
Abstract
A series of MoO3/γ-Al2O3 catalysts with different Mo surface densities (Mo atoms/nm2) has been prepared by incipient wetness impregnation method. Structural characteristics of the prepared catalysts were investigated by atomic absorption spectroscopy, X-ray diffraction, Fourier Transform Infrared spectroscopy, N2 adsorption at −196 °C, and temperature-programmed reduction (TPR). The catalytic activities of the prepared catalysts were tested by cyclohexene conversion between 200 and 400 °C. XRD results indicated that molybdenum oxide species were dispersed as a monolayer on the support up to 4.04 Mo atoms/nm2, and the formation of crystalline MoO3 was observed above this loading. FTIR and TPR results showed that molybdenum oxide species were present predominantly in tetrahedral form at lower loading, and polymeric octahedral forms were dominant at higher loading. Cyclohexene conversion reaction proceeded mainly through the simple dehydrogenation pathway in the studied temperature range 200–400 °C and was found to be highly dependent on MoO3 dispersion.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Al2O3-supported MoO3 catalysts have been widely used in a number of catalytic reactions such as hydrogenolysis and hydrodesulfurization of crude oil [1], benzene hydrogenation [2], alkene metathesis reactions [3], and oxidative dehydrogenation of alkanes [4]. The structures of the supported Mo have been extensively investigated utilizing a variety of structural characterization techniques [1, 5–9]. Mo/Al2O3 catalysts are often referred to as monolayer systems, due to the tendency of the active phase to spread over the support at elevated temperature. In this way, an overlayer of MoOx is formed on the support surface, and the surface free energy of the system is minimized. As Mo surface densities exceed “monolayer” coverage, crystalline MoO3 forms at low treatment temperatures (<873 K) and Al2(MoO4)3 at higher temperatures [5].
Cyclohexene (CHE) conversion is an important probe catalytic reaction. CHE undergoes various transformations depending on the catalyst composition and the experimental conditions [10, 11]. In the absence of hydrogen, disproportionation and dehydrogenation take place. Disproportionation of CHE gives benzene (BZ) and cyclohexane (CHA). This reaction is termed hydrogen transfer when carried out catalytically, since CHE behaves as both hydrogen donor and acceptor [12, 13].
In this work, a systematic study of a series of MoO3/Al2O3 catalysts with different Mo surface densities was performed. A detailed characterization (atomic absorption spectroscopy, XRD, N2 physisorption, FTIR, TPR) of their textures and structures was performed. The catalytic performance of the MoO3/Al2O3 catalysts for CHE conversion at 200–400 °C was carried out using pulse technique in the absence of hydrogen and was correlated to the dispersion and the surface structure of MoO3/Al2O3 catalysts.
Experimental
Catalyst preparation
A series of MoO3/γ-Al2O3 catalysts was prepared by incipient wetness impregnation of γ-Al2O3 (treated at 500 °C for 3 h before use, 162.9 m2g−1, 0.32 cm3g−1) with an aqueous solution of ammonium heptamolybdate, followed by drying at 120 °C for 12 h and calcination at 450 °C in air for 4 h. The catalysts thus prepared were designated as MoAlx where x refers to the Mo surface density in Mo atoms/nm2. MoAl0 refers to the γ-Al2O3 support impregnated with deionized water and subjected to the same treatment used to prepare MoO3/γ-Al2O3 catalysts.
Characterization
The molybdenum content was determined by atomic absorption spectroscopy (AAS, Perkin-Elmer 2380 apparatus). The XRD patterns were recorded on a Philips diffractometer model PW 150 with CuKα radiation at 30 kV and 40 mA. The specific surface area (S BET, m2g−1) of catalyst samples was measured by the BET method with a Quantachrome Nova 3200 S automates gas sorption apparatus using nitrogen adsorption at −196 °C. Prior to the adsorption measurements, the samples were degassed at 300 °C for 2 h under vacuum. The corrected surface area (S corr., m2g−1) was calculated assuming that the surface area of the catalyst is only due to the γ-Al2O3. It is obtained by determining the amount of γ-Al2O3 per g catalyst from chemical composition and normalizing the surface area to that amount [14]. The FT-IR spectra of the samples were recorded with a Shimadzu FTIR spectro-photometer in KBr matrix in the range of 4,000–400 cm−1. Temperature-programmed reduction (TPR) experiments were conducted on a CHEM-BET 3000 TPR instrument. TPR profiles were taken from ambient temperature to 1,000 °C (10 °C/min). A 10% vol. H2 in Ar mixture at a flow rate of 50 mL min−1 was used as the reducing gas.
Catalyst performance studies
Catalytic dehydrogenation of cyclohexene (CHE) was carried out in a pulse fixed-bed, vertical flow type reactor attached in line with a gas chromatograph (Clarus 500 gas chromatograph) with FID detector equipped with a column made of a stainless steel cylinder of 3 mm inside diameter and 4 m long and packed with 5 wt% Bentone-34 and 5 wt% diisodecyl-phthalate on chemsorb (35–80 mesh), 1 μL of CHE was injected over 0.1 g of the preactivated catalyst at 400 °C using N2 as a carrier gas (50 mL min−1). The activities of molybdena-supported catalysts were estimated by varying the temperature from 200 to 400 °C, with 50 °C decrements starting from 400 °C downwards.
Results and discussion
Catalyst characterization
Atomic absorption spectroscopy (AAS)
The actual Mo loadings determined by AAS are given in Table 1. The experimental values were found to be in good agreement with the theoretical ones.
The Mo surface densities of the supported MoO3/γ-Al2O3 catalysts are expressed as the number of Mo atoms per square nanometer of surface area (Mo atoms/nm2) [15]. They were calculated from the actual molybdena content and the corrected surface area of the alumina support and are given in Table 1.
X-ray diffraction (XRD)
The XRD patterns of the MoO3/γ-Al2O3 catalysts with different Mo surface densities are shown in Fig. 1; bulk MoO3 and γ-Al2O3 are also given for comparison. All samples exhibited broad peaks corresponding to microcrystalline γ-Al2O3 (JCPDS No. 10–425). No additional diffraction lines corresponding to crystalline molybdenum containing compounds were detected in samples with Mo surface density up to 4.04 Mo atoms/nm2, indicating that molybdenum species were either highly dispersed or present as MoO3 crystallites having sizes less than 4 nm, which is beyond the detection capacity of XRD. On the other hand, higher Mo surface densities led to the appearance of diffraction lines at 2θ of 27.3°, 25.7°, and 23.3° which are attributed to the [021], [040], and [110] crystallographic planes of the orthorhombic MoO3 phase (JCPDS No. 35–609). The intensity of the diffraction lines ascribed to MoO3 increases with Mo surface density, indicating the formation of crystalline MoO3 at high Mo surface density.

XRD patterns for the series of MoO3/γ-Al2O3 catalysts
The dispersion capacity of MoO3 on the surface of γ-Al2O3 was measured by XRD quantitative phase analysis. XRD quantitative analysis was carried out by plotting the area ratio of crystalline MoO3 peak at 27.3° and support peak at 67° \( \left( {{{I_{{{\text{MoO}}_{3} }} } \mathord{\left/ {\vphantom {{I_{{{\text{MoO}}_{3} }} } {I_{{{\text{Al}}_{2} {\text{O}}_{3} }} }}} \right. \kern-\nulldelimiterspace} {I_{{{\text{Al}}_{2} {\text{O}}_{3} }} }}} \right) \) as a function of Mo surface density (Fig. 2). The resultant straight line gives an intercept with the horizontal axis corresponding to the dispersion capacity of MoO3 on the surface of γ-Al2O3 [16]. The result indicated that the dispersion capacity of molybdena on γ-Al2O3 is about 4.04 Mo atoms/nm2.

Determination of dispersion capacity of MoO3 on γ-Al2O3 through quantitative XRD
Specific surface areas
The computed values of S BET of γ-Al2O3 and MoO3/γ-Al2O3 catalysts and the corrected surface area of the γ-Al2O3 support are cited in Table 1. The corrected surface areas remained constant up to 4.04 Mo atoms/nm2 and then decreased with further increasing of Mo surface density. This indicated that Mo incorporation up to 4.04 Mo atoms/nm2 did not lead to the formation of large bulky species, which may block the fine pores of the alumina support; in other words, oxide metal distribution is well dispersed on the surface of support probably as monolayer. This agrees with the absence of XRD lines of crystalline molybdenum species on these catalysts. However, at higher Mo surface densities, formation of crystalline MoO3 phase which partially blocks up the pores of alumina could be responsible of the decrease in the corrected surface areas of the catalysts. For these catalysts, the XRD profiles provided evidence of the presence of MoO3 crystals on the support surface.
FTIR spectroscopy
The FTIR spectra for MoO3/γ-Al2O3 catalysts are shown in Fig. 3. Examination of Fig. 3 shows the following: (i) for MoAl1.0 sample, the band at 914 cm−1 characterizes the stretching mode of the Mo=O bond in surface-bound Mo species. These species may be either isolated tetrahedral or octahedral polymolybdate species. The absence of a band due to bridged bonds in the lower wave number region indicates that these species are isolated tetrahedral species [1, 17], (ii) for catalyst samples MoAl2.0, MoAl3.0, and MoAl4.0, the band due to stretching is observed at 950 cm−1. The appearance of a new band at 650 cm−1 is ascribed to the characteristic bridged Mo–O–Mo bonds indicates the formation of octahedral polymolybdate species in addition to the isolated tetrahedral species [1, 17, 18]. The increase in the intensity of this band with MoO3 loading indicates the growth of the polymolybdate species. The dispersed nature of MoO x species at these low surface densities is consistent with the absence of crystalline structures in their XRD patterns, (iii) at higher loadings, the bands due to microcrystallites MoO3 appears at 600, 830, and 880 cm−1 [17]. The intensity of these bands increased with increasing molybdenum loading. These results are in line with XRD analysis, which indicates the formation of microcrystallites MoO3 at Mo loadings exceeding monolayer content.

FTIR spectra of catalysts with different MoO3 contents: a MoAl1.0; b MoAl2.0; c MoAl3.0; d MoAl4.0; e MoA5.6 l; f MoAl7.2; g MoAl9.3
Temperature-programmed reduction (TPR)
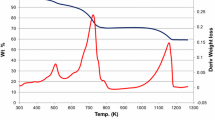
The TPR profiles of γ-Al2O3 as well as ΜoAlx catalysts are shown in Fig. 4. The support has very low reducibility. It gives a very small hump at around 529 °C, indicating that the support contribution to the reduction profiles is negligible throughout the temperature range studied.

TPR profiles of γ-Al2O3 and supported molybdenum oxide catalysts
As can be seen in Fig. 4, in the case of supported molybdenum catalysts, two main reduction peaks are easily distinguished. It is well known that at low Mo surface densities, molybdenum oxide is present predominantly as tetrahedral species which are difficult to reduce due to strong interaction with the support [20, 21]. As the Mo surface density increases, both tetrahedral and octahedral species coexist on the support surface. It is also known that octahedral and other higher polyhedral species are reduced relatively easily [20, 22]. The less polarized bonds of polymolybdates are more easily reduced compared to those directly bonded to alumina [23]. Further at very high Mo surface density, in addition to the two above-mentioned species, crystalline MoO3 is present [23]. It has been found by FTIR that the polymolybdates are reduced in H2 previously to free MoO3. Consequently, the low temperature peak (419–485 °C) could be assigned to the partial reduction of Mo+6 to Mo+4 (MoO3→MoO2) in dispersed octahedral species, whereas the high temperature peak (787–850 °C) may be due to a further progress in the reduction of partially reduced MoOx species formed in the first reduction peak Mo4+ to Mo0 (MoO2→Mo0), together with the partial reduction of tetrahedrally coordinated Mo species. The third small peak in the TPR plot of MoAl5.6 is attributed to the reduction of small MoO3 crystallites.
The peak positions and quantitative hydrogen consumption data of MoO3/γ-Al2O3 catalysts are shown in Table 2. It can be noted that: (i) the hydrogen consumption per gram of catalyst as well as per gram of Mo was found to increase with increase in molybdena loading, indicating an increase in the reducibility of molybdenum with increased Mo loading, (ii) the H2/MoO3 molar ratio was found to be lower than the theoretical value (3), indicating that the reduction is not complete under the conditions employed. However, in the case of high loadings, the value increases but is still lower than the theoretical value indicating that some metallic-oxo species are partially sublimated (i.e., melting point of MoO3 is 795 °C) or not completely reduced during the TPR analysis because of strong interaction with the support [24].
Catalyst performance studies
The catalytic dehydrogenation of CHE was carried out using MoO3/γ-Al2O3 catalysts with different Mo surface densities in the temperature range of 200–400 °C. The conversion and product distributions of catalysts at different reaction temperatures are given in Table 3. According to the catalytic activity measurements, BZ was the major product, meanwhile CHA, 1-methyl cyclopentene (1-MCP), and low molecular weight hydrocarbons were also observed due to CHE disproportionation, isomerization, and hydrogenolysis, respectively. Moreover, BZ was found to be the sole dehydrogenation product of CHE under the reaction conditions. No other dehydrogenation products such as 1,3- or 1,4- cyclohexadiene were detected.
As can be seen from Table 3, both the conversion of CHE and the BZ yield were found to increase with temperature and reached about 85.6 and 67.2%, respectively, at 400 °C when using MoAl4.0 as the catalyst. They also increased with increasing Mo surface density up to 4.40 Mo atom/nm2 and then decreased with further increase in Mo surface density. The highest activity, where the CHE conversion and the BZ yield were, was obtained with catalyst, which contained an amount of molybdenum closely corresponding to a monolayer on the alumina surface (MoAl4.0). When pure alumina was used as a catalyst for the reaction, only 15.58% CHE conversion was obtained with 9.9% BZ yield at 400 °C. When MoAl4.0 was used as catalyst, 85.62% CHE conversion was obtained with 67.2% BZ yield at 400 °C. The role of catalyst in this reaction is thus clearly evident. This suggests that nanosized molybdenum-oxo species uniformly dispersed on alumina are the main active species for CHE dehydrogenation to BZ. The decrease in activity of the catalysts beyond 4.40 Mo atom/nm2 is due to the decrease in the dispersion of the metal species and the formation of bulk MoO3 crystallites. XRD, FTIR, and TPR also supported the activity results. This agrees with Rioux et al. [25] that the dehydrogenation of CHE is preferred on small particles and is predominant at high dispersion.
Beside BZ, CHA was also produced due to CHE disproportionation. It has been reported that, in the absence of hydrogen, CHE will undergo two competitive pathways: disproportionation to BZ and CHA [Eq. 1]; this reaction is termed as hydrogen transfer reaction (HT), since CHE acts as hydrogen donor and acceptor. In this reaction, hydrogen redistributes among three CHE molecules to form two molecules of CHA and one of BZ [12, 13]. The second pathway is the dehydrogenation of CHE to BZ and hydrogen [Eq. 2]. 

Both pathways can occur simultaneously to a varying extent depending upon the catalyst type and the experimental conditions [13].
Our results clearly showed that CHE conversion proceeded predominately through the simple dehydrogenation pathway for all these catalysts over the entire temperature range (200–400 °C) as revealed from the high yield of BZ when compared to that of CHA (Table 3). Moreover, dehydrogenation increased but disproportionation decreased continually with increasing temperature (Table 3), indicating that the degree of prevalence of dehydrogenation over disproportionation increases continually with temperature. These results are in accordance with those obtained by Ahmed and Chowdhury [26] who investigated the dehydrogenation reaction of CHE on Pt/alumina catalyst in the temperature range 200–340 °C and found that dehydrogenation was the main reaction in this temperature range, and a small fraction of CHE was disproportionating.
Aramendïa et al. [11] studied the transformation of CHE on palladium catalysts over a broad temperature range. They found that CHE transformation took place via two competitive processes: disproportionation and dehydrogenation, where disproportionation prevailed at low temperatures and dehydrogenation at high temperatures.
The prevalence of dehydrogenation over disproportionation can be interpreted as follows: hydrogen produced from CHE dehydrogenation to BZ must be removed from the catalyst surface to complete the catalytic cycle [27]. There are three possible pathways for hydrogen removal [27]: release of hydrogen molecule (pure dehydrogenation reaction), hydrogenation of CHE to CHA (the overall reaction is termed as disproportionation reaction), and formation of smaller hydrocarbons (hydrogenolysis of CHE). So, it can be said that the disproportionation of CHE to CHA and BZ consists of two successive processes: the dehydrogenation of CHE into BZ and the following hydrogenation of another CHE [27]. In other words, in disproportionation, CHE is dehydrogenated to BZ, and the hydrogen formed does not desorb but is consumed in CHE hydrogenation to CHA. Therefore, the role of the catalyst is hydrogen transportation [28]. Temperature is the most important factor governing the catalyst ability toward hydrogen utilization in the hydrogenation of CHE to CHA. Rebhan and Haensel [13] studied CHE conversion using group VIII metals and observed a critical temperature; they called it the “transition temperature”, below which hydrogen would adsorb irreversibly and be utilized to the fullest extent in the hydrogenation of CHE to CHA. Thus, below this temperature, no free hydrogen was detected in the gas phase. Above the critical temperature, hydrogen adsorption would occur reversibly and be in equilibrium with the gas phase. Thus, with increasing temperature, the utilization of hydrogen would drop, consequently, disproportionation declines. Triwahyono et al. [29] studied hydrogen adsorption over Pt/MoO3 and Pt-free MoO3. They found that the hydrogen uptake was scarcely appreciable on Pt-free MoO3 where the presence of specific sites such as Pt is required in the hydrogen adsorption on MoO3 type catalyst and that hydrogen uptake increased with the increase in the adsorption temperature up to 150 °C and decreased at the adsorption temperatures above 150 °C. Resuming above, the prevalence of dehydrogenation over disproportionation of CHE over ΜoΟ3/γ-Al2O3 catalysts may result from their low ability toward hydrogen adsorption and the high temperature range used throughout the reaction experiments.
There could be another reason, in addition to those mentioned above, for the low yield of CHA compared to that of BZ in the studied temperature range. It is also possible that CHA originally formed from disproportionation of CHE was dehydrogenated to BZ [30]. This can be supported by the results of Riad and Mikhail [31] who found CHA dehydrogenation to take place over molybdenum catalysts supported on alumina using the same pulse microcatalytic reactor in a temperature range comparable to that used in the present study.
In addition to BZ and CHA, small amounts of 1-MCP were also formed. 1-MCP formation was attributed to skeletal isomerisation of CHE. Skeletal isomerisation of CHE passes through carbonium ion formation, thus depends on the catalyst acidity [32]. Brønsted acid sites are mainly responsible for the CHE skeletal isomerization reaction [33]. Since γ-Al2O3 is used as a support for the catalysts investigated throughout, this work has low surface acidity; accordingly, 1-MCP yield is low. Figure 5 represents CHE skeletal isomerisation mechanism. For all catalyst samples, the 1–3 hydride shift that changes the tertiary carbenium ion I into a secondary ion (IV) is not as easy as corresponds to their low surface acidity [34]. So, the 3-MCP formation on the catalyst samples is difficult and thus is not observed in the present experimental conditions. The data in Table 3 showed that the yield of 1-MCP increases with increasing reaction temperature to reach a maximum at temperature of about 300 °C, beyond which it decreases with a further increase in temperature. From the Table, it is also clear that 1-MCP yield increased continually with increasing Mo surface density; this may be attributed to the generation of Brønsted acid sites in catalysts with high molybdenum surface density. Formation of Brønsted acid sites in alumina-supported catalysts with high molybdenum oxide content was observed previously by Rodriguez-Ramos et al. [35].

Cyclohexene skeletal isomerisation mechanism [34]
During the dehydrogenation of CHE, lower molecular weight hydrocarbons such as methane, ethane and propane have been produced due to hydrogenolysis reactions, and their amounts depended on the reaction temperature (Table 3). The hydrogenolysis activities of the catalysts are low as corresponds to the low acidity of the γ-Al2O3 used as a support. The yield of hydrogenolysis products increased with Mo surface density due to the increased acidity of the catalysts.
Conclusions
The activity of Mo catalysts supported on alumina with different molybdenum surface densities was studied in the dehydrogenation of cyclohexene at different reaction temperatures. The results indicated that the activity of the catalysts increased with molybdenum surface density up to 4.04 Mo atom/nm2. The highest conversion and BZ yield were obtained when the catalyst contained an amount of molybdenum closely corresponding to a monolayer on the alumina surface. XRD results suggest that below monolayer level MoO3 exists in a highly dispersed amorphous state and above this loading crystalline MoO3 can be seen. FTIR spectroscopy results suggested that the tetrahedral molybdenum-oxo species is present at lower loadings, and the octahedral and crystalline MoO3 particles are present at higher MoO3 loading in accordance with XRD data. CHA yield was found to be low due to low disproportionation reaction as a result of high temperature. The yields of isomerization/hydrogenolysis products were also low due to low acidity of these catalysts.
References
Duan A, Wan G, Zhao Z, Xu C, Zheng Y, Zhang Y, Dou T, Bao X, Chung K (2007) Catal Today 119:13–18
Quincy RB, Houalla M, Proctor A, Hercules DM (1990) J Phys Chem 94:1520–1526
Handzlik J, Shiga A, Kondziołka J (2008) J Mol Catal A 284:8–15
Haddad N, Bordes-Richard E, Barama A (2009) Catal Today 142:215–219
Bergwerff JA, Jansen M, Leliveld B(R)G, Visser T, de Jong KP, Weckhuysen BM (2006) J Catal 243:292–302
Heracleous E, Lee AF, Vasalos IA, Lemonidou AA (2003) Catal Lett 88:47–53
Abello MC, Gomez MF, Ferretti O (2001) Appl Catal A 207:421–431
Hensen EJM, Kooyman PJ, van der Meer Y, van der Kraan AM, de Beer VHJ, van Veen JAR, van Santen RA (2001) J Catal 199:224–235
Gong J, Ma X, Wang S, Yang X, Wang G, Wen S (2004) React Kinet Catal Lett 83:113–120
Aboul-Gheit AK, Abdel-Hamid SM, Aboul-Fotouh SM, Aboul-Gheit NAK (2006) J Chin Chem Soc 53:793–802
Aramendïa MA, Boráu V, García IM, Jiménez C, Marinas A, Marinas JM, Urbano FJ (2000) J Mol Catal A 151:261–269
Boudart M, McConica CM (1989) J Catal 117:33–41
Rebhan DM, Haensel V (1988) J Catal 111:397–408
Chen X, Clet G, Thomas K, Houalla M (2010) J Catal 273:236–244
Nasser H, Rédey Á, Yuzhakova T, Kovács J (2009) J Therm Anal Calorim 95:69–74
Wang X, Zhao B, Jiang D, Xie Y (1999) Appl Catal A 188:201–209
Imamura S, Sasaki H, Shono M, Kanaiy H (1998) J Catal 177:72–81
Mortimer R, Powell JG, Greenblatt M, McCarroll WH, Ramanujachary KV (1993) J Chem Soc 89:3603–3609 Faraday Trans
Kumaran GM, Garg S, Soni K, Kumar M, Sharma LD, Rao KSR, Dhar GM (2007) Ind Eng Chem Res 46:4747–4754
Aberuagba F, Kumar M, Muralidhar G, Sharma LD (2004) Pet Sci Technol 22:1287–1298
Chary KVR, Reddy KR, Kishan G, Niemantsverdriet JW, Mestl G (2004) J Catal 226:283–291
Maity SK, Rana MS, Srinivas BN, Bej SK, Muralidhar G, Rao TSRP (2000) J Mol Catal A 153:121–127
Damyanova S, Petrov L, Centeno MA, Grange P (2002) Appl Catal A 224:271–284
Gonzàlez-Cortés SL, Xiao T, Lin T, Green MLH (2006) Appl Catal A 302:264–273
Rioux RM, Hsu BB, Grass ME, Song H, Somorjai GA (2008) Catal Lett 129:10–19
Ahmed K, Chowdhury HM (1992) Chem Eng J 50:165–168
Amano H, Sato S, Takahashi R, Sodesawa T (2001) Phys Chem Chem Phys 3:873–879
Díaz E, Adrio G, Ordóñez S, Vega A, Coca J (2004) Catal Lett 96:169–175
Triwahyono S, Abdul Jalil A, Timmiati SN, Ruslan NN, Hattori H (2010) Appl Catal A 372:103–107
Aramendïa MA, Borau V, Jiménez C, Marinas JM, Sempere ME (1987) J Catal 108:487–490
Riad M, Mikhail S (2008) Catal Comm 9:1398–1403
Keller V, Barath F, Maire G (2000) J Catal 189:269–280
Navio JA, Colo′n G, Macias M, Campelo JM, Romero AA, Marinas JM (1998) J Mol Catal 135:155–162
Campelo JM, Garcia A, Gutierredz JM, Luna D, Marinas JM (1984) Can J Chem 62:1455–1458
Rodriguez-Ramos I, Guerrero-Ruiz A, Horns N, Rtirez de la Piscina P, Fierro JLG (1995) J Mol Catal 95:147–154
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
El Kady, F.Y.A., Shaban, S.A. & Abo El Naga, A.O. Catalytic dehydrogenation of cyclohexene over MoO3/γ-Al2O3 catalysts. Transition Met Chem 36, 237–244 (2011). https://doi.org/10.1007/s11243-011-9461-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-011-9461-7