O)/n(S)), were also investigated in detail. The results may bring some improvement in the application of oxidative desulfurization.
Experimental
Chemicals
The block copolymer EO20PO70EO20 (Pluronic P123) was purchased from Aldrich Corporation (USA). Tetraethoxysilane (TEOS), dibenzothiophene (DBT, 99 %) and 3-aminopropyltriethoxysilane were purchased from Acros Organics (USA). Disodium phosphate dodecahydrate (Na2HPO4·12H2O), hydrogen peroxide (H2O2), sodium metavanadate (NaVO3), sodium molybdate dihydrate (Na2MoO4·2H2O), sulfuric acid, diethyl ether,toluene and acetonitrile were purchased from Sinopharm Chemical Reagent Co., Ltd. All chemicals were of analytical grade and used without any further purification.
Catalyst preparation
Synthesis of molybdovanadophosphoric acid and SBA-15
The pure siliceous SBA-15 and molybdovanadophosphoric acid were synthesized according to the procedures described previously, respectively [24, 25].
Synthesis of SBA-15-NH2
Amine-functionalized SBA-15 was synthesized by the reaction of SBA-15 with 3-Aminopropyltriethoxysilane (APTES) using a grafting method [26]. In a typical preparation, freshly activated SBA-15 (1.0 g) was reacted with APTES in 50 mL toluene (distilled over sodium and dried) under reflux for 8 h. The resultant white solid was filtered off, washed with dried toluene and then dried under vacuum at room temperature for 12 h. The product was designated as SBA-15-NH2.
Immobilization of PMoV2 on mesoporous support
Molybdovanadophosphoric acid was impregnated on mesoporous SBA-15 support via incipient wetness technique. Mesoporous support (SBA-15, SBA-15-NH2, 1.5 g) was added to an aqueous solution of PMoV2 (1.0 g) with vigorously stirring for 24 h at room temperature. The mixtures were subjected to evaporation and dried to get the solid products. The solid was then dried at room temperature and designated as PMoV2/SBA-15 and PMoV2/SBA-15-NH2.
Characterization of the samples
IR spectra of samples (KBr pellets) were recorded on a Perkin-Elmer infrared spectrometer in the range of 400–4,000 cm−1. X-ray diffraction (XRD) measurements were performed on a D/MAX-RB X-ray diffractometer using a Cu Kα radiation (1.5406 Å) source at 40 kV and 30 mA, from 0.5 to 10° with a scan rate of 0.5°/min. Nitrogen adsorption–desorption data were measured with a BELSORP-Mini II analyzer at 77 K. Prior to the measurement, all samples were degassed under nitrogen atmosphere for 3 h at 200 °C. The surface area was calculated by the Brunauer–Emmett–Teller (BET) method.
Catalytic experiments
Model oil was prepared by dissolving DBT in n-hexane containing 1,000 ppm corresponding to S-content. Catalytic oxidative desulfurization was performed in a round bottom flask in an oil bath under magnetic stirring at atmospheric pressure. The oxidative reaction was carried out with 20 mL of model oil, a certain amount of H2O2 as oxidant and 5 mL of acetonitrile as the extractant, using PMoV2/SBA-15 and PMoV2/SBA-15-NH2 as catalysts, respectively. After the reaction, the mixture was cooled down to room temperature and then the catalyst was centrifuged. The upper reaction samples were withdrawn and subjected to GC-FID analysis. The catalyst was washed with acetonitrile three times, dried and used in the next run.
Results and discussion
Characterization
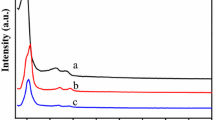
FTIR spectra are quite useful to identify structural and bonding changes in the Keggin unit present in the supported catalysts (The basic Keggin unit is composed of a central XO4 tetrahedron (X = P or Si) surrounded by 12 MO6 octahedra (M = Mo or W)) [27]. As can be seen from Fig. 1, the four IR bands characteristic of neat PMoV2 appeared at 1,050–1,100 cm−1 (P–O stretching), 900–1,000 cm−1 (Mo–O stretching), 850–900 cm−1 (stretching of Mo–O–Mo inter bridges between corner-sharing MoO6 octahedra) and 750–800 cm−1 (stretching of Mo–O–Mo intra bridges between edge-sharing MoO6 octahedral) [28]. For pure SBA-15, a strong absorption band at 1,078 cm−1 assigned to Si–O–Si asymmetric vibrations was observed. Owing to the overlap of Mo–O–Mo and P–O bands with those of Si–O–Si stretching vibrations of mesoporous supports, only the bands corresponding to (Mo–O–Mo and Mo=O) were discernible after immobilization within the supports. As shown in Fig. 1, the reflections related to bulk PMoV2 revealed that the Keggin structure was retained after immobilization. However, the bands at 958 and 859 cm−1 shifted to 938 and 796 cm−1, respectively, after supporting HPA on amine-modified SBA-15. And a new band appeared at 938 cm−1 in PMoV2/SBA-15-NH2 compared with SBA-15-NH2. All of these may be related to the interaction of HPA with the amine groups in the functionalized SBA-15.

IR spectra of various samples: a PMoV2, b SBA-15, c PMoV2/SBA-15, d SBA-15-NH2, e PMoV2/SBA-15-NH2
Small-angle XRD patterns of catalyst samples are illustrated in Fig. 2. As can be seen from Fig. 2a, the three distinct Bragg diffractions, (100), (110) and (200), are characteristics of a highly ordered two-dimensional hexagonal mesoscopic structure of pure SBA-15 [24]. All three reflections are still detectable in Fig. 2b after immobilization of PMoV2, suggesting that the hexagonal pore structure of the support is retained. However, intensities of these characteristic peaks are lower than those of pure SBA-15. This may be caused by the interaction between the mesoporous SBA-15 and PMoV2 Keggin units. A similar pattern is observed for PMoV2 interaction within SBA-15-NH2 (Fig. 2d). This is in agreement with the results in the previous literature [29].

Small-angle XRD patterns of a SBA-15, b PMoV2/SBA-15, c SBA-15-NH2, d PMoV2/SBA-15-NH2
Evaluation of performance of various catalysts
The activities of various catalysts for oxidative desulfurization are compared in Table 1. All reactions were performed with hydrogen peroxide (H2O2) in Table 1. The difference between each entry was the catalyst. H2O2 is essentially used as the oxidant and PMoV2 is the catalyst. In the first three entries (entry 1, 2 and 3), the conversions are similar to each other, and this could also illuminate that the supports actually made no more contributions for catalytic activity. Similar results were found in the previous literatures [31, 32]. The supported catalyst is made to increase the catalytic activity. After being supported on SBA-15, PMoV2/SBA-15 showed lower catalytic activities than PMoV2 which is due to the decrease in surface area. Compared with PMoV2/SBA-15, PMoV2/SBA-15-NH2 exited a similar catalytic activity (98.1 % vs. 97.8 %). Considering the much lower surface area of PMoV2/SBA-15-NH2 than that of PMoV2/SBA-15, it is certain that PMoV2/SBA-15-NH2 showed a higher activity per square meter of catalyst surface than PMoV2/SBA-15.
Catalytic stability and reusability
To assess the stability and reusability, the two different supported catalysts in the oxidative desulfurization were separated by centrifugation, washed off several times after completion of the reaction and dried before being applied to the subsequent run. It can be seen in Fig. 3, during the second run with PMoV2/SBA-15, an 8.7 % reduction of DBT conversion was obtained comparing with the fresh PMoV2/SBA-15. This may be attributed to the leaching of unstable immobilized PMoV2, which caused the reduction in active sites. There is a further slight activity loss in the second recycle using PMoV2/SBA-15-NH2 as the catalyst. However, this significant activity loss is not evidenced with PMoV2/SBA-15-NH2 in the following recycles. It illustrated that PMoV2/SBA-15-NH2 is more durable than PMoV2/SBA-15 in the oxidative desulfurization. The silanol groups, which are present at the surface and inside the channels of the mesoporous SBA-15, reacted with aminosilane molecules and ≡ Si(CH2)3NH3 + was formed [33, 34]. Then, after PMoV2 was supported on the amino-functionalized SBA-15, the salt ≡Si(CH2)3NH3·PMoV2 was formed. It was the chemical interaction between ≡Si(CH2)3NH3 + and PMoV2 that made PMoV2/SBA-15-NH2 more stable than PMoV2/SBA-15. Therefore, PMoV2/SBA-15-NH2 was chosen as the catalyst in the following investigation into the oxidative desulfurization.

Influence of recycle times on the DBT conversion. Reaction conditions: 20 mL model oil, 60 °C, 120 min, n(O)/n(S) = 16, 0.32 of molar ratio of [P]/DBT
Effect of reaction temperature
Reaction temperature is critical to the DBT conversion. Therefore, influence of the reaction temperature on the DBT conversion over the catalyst PMoV2/SBA-15-NH2 was investigated (Fig. 4). Increasing the reaction temperature could enhance the oxidation of DBT; however, this could also accelerate the decomposition of hydrogen peroxide. As can be seen in Fig. 4, the reaction rate increased with temperature. When the temperature was elevated from 30 to 60 °C, a drastic change in the rate of the reaction was found. Obviously, this may be ascribed to the rate of reaction governed by the temperature below 60 °C. However, if the temperature was higher than 60 °C, the DBT conversion rarely changed with further reaction time. This may be related to the decomposing of H2O2 paralleled with the increase in reaction temperature. Therefore, 60 °C was chosen as the reaction temperature.

Influence of reaction temperature on DBT conversion. Reaction conditions: 20 mL model oil, 120 min, n(O)/n(S) = 16, 0.32 of molar ratio of [P]/DBT
Effect of reaction time
Figure 5 shows the influence of reaction time on the DBT conversion over PMoV2/SBA-15-NH2. It was obvious that the conversion increased greatly with the increase in reaction time up to 120 min, and after that, the DBT conversion increased comparatively slowly along with the increase in reaction time. This can be ascribed to the changes in reaction rate with the reaction time. The reaction rate increased quickly during the initial stage of reaction because of the higher concentration of DBT and H2O2. Then with the reaction continued, the reaction rate decreased with the concentration. A slow change could be found between the reaction time of 80 and 120 min. And after the reaction continued for 120 min, the reaction rate was no longer changed, perhaps because hydrogen peroxide was used up. The DBT conversion reached the maximum and did not increase further. Therefore, 120 min was chosen as the optimal reaction time.

Influence of reaction time on DBT conversion. Reaction conditions: 20 mL model oil, 60 °C, n(O)/n(S) = 16, 0.32 of molar ratio of [P]/DBT
Effect of H2O2 amount
The amount of oxidant played an important role in the oxidative desulfurization of DBT. To investigate the influence of H2O2 amount on DBT conversion, oxidative desulfurization of DBT under various n(O)/n(S) was carried out. As can be seen from Fig. 6, the DBT conversion plot curve increased with n(O)/n(S) up to 16 and then the plot curve turned smoothly, which indicated that the DBT conversion slightly changed. However, as shown in scheme 1, in order to oxidize the sulfur-containing compounds to the corresponding sulfones using H2O2 as the oxidant, 2 mol of H2O2 is consumed for 1 mol of sulfur-containing compounds according to the stoichiometric reaction. The optimal n(O)/n(S) is much higher than stoichiometric n(O)/n(S) in DBT oxidation. The large excess of hydrogen peroxide, on the one hand, can be attributed to the thermal decomposition during the reaction. On the other hand, the excess of oxidant could increase the reaction rate [5, 18]. Therefore, with the economy and reaction rate taken into consideration, 16:1 molar ratio of O/S was chosen for further investigation.

Effect of the n(O)/n(S) on DBT conversion. Reaction conditions: 20 mL model oil, 60 °C, 120 min, 0.32 of molar ratio of [P]/DBT

The process of oxidative reaction of DBT using H2O2 as the oxidant
Effect of catalyst dosage
To assess the influence of catalyst dosage on DBT conversion, different catalyst dosages were used and the results are shown in Fig. 7. It was found that the DBT conversion increased with the increasing of molar ratio of catalyst to DBT ([P]/DBT). This may be related to the increase in the total number of catalyst active sites. It was found that 97.8 % DBT conversion was obtained, when the molar ratio reached to 0.32. When [P]/DBT is lower than 0.32, it is unfavorable for the reaction rate. However, if [P]/DBT is further increased to higher than 0.32, the DBT conversion increased comparatively slowly. From an economic point of view, this would be unconducive to the industrial application. Therefore, 0.32 of [P]/DBT was chosen in the investigation.

Influence of catalyst amount on the DBT conversion. Reaction conditions: 20 mL model oil, 60 °C, 120 min, n(O)/n(S) = 16
Conclusions
In this work, the catalyst PMoV2/SBA-15-NH2 was obtained by grafting the molybdovanadophosphoric acid onto the 3-aminopropyltrimethoxy silane-functionalized SBA-15. Such materials can be efficiently used for environmentally friendly heterogeneous catalysis. Compared with PMoV2/SBA-15, PMoV2/SBA-15-NH2 shows good reusability during a six-cycle test due to the strong interaction of PMoV2 and functionalized SBA-15. The results may bring improvements in the application of oxidative desulfurization.
References
Song C, Ma X (2003) New design approaches to ultra-clean diesel fuels by deep desulfurization and deep dearomatization. Appl Catal B: Environ 41:207–238
Song Y, Tsunashima R (2012) Recent advances on polyoxometalate-based molecular and composite materials. Chem Soc Rev 41:7384–7402
Otsuki S, Nonaka T, Takashima N, Qian W, Ishihara A, Imai T, Kabe T (2000) Oxidative desulfurization of light gas oil and vacuum gas oil by oxidation and solvent extraction. Energ Fuel 14:1232–1239
Babich IV, Moulijn JA (2003) Science and technology of novel processes for deep desulfurization of oil refinery streams: a review. Fuel 82:607–631
Collins FM, Lucy AR, Sharp C (1997) Oxidative desulphurisation of oils via hydrogen peroxide and heteropolyanion catalysis. J Mol Catal A: Chem 117:397–403
Li X, Yang Y, Huang S, Xu Q (2009) Preparation of WO3-SBA-15 mesoporous molecular sieve. Trans Met Chem 34:943–947
Kim JH, Ma X, Zhou A, Song C (2006) Ultra-deep desulfurization and denitrogenation of diesel fuel by selective adsorption over three different adsorbents: a study on adsorptive selectivity and mechanism. Catal Today 111:74–83
Zhang S, Zhang Q, Zhang ZC (2003) Extractive desulfurization and denitrogenation of fuels using ionic liquids. Ind Eng Chem Res 43:614–622
Soleimani M, Bassi A, Margaritis A (2007) Biodesulfurization of refractory organic sulfur compounds in fossil fuels. Biotechnol Adv 25:570–596
Lü H, Gao J, Jiang Z, Yang Y, Song B, Li C (2007) Oxidative desulfurization of dibenzothiophene with molecular oxygen using emulsion catalysis. Chem Commun:150–152
Yazu K, Furuya T, Miki K, Ukegawa K (2003) Tungstophosphoric acid-catalyzed oxidative desulfurization of light oil with hydrogen peroxide in a light oil/acetic acid biphasic system. Chem Lett 32:920–921
Ali SH, Hamad DM, Albusairi BH, Fahim MA (2009) Removal of dibenzothiophenes from fuels by oxy-desulfurization. Energ Fuel 23:5986–5994
Li C, Gao J, Jiang Z, Wang S, Lu H, Yang Y, Jing F (2005) Selective oxidations on recoverable catalysts assembled in emulsions. Top Catal 35:169–175
Ishihara A, Wang D, Dumeignil F, Amano H, Qian EW, Kabe T (2006) Oxidative desulfurization and denitrogenation of a light gas oil using an oxidation/adsorption continuous flow process. Appl Catal A: Gen 279:279–287
Chan N, Lin T, Yen T (2008) Superoxides: alternative oxidants for the oxidative desulfurization process. Energ Fuel 22:3326–3328
Zaykina RF, Zaykin YA, Yagudin SG, Fahruddinov IM (2004) Specific approaches to radiation processing of high-sulfuric oil. Radiat Phys Chem 71:467–470
Tam PS, Kittrell JR, Eldridge JW (1990) Desulfurization of fuel oil by oxidation and extraction. 1. Enhancement of extraction oil yield. Ind Eng Chem Res 29:321–324
Te M, Fairbridge C, Ring Z (2001) Oxidation reactivities of dibenzothiophenes in polyoxometalate/H2O2 and formic acid/H2O2 systems. Appl Catal A: Gen 219:267–280
Mei H, Mei B, Yen T (2003) A new method for obtaining ultra-low sulfur diesel fuel via ultrasound assisted oxidative desulfurization. Fuel 82:405–414
Yan X, Mei P, Lei J, Mi Y, Xiong L, Guo L (2009) Synthesis and characterization of mesoporous phosphotungstic acid/TiO2 nanocomposite as a novel oxidative desulfurization catalyst. J Mol Catal A: Chem 304:52–57
Yang L, Qi Y, Yuan X, Shen J, Kim J (2005) Direct synthesis, characterization and catalytic application of SBA-15 containing heteropolyacid H3PW12O40. J Mol Catal A: Chem 229:199–205
Nowinska K, Kaleta W (2000) Synthesis of Bisphenol-A over heteropoly compounds encapsulated into mesoporous molecular sieves. Appl Catal A: Gen 203:91–100
Verhoef MJ, Kooyman PJ, Peters JA, van Bekkum H (1999) A study on the stability of MCM-41-supported heteropoly acids under liquid- and gas-phase esterification conditions. Micropor Mesopor Mat 27:365–371
Zhao D, Huo Q, Feng J, Chmelka BF, Stucky GD (1998) Nonionic triblock and star diblock copolymer and oligomeric surfactant syntheses of highly ordered, hydrothermally stable, mesoporous silica structures. J Am Chem Soc 7863:6024–6036
Tsigdinos GA, Hallada CJ (1968) Molybdovanadophosphoric acids and their salts. I. Investigation of methods of preparation and characterization. Inorg Chem 7:437–441
Johnson BJ, Stein a (2001) Surface modification of mesoporous, macroporous, and amorphous silica with catalytically active polyoxometalate clusters. Inorg Chem 40:801–808
Ganapathy S, Fournier M, Paul JF, Delevoye L, Guelton M, Amoureux JP (2002) Location of protons in anhydrous Keggin Heteropolyacids H3PMo12O40 and H3PW12O40 by 1H{31P}/31P{1H} REDOR NMR and DFT quantum chemical calculations. J Am Chem Soc 124:7821–7828
Mizuno N, Misono M (1998) Heterogeneous catalysis. Chem Rev 98:199–218
Lim MH, Blanford CF, Stein A (1998) Synthesis of ordered microporous silicates with organosulfur surface groups and their applications as solid acid catalysts. Chem Mater 10:467–470
Okuhara T, Mizuno N, Misono M (1996) Catalytic chemistry of heteropoly compounds. Adv Catal 41:113–252. doi:10.1016/S0360-0564(08)60041-3
Al-Shahrani F, Xiao T, Llewellyn SA, Barri S, Jiang Z, Shi H, Martinie G, Green MLH (2007) Desulfurization of diesel via the H2O2 oxidation of aromatic sulfides to sulfones using a tungstate catalyst. Appl Catal B: Environ 73:311–316
Jiang X, Li H, Zhu W, He L, Shu H, Lu J (2009) Deep desulfurization of fuels catalyzed by surfactant-type decatungstates using H2O2 as oxidant. Fuel 88:431–436
Bordoloi A, Lefebvre F, Halligudi S (2007) Selective oxidation of anthracene using inorganic–organic hybrid materials based on molybdovanadophosphoric acids. J Catal 247:166–175
Kaleta W, Nowinska K (2001) Immobilisation of heteropoly anions in Si-MCM-41 channels by means of chemical bonding to aminosilane groups. Chem Commun:535–536
Acknowledgments
We gratefully acknowledge the financial support of the Nature Science Foundation of Hubei Province of China (2011CHB044) and the Foundation of the Educational Commission of Hubei Province of China (D20121401).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, J., Hu, B., Tan, J. et al. Deep oxidative desulfurization of fuels catalyzed by molybdovanadophosphoric acid on amino-functionalized SBA-15 using hydrogen peroxide as oxidant. Transition Met Chem 38, 495–501 (2013). https://doi.org/10.1007/s11243-013-9716-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-013-9716-6