
Abstract
A mononuclear complex [CuL] (1), a binuclear complex [Cu2LCl2(H2O)] (2), a trinuclear complex [Cu3L2](ClO4)2 (3) involving o-phenylenediamine and salicylaldehyde and another binuclear complex of a tridentate ligand (H2L1) [Cu2L 12 ](CH3COO)2 (4) involving o-phenylenediamine and diacetylmonoxime have been synthesized, where H2L = N,N′-o-phenylenebis(salicylideneimine) and H2L1 = 3-(2-aminophenylimino)butan-2-one oxime. All the complexes have been characterized by elemental analyses, spectral and magnetic studies. The binuclear complex (2) was characterized structurally where the two Cu(II) centers are connected via an oxygen-bridged arrangement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Present trends in the design, synthesis, isolation and crystal structure characterization of unsymmetrical Schiff base type complexes of transition metals stem from the realization that ligands systems around the central metal ions in many living systems are unsymmetrical. The use of unsymmetrical ligands is more advantageous over their symmetrical analogues to explain the metal ion–binding site in metalloproteins and in efforts to mimic the enzymatic efficiency and selectivity of natural systems with synthetic materials [1–6]. The work in our laboratory with unsymmetrical ligands stems from the above observations [7–11]. Unsymmetrical ligands involving an oxime functionality exhibit an interesting chelating property in the area of complex chemistry [11–14]. The importance of oxime moiety in the ligand environment has been described in detail in our earlier publications where we have introduced an oxime function in the unsymmetrical ligand environment [11, 12]. Nevertheless, we have noted that unsymmetrical Schiff base ligands containing an oxime moiety are quite rare to date, more so with aromatic diamines [15, 16], our interest arises therefore to synthesize an unsymmetrical Schiff base ligand involving o-phenylenediamine as aromatic amine and diacetylmonoxime with salicylaldehyde as the other carbonyl compound. The process applied to synthesize this unsymmetrical ligand was via the single condensation of o-phenylenediamine with salicylaldehyde following the method mentioned in our earlier communication [8].
The free amine group of the single condensed product was then reacted with diacetylmonoxime.
However, it was observed that any attempt to synthesize a Cu(II) complex of the unsymmetrical Schiff base derived from one mole of the said amine with the two above carbonyl compounds, yielded only the symmetrical complex of the Schiff base (H2L) as the sole product. Thus, the present paper is devoted to the synthesis, structural features and spectroscopic characterizations of some Cu(II) complexes of symmetrical bis-Schiff base ligands (H2L).
We had attempted to synthesize the unsymmetrical Schiff base Cu(II) complex involving both the diacetylmonoxime and salicylaldehyde via the high dilution technique in chloroform. In this case also, only complex 2 was isolated. This same compound was isolated as expected involving o-phenylenediamine and salicylaldehyde in 1:2 proportion taking ethanol as solvent. Complex 2 is binuclear with two metal centers brought into combination via an oxygen-bridged structure. Irrespective of the nature of co-anion present, complex 2 separated out when the unsymmetrical variety was attempted to be isolated via single condensation in chloroform medium. But with ethanol as solvent, only the neutral Cu(II) complex 1 separated out as the most stable species with bromide, perchlorate and acetate as counter anion. A trinuclear complex 3 involving an analogous metal environment has been synthesized using a similar method, but with noncoordinating anion, namely perchlorate in place of the halogen atom in stoichiometric molar proportion. Spectrophotometric and elemental analyses support the proposed structures of complex 1, 2 and 3. Several attempts to obtain suitable single crystals of 1 and 3 for structural analysis were futile. Our continuing study reveals that when complex 1 was reacted with CuCl2, again the binuclear complex 2 was synthesized as expected. But several approaches to prepare binuclear bromo variety were unsuccessful.
Results and discussion
Though the initial aim of this work was to synthesize an unsymmetrical Schiff base ligand involving o-phenylenediamine and the corresponding Cu(II) complexes, the products obtained were complexes of a symmetrical Schiff base ligand. The two different carbonyl compounds chosen to synthesize an unsymmetrical Schiff base were salicylaldehyde and diacetylmonoxime. In all the attempts where the above two carbonyl moieties were allowed to react with the diamine, namely o-phenylenediamine, no oxime function gets incorporated into the ligand moiety. However, in the absence of salicylaldehyde, when o-phenylenediamine was reacted with diacetylmonoxime, the tridentate Schiff base with the oxime function (H2L1) could be synthesized yielding a binuclear copper(II) complex 4 (Scheme 1). The composition of this complex was confirmed by mass spectrometric study (found m/z (M+H)+ = 508.0; M calcd = 507.7).
It is therefore observed that the symmetrical tetradentate Schiff base complex 2 with salicylaldehyde was the most stable product that could be isolated as the one and only variety even in the presence of diacetylmonoxime. The reactivity of different carbonyl functions with diamines has been studied by Tan et al. [17], who carried out the synthesis of unsymmetrical Schiff base complexes by the displacement of one carbonyl function from the complexes replacing it with a new carbonyl moiety. We tried in our laboratory to get the unsymmetrical complex from the symmetrical Schiff base complex 1 by replacing salicylaldehyde with diacetylmonoxime, but the symmetrical complex was reluctant to react with diacetylmonoxime.
We have isolated a binuclear complex 2 and a trinuclear complex 3 using CuCl2 and Cu(ClO4)2 as the metal salts, respectively. A 1:1 molar proportion of the ligand and copper(II) perchlorate yielded the mononuclear complex 1, but reaction in 2:3 molar (ligand:metal) ratio gave the trinuclear compound 3. On the other hand, CuBr2 and Cu(CH3COO)2 always yielded the mononuclear variety and no dinuclear or trinuclear species could be isolated even if the reaction was carried out with different stoichiometry of ligand and metal and varying the solvent medium. The dinuclear CuCl2 complex 2 was isolated in chloroform even when Cu(CH3COO)2 was used as the starting material. Probably, the hydrochloric acid present in the undistilled chloroform gave the chloro complex. This was substantiated by using distilled, pure chloroform that yielded monomeric complex 1 with copper(II)acetate. The monomeric complex 1 isolated from CuBr2 and Cu(CH3COO)2 was allowed to react with CuCl2 that yielded the binuclear complex 2 (Scheme 2). This reaction failed to give a similar binuclear complex when reacted with CuBr2 and Cu(CH3COO)2. Therefore, the mononuclear complex was acting as a bidentate ligand with respect to CuCl2 or Cu(ClO4)2 but not with CuBr2 or Cu(CH3COO)2. The structural pattern of the monomeric moiety plays a significant role for further reaction with another metal center. Several compounds of this type were extensively studied by Gruber et al. [18] in particular Cu-complexes involving ethylenediamine and 1,2-diaminopropane but none were structurally characterized. To the best of our knowledge, the symmetrical Schiff base complex 2 involving o-phenylenediamine has not been structurally characterized untill now. Complex 1, which acts as a bidentate complex ligand to form adducts with CuCl2 and Cu(ClO4)2, failed to do so with CuBr2 or Cu(CH3COO)2. Gruber et al. [18] observed that if the complex ligand is planar, then the metal to which it coordinates is unlikely to have a planar configuration. Two adjacent metal atoms are unlikely to be bridged together as perfect tetrahedra, because of the very short metal to metal separation that would be required. There is a greater distortion from planarity of the bromine-bonded copper atom than the chlorine-bonded one. This distortion toward tetrahedral configuration brings the two copper atoms too close, leading to instability. As the deviation from planarity of the metal outside the complex ligand in a binuclear complex increases, more distortion from planarity should be observed in the complex ligand itself. The bridging therefore is controlled by the stereochemistry of the metal outside the Schiff base complex. For noncoordinating anion, e.g. ClO4 −, where the anion is not positioned inside the metal complex, a trinuclear variety separated out since the copper atom at the center coordinated to the two complex ligands do not enforce any steric requirement of the copper centers at the complex ligands. Though it is suggested in the absence of structural studies that the three metal equatorial planes cannot be coplanar, but should deviate from planarity either in the complex ligand or the metal ion at the center, it seems probable that since acetate-bonded copper atom also is distorted from planarity to a pseudo tetrahedral center, a dinuclear or trinuclear compound should be unstable since the metal atoms come too close to form a stable complex (Scheme 2, 3). The steric requirement of the acetate ion should be more or less like the bromide.

Schematic representation for the preparation of complex 4

Preparation of the complexes 1, 2 and 3

Synthetic scheme of alternate way to prepare complex 2
Spectroscopic studies
The IR spectra for complexes 1, 2 and 3 show no bands for υ(NH2) indicating the condensation of free amine groups but the stretching frequency of free amine group appears at 3,228 cm−1 along with NH2 bending at 1,660 cm−1 for complex 4. The bands for azomethine (C=N) groups present in all the compounds are distinct and occur within 1,639–1,603 cm−1. General lowering of this band in all the compounds substantiates the coordination of the Cu(II) centers with the azomethine nitrogen. Usually, stretching vibration for water appears above 3,400 cm−1. Hence, the appearance of a broad band in the range of 3,430 cm−1 for complex 2 indicates the presence of a water molecule. This nature and lowering of the position indicates that the H atoms are involved in an intermolecular H-bond network. The appearance of a sharp intense band at 1,090 cm−1 for complex 3 is consistent with the proposed structure. The mononuclear compound 1 obtained by using Cu(ClO4)2·6H2O as the starting material does not show any band in this region, indicating that it is the same compound obtained when chloride and acetate are used as the counter anions.
The electronic spectra of the complexes are consistent with the square-planar geometry around the Cu(II) center. Two d–d transitions are expected to appear between 500 and 430 nm absorption around 430 nm with a shoulder around 400 nm. These two bands can be assigned to 2B1g-2A1g and 2B1g-2Eg [19]. More intense bands from 300 to 200 nm are charge transfer in origin. The broadness of the d–d band at 430 nm for the binuclear compound 2 indicates an overlapping of the d–d band in trigonal bipyramidal geometry in this region. The intense band around 250 nm for the binuclear compound, which is absent in the other two compounds probably accounts the difference in color. The distortion in the trigonal bipyramidal geometry from regular square-planar configuration can account for a ligand to metal charge transfer, which should be absent in the other two complexes. The other higher energy bands for the three complexes occur in the same region.
Crystal structure results
Complex 2
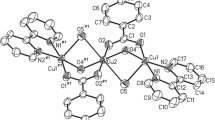
The structure determination reveals that complex 2 represents a binuclear species [Cu2LCl2(H2O)] as shown in Fig. 1, and the selected bond lengths and angles are given in Table 1. The Cu(2) atom is four-coordinated being bonded to the four donor atoms O(11), O(25), N(19) and N(26) of the symmetrical tetradentate ligand. The four donor atoms are planar with a r.m.s. deviation of 0.003 Å. The copper atom deviates from the mean plane by 0.033(3) Å. The tetrahedral distortion is not so apparent since all the four donor atoms lie almost in the same plane. The dihedral angles between the two planes [N(l9)–Cu(2)–N(26) and O(25)–Cu(2)–O(11)] is 2.70° compared with 0° for a perfectly square-planar arrangement and 90°for a perfect tetrahedral arrangement. The two oxygen atoms O(11) and O(25) in the ligand also bridge to Cu(1). Cu(1) is five-coordinated with a distorted square-pyramidal geometry. The basal plane is occupied by two chlorine atoms with equivalent bond lengths, Cu(1)–Cl(1) 2.262(2) Cu(1)–Cl(2) 2.266(2) Å, a water molecule Cu(1)–O(100) 1.980(4) Å and a bridging oxygen atom Cu(1)–O(11) 1.966(4) Å. The apical position is occupied by a second bridging oxygen atom that bonds less strongly, Cu(1)–O(25) 2.354(5) Å. The four donor atoms in the basal plane show a r.m.s. deviation of 0.261 Å. The copper atom Cu(1) is deviated from the mean plane by 0.171(2) Å in the direction of the axial atom O(25). The extent of distortion of coordination polyhedron from the square pyramid to the trigonal bipyramid has been calculated applying the Addison parameter (τ) [20] as an index of the degree of trigonality, and τ is defined as (β − α)/60 where β and α are the two trans-basal angles. For a perfectly square-pyramidal geometry, τ is equal to zero, while it becomes unity for perfectly trigonal–bipyramidal geometry. The Addison parameter (τ = 0.278) for the complex indicates that the penta coordinated geometry is square pyramidal with a considerable distortion toward trigonal bipyramid. Although the hydrogen atoms on the water molecule could not be located, it seems likely that O(100) is involved in intermolecular hydrogen-bonded chains with Cl(1′)(x + 1, y, z) and Cl(2′)(1.5 − x, y − 0.5, 0.5 − z) at 3.175 Å and 3.175 Å, respectively (Fig. 2).

ORTEP-3 picture for complex 2 with a limited atom numbering scheme. Thermal ellipsoids are shown at 30% probability

Proposed H-bonded chains of complex 2
Variable temperature magnetic study
Variable temperature magnetic study was done with complex 2. According to the structural data, complex 2 is a binuclear complex, in which the two Cu(II) ions are bridged by two phenoxo-oxygen atoms. The magnetic properties of the complex are very simple and correspond to an almost isolated binuclear entity, in line with the structural data. The magnetic susceptibility data were collected on polycrystalline samples over the temperature range of 300–2 K using an applied magnetic field of 10 Oersted. The plot of temperature dependence of χ M (χ M is the molar magnetic susceptibility for 2 Cu(II) ions) is shown in Fig. 3. To fit the temperature dependence magnetic susceptibility data, we used the dimeric model considering inter-dimer interaction. The data fitted reasonably well using the following Bleaney–Bowers expression considering small paramagnetic impurity.

Plot of χM versus T data for complex 2. Solid line represents the fitting using the inter-dimer model
The best-fit parameters obtained are the following: J = −217.095 cm−1, g = 2.09889 and R 2 = 2.003 × 10−5 R = 1.158 × 10−5 (R is the agreement factor defined as R = Σ[(χm)obs − (χm)calcd]2/Σ[(χm)obs]2). ρ (% of paramagnetic impurity) = 0.48682. χdia (diamagnetic contribution) = 0.00026 cm3 mol−1.
From the structural data, it is clear that the complex 2 is a phenoxo-bridged dimer in which the magnetic orbital, d 2x 2−y of two Cu(II) ions interact via the two bridging phenoxo groups. Magnetostructural correlation of such complexes has been reported [9, 21, 22]. The average bridging Cu–O–Cu angle in this complex is 100.81°(Cu(2)–O(25)–Cu(1) = 94.70° and Cu(2)–O(11)–Cu(1) = 106.92°) and the Cu2O2 ring is nearly planar (no atom deviates more than 0.06 Ǻ from the least square mean plane). The Cu–Cu distance is 3.126 Ǻ. All these data are in agreement with the strong antiferromagnetic coupling in the complex.
Experimental
Elemental analyses (carbon, hydrogen and nitrogen) were performed using a Perkin–Elmer 240C elemental analyzer. IR spectra in KBr (4,500–500 cm−1) were recorded using a Perkin–Elmer RXI FT-IR spectrophotometer. Electronic spectra in acetonitrile (1,200–350 nm) were recorded on a Hitachi U-3501 spectrophotometer. The magnetic susceptibility measurements were done with a MPMS XL5 SQUID magnetometer (QUANTUM DESIGN) and diamagnetic corrections were made using Pascal’s constants.
All chemicals were of reagent grade and used without further purification.
Caution! Metal perchlorates in the presence of organic ligands are potentially explosive. Only a small amount of the material should be prepared, and it should then be handled with care.
Synthesis of the [CuL] (1)
Salicylaldehyde (10 mmol, 1.1 mL) was added to a solution of o-phenylenediamine (5 mmol, 0.54 g) in ethanol (25 mL) with stirring. To the resulting mixture, an ethanolic solution (20 mL) of CuBr2·2H2O (5 mmol, 1.30 g) was added dropwise. This was stirred continuously for 3 h. Gradually, the color of the solution changed to deep blue. A deep blue compound precipitated out and was filtered off. Due to its highly insoluble nature, no single crystals suitable for X-ray diffraction were obtained. Yield: (~90%). Anal. found: C, 62.9; N, 6.9; H, 3.7; Cu, 16.5. Calc. for C20N2O2H14Cu: C, 63.6; N, 7.4; H, 3.7; Cu, 16.8%. HRMS (ESI): found m/z (M+H)+ = 395.9; M calcd = 396.5; magnetic moment (at 25 °C) μ eff = 1.7 BM.
The same product was obtained when Cu(ClO4)2·6H2O (5 mmol, 1.89 g) or Cu(CH3COO)2·H2O (5 mmol, 1.00 g) were used instead of CuBr2·2H2O.
[Cu2LCl2(H2O)] (2)
Complex 2 was prepared by the same procedure as complex 1 but using CuCl2·2H2O (5 mmol, 0.85 g) instead of CuBr2·2H2O. Gradually, the color of the solution changed to reddish brown. A reddish brown compound precipitated out and was filtered off, and the filtrate was kept at room temperature. Overnight, brown-colored single crystals suitable for X-ray diffraction were obtained. Yield: (~70%). Anal. found: C, 44.9; N, 4.9; H, 2.9; Cu, 23.5. Calc. for C20N2O3H16Cu2Cl2 : C, 45.3; N, 5.3; H, 3.0; Cu, 23.9%.
The same product was obtained by the following process in the presence of diacetylmonoxime.
A solution of salicylaldehyde (10 mmol, 1.1 mL) in chloroform (50 mL) was added dropwise to a solution of o-phenylenediamine (10 mmol, 1.08 g) in chloroform (50 mL). After completion of the addition, the solution was stirred for an additional 3 h, and chloroform was evaporated under reduced pressure, yielding the monocondensed 2-[(2-amino-phenylimino)-methyl]-phenol as a viscous liquid. It was dissolved in methanol (10 mL). Solid diacetylmonoxime (10 mmol, 1.011 g.) was added, and the mixture was stirred for 24 h. A solution of Cu(CH3COO)2·H2O(10 mmol, 1.99 g) in methanol (20 mL) was added to the resulting solution with continuous stirring to precipitate a brown-red solid. It was recrystallized from ethanol solution to obtain single crystals of 2 suitable for X-ray diffraction. Yield: (~70%).
[Cu3L2](ClO4)2 (3)
Salicylaldehyde (10 mmol, 1.1 mL) was added to a solution of o-phenylenediamine (5 mmol, 0.54 g) in ethanol (25 mL) with stirring. An ethanolic solution (10 mL) of Cu(ClO4)2·6H2O (15 mmol, 5.67 g) was added dropwise to the resulting mixture. This was stirred continuously for 3 h. Gradually, the color of the solution changed to deep green. A deep green crystalline compound precipitated out and was filtered off and recrystallised from ethanol. Unfortunately, no single crystals suitable for X-ray diffraction could be obtained. Yield: (~90%). Anal. found: C, 46.9; N, 5.4; H, 2.7; Cu, 18.5. Calc. for C40N4O12H28Cu3Cl2: C, 47.2; N, 5.5; H, 2.8; Cu, 18.7%. HRMS (ESI): found m/z (M+H)+ = 819.4; M calcd = 819.5. Magnetic moment (at 25 °C) μ eff = 1.63 BM.
[Cu2L 12 ](CH3COO)2 (4)
A mixture of diacetylmonoxime (5 mmol, 0.505 g) and o-phenylenediamine (5 mmol, 0.54 g) in ethanol (25 mL) was refluxed for about 3 h. An ethanolic solution (10 mL) of Cu(CH3COO)2·H2O (5 mmol, 1.00 g) was added dropwise to the resulting mixture. This was stirred continuously for 3 h. Gradually, the color of the solution changed to brown. A brown crystalline compound precipitated out and was filtered off and recrystallised from ethanol but no single crystals suitable for X-ray diffraction could be obtained. Yield: (~80%). Anal. found: C, 45.9; N, 13.4; H, 4.7; Cu, 19.9. Calc. for C24N6O6H30Cu2: C, 46.1; N, 13.4; H, 4.8; Cu, 20.3%.
X-ray crystallography
Crystal data are given in Table 2. A total of 4,892 independent reflections were collected with Mo Kα radiation at 150 K using the Oxford Diffraction X-Calibur CCD System. The crystal was positioned at 50 mm from the CCD. Three hundred twenty-one frames were measured with a counting time of 10 s. Data analysis was carried out with the CrysAlis program [23]. An empirical absorption correction was carried out using Abspack [24]. The structure was solved using direct methods with the Shelxs97 program [25]. The nonhydrogen atoms were refined with anisotropic thermal parameters. The hydrogen atoms bonded to carbon were included in geometric positions and given thermal parameters equivalent to 1.2 times those of the atom to which they were attached. The hydrogen atoms on the water molecule were not located. The structure was refined using all data on F 2 using Shelxl97 [25] to R1 0.0721, wR2 0.0775 for 1,411 reflections with I > 2σ(I) and R1 0.0979, wR2 0.2598 for all data.
Concluding remarks
Three symmetrical Schiff base complexes have been studied and one of them has been characterized structurally. It was our aim to synthesize unsymmetrical ligands involving o-phenylenediamine, salicylaldehyde and diacetylmonoxime, but we could not isolate the desired unsymmetrical Schiff base complexes. In the presence of diacetylmonoxime, only the symmetrical complex was isolated. Mononuclear, binuclear and trinuclear compounds were obtained by varying the counter anions. It seems probable, therefore, that the salicylaldehyde symmetrical Schiff base complex was the most stable species that separated out even in the presence of diacetylmonoxime. The Schiff base compound derived from diacetylmonoxime could only be isolated in the absence of salicylaldehyde.
References
Sigel H (1980) Inorg Chem 19:1411
Elder RC, Blubaugh EA Jr, Heinmann WR, Bruke PJ, McMillian RD (1983) Inorg Chem 22:2771
Gaykema WPJ, Volveda A, Hol WJH (1985) J Mol Biol 187:2255
Gaykema WPJ, Hol WGH (1989) J Mol Biol 209:249
Bu XR, Jackson CR, Derveer DV, You XZ, Meng QJ, Wang RX (1997) Polyhedron 17:2991–3001
Ray MS, Mukhopadhyay G, Bhattacharya R, Chaudhuri S, Righi L, Bocelli G, Ghosh A (2003) Polyhedron 22:617
Chattopadhyay S, Ray MS, Chaudhuri S, Mukhopadhyay G, Bocelli G, Cantoni A, Ghosh A (2006) Inorg Chim Acta 359:1367
Ray MS, Ghosh A, Bhattacharya R, Mukhopadhyay G, Drew MGB, Ribas J (2004) Dalton Trans 252
Ray MS, Mukhopadhyay G, Drew MGB, Lu T-H, Chaudhuri S, Ghosh A (2003) Inorg Chem Commun 6:961
Ray MS, Ghosh A, Chaudhuri S, Drew MGB, Ribas J (2004) Eur J Inorg Chem 15:3110
Maity D, Mukherjee P, Ghosh A, Drew MGB, Mukhopadhyay G (2008) Inorg Chim Acta 361:1515
Maity D, Chattopadhyay S, Ghosh A, Drew MGB, Mukhopadhyay G (2009) Polyhedron 28:812–818
Kanderal OM, Kozlowski H, Dobosz A, Kozlowska JS, Meyer F, Fritsky OI (2005) J Chem Soc Dalton Trans 1428
Costes JP, Dahan F, Dupuis A, Laurent JP (1998) J Chem Soc Dalton Trans 1307
Kukushkin VY, Tudela D, Pombeiro AJL (1996) Coord Chem Rev and references cited therein. 156: 333–362
Chaudhuri P (2003) Coord Chem Rev and references cited therein. 243: 143–190
Tan S-F, Ang K-P (1988) Transition Met Chem 13:64
Gruber SJ, Harris CM, Sinn E (1968) J Inorg Nucl Chem 30:1805
Lever ABP (1984) Inorganic electronic spectroscopy, 2nd edn. Elsevier Science Publishers, Amsterdam
Addison AW, Rao TN, Reedijk J, van Rijn J, Verschoor GC (1984) J Chem Soc Dalton Trans 1349
Thompson LK, Mandal SK, Tandon SS, Bridson JN, Park MK (1996) Inorg Chem 35:3117
Ruiz E, Alemany P, Alvarez S, Cano J (1997) Inorg Chem 36:3683
CrysAlis (2008) Oxford Diffraction Ltd., Abingdon, UK
Abspack Program (2007) Oxford Diffraction Ltd., Abingdon, UK
Sheldrick GM, Shelxs97 and Shelxl97, Programs for Crystallographic solution and refinement Acta Crystallogr A64:112 (2008)
Acknowledgments
We thank EPSRC and the University of Reading for funds for the X-Calibur system. Thanks are also due to Prof. Ashutosh Ghosh, Department of Chemistry, University College of Science, University of Calcutta, 92, A.P.C. Road, Kolkata-700 009, India, for his valuable suggestions during preparation of the manuscript and Sri Sanjib Giri of Indian Association for the Cultivation of Science, Jadavpur, Kolkata-700 032, India, for the magnetic measurements.
Author information
Authors and Affiliations
Corresponding author
Appendix A. Supplementary data
Appendix A. Supplementary data
Crystallographic data for the analysis have been deposited with the Cambridge Crystallographic data Centre, CCDC No 721696 (complex 2). Copies of this information may be obtained free of charge from CCDC, 12 Union Road, Cambridge, CB21EZ, UK (fax:+441223336033; email:deposit@ccdc.cam.ac.uk or www: http://www.ccdc.cam.ac.uk).
Rights and permissions
About this article
Cite this article
Maity, D., Drew, M.G.B., Godsell, J.F. et al. Synthesis and characterization of Cu(II) complexes of tetradentate and tridentate symmetrical Schiff base ligands involving o-phenelenediamine, salicylaldehyde and diacetylmonoxime. Transition Met Chem 35, 197–204 (2010). https://doi.org/10.1007/s11243-009-9314-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11243-009-9314-9