
Abstract
Ultraviolet (UV) emerging with the sun light plays crucial role in plants ontogenesis as the one of the main environmental stresses. Prolonged exposure to UV causes damage to DNA, proteins and membranes, and the inhibition of protein synthesis and photosynthetic reactions. Moreover, UV radiance is known to induce metabolic modifications in plants, particularly to secondary metabolite biosynthesis. Multitude of the studies published to date considering exposure of grape cells to UV revealed enhancement in stilbene compounds production, and many of which possess unique biological and pharmacological properties. However, the epigenetic mechanisms regulating expression of the genes involved in stilbene biosynthesis in response to UV exposure were not studied. In this study as model objects were used grape Vitis amurensis plant cell cultures capable to produce trans-resveratrol (t-resveratrol). T-resveratrol, a naturally occurring plant phenol, has been reported to exhibit a wide range of valuable biological and pharmacological properties. T-resveratrol synthesized via the phenylpropanoid pathway, where phenylalanine ammonia-lyase (PAL) is the first enzyme in this pathway and stilbene synthase (STS) is the enzyme that directly catalyzes the reaction of t-resveratrol formation. Exposure of V. amurensis cells to UV of C type significantly increased t-resveratrol production and certain STS and PAL expression. Using bisulfite sequencing, we demonstrated that methylation level of the VaSTS2, VaSTS6 and VaSTS10 genes significantly decreased, while methylation level of constitutively expressed VaSTS1 gene did not changed significantly.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Being essential part of the solar irradiation ultraviolet (UV) subdivided into three bands termed UV-A (315–400 nm), UV-B (280–315 nm) and UV-C (100–280 nm) according to its wave length and effects on the living organisms. Invisible for the human eyes UV radiance has a profound effect on the sessile organisms including plants unable to avoid such type of stress physically. Being very stable to environmental stressors plant genome is still sensitive to spontaneous mutations caused directly by UV exposure and reactive oxygen species (ROS) as a side effect (Tuteja et al. 2009; Rastogi et al. 2010).
Among the other UV radiation types UV-A has a less pronounced effect on plant genome stability because it cannot be absorbed by DNA molecule itself. However, UV-A is able to damage DNA via indirect photosensitizing reactions-mediated ROS generation (Alscher et al. 1997). During the long time effect of UV-A on plant physiology was underestimated, nevertheless recent studies on eggplant Solanum melongena L., pepper Capsicum annuum L. and pea Pisum sativum L. showed significant alternation in morphology and secondary metabolites production in studied species in response to prolonged UV-A exposure (Dáder et al. 2014; Siipola et al. 2015). In contrary to UV-A the effects of UV-B have been analyzed on diverse plants species and vary depending on duration and wavelength of the UV-B treatment. Exposure to UV-B over a long period of time causes tissue necrosis and induces the expression of many genes normally involved in defense, wounding, or general stress responses (Brosché and Strid 2003; Casati and Walbot 2003; Frohnmeyer and Staiger 2003). Moreover, several studies have reported damage to DNA, proteins and membranes and the inhibition of protein synthesis and photosynthetic reactions as the result of prolonged UV-B exposure (Rozema et al. 1997; Jansen et al. 1998; Tuteja et al. 2001; Takahashi et al. 2011). The Earth atmosphere being transparent for UV of A and B types, almost entirely absorbs radiance corresponding to UV-C spectrum. Thus, it was proposed that UV-C radiation has insignificant effect on biota and cannot modulate plant physiology as plants lack of the receptors responsible for sensing of UV-C radiance. However, further studies showed that plant species including Arabidopsis thaliana L., Vitis vinifera L., Arachis hypogaea L. and other are also acquisitive to UV-C stress having a negative impact on crop yield by altering a gain in biomass and affecting seed set (Kuo-Lung et al. 2005; Belchi-Navarro et al. 2012; Migicovsky and Kovalchuk 2014). Moreover, mentioned studies suggested that exposure to UV-C of A. thaliana plants also influences the response of the progeny with possible involvement of epigenetic factors to coordinate this process (Migicovsky and Kovalchuk 2014). Thus, being the less studied the mechanism of signal transduction within plant cells during UV-C response has to be described for our better understanding of regulatory networks.
Depended from the sun light, plants developed the strategy aimed to defend genome integrity and prevent devastating effect of UV irradiance based on the production of secondary metabolites able to prevent effects of this negative factor. Phytochemical investigations of plant responses to UV stress have revealed the induction of phenolics such as stilbenoids in various Vitis sp., presumably antioxidants, which may protect cells against UV-induced oxidative damage (Kostyuk et al. 2008; Pezet et al. 2003). Studies conducted on soybean Glycine max, sweet wormwood Artemisia annua L., apple Malus spp., centella Centella asiatica L., cannabis Cannabis sativa L., kangaroo vine Cissus antarctica Vent. and many others revealed up-regulation of flavonoids, phenolics and anthocyanins content, as UV screens and stilbene-related compounds, isoprenoid-derived terpenes, different phenolic compounds content possessing antioxidant activity in response to UV exposure (Marti et al. 2014; Lu et al. 2015; Pandey and Pandey-Rai 2014; Sullivan et al. 2014; Bidel et al. 2015).
However, a great number of the secondary metabolites induced by UV treatment have a wide spectrum of important biological and pharmacological properties including 3,5,4′-trihydroxy-trans-resveratrol (t-resveratrol) synthesized by Vitaceae, Fabaceae, Polygonaceae and species from other families (Kiselev 2011). T-resveratrol has been shown to interfere with oxidative modifications of lipids (Frankel and Waterhouse 1993). Moreover, t-resveratrol possesses anti-neoplastic activity that inhibits tumor establishment and growth and the formation of metastases (Aggarwal et al. 2004). It is known that t-resveratrol is synthesized via the phenylpropanoid pathway (Langcake and Pryce 1977), where phenylalanine ammonia-lyase (PAL, EC 4.3.1.24) is the first enzyme in this pathway and stilbene synthase (STS, EC 2.3.1.95) is the enzyme that directly catalyzes the reaction of t-resveratrol formation. Furthermore, t-resveratrol being toxic for plant cells can be glycosylated to form di-glucoside t-resveratrol and t-piceid or condensed into ε-viniferin and δ-viniferin known to have less pronounced biological effect (Langcake and Pryce 1977). It is known, that PAL and STS exist as a multi-gene family in most stilbenoid-producing species due to functional divergence between certain gene representatives (Kiselev 2011; Parage et al. 2012). Nevertheless, the process of regulation of such a redundant gene family counting up to 48 STS representatives in genome of V. vinifera (cv PN40024) remains to stay unclear (Parage et al. 2012).
Studies conducted on V. vinifera concerning the mechanism of UV response revealed 238 genes up-regulated more than fivefold by UV-C light (Suzuki et al. 2015; Xu et al. 2015). Among the others, genes encoding enzymes responsible for phenylpropanoids production and stilbenes biosynthesis were activated in a greater degree in V. vinifera cv. Malbec leafs being exposed to UV-B (Pontin et al. 2010). Experiments on UV-C treatment of V. vinifera cv. Pinot noir leafs conducted by Vannozzi et al. (2012) revealed differences in certain STS genes activation, showing only 22 STS representatives greatly increasing in their expression. According to the literature data up-regulation of STS gene expression in response to UV-C treatment of V. vinifera leafs was coordinated by MYB-R2R3 transcription factors VvMyb14 and VvMyb15 probably working as positive regulators (Höll et al. 2013). However, this observation leaves unexplained differential nature of STS genes regulation. Furthermore, in most cases gene expression is regulated by epigenetic factors including cytosine DNA methylation as one of the most significant mechanism in plant development.
In higher plants, DNA methylation primarily functions in defense reactions of the cells and maintaining genomic stability. Cytosine DNA methylation plays a crucial role in the mechanisms of transcriptional gene silencing. DNA methylation within a gene promoter region or so-called body methylation of a protein-coding nucleotide sequence is known to modulate gene expression at different stages of ontogenesis (Law and Jacobsen 2010). Cytosine methylation within a promoter or 5′- and 3′-end areas has more pronounced effect on gene expression. Moreover, a broad range of constantly expressing genes are methylated within the central area of their protein-coding sequences (Messeguer et al. 1991; Lister et al. 2008). DNA methylation is a relatively stable and heritable modification that controls gene expression, cellular differentiation, genomic imprinting, paramutation, transposon inactivation and embryogenesis (Finnegan et al. 1998; Bird 2002; Vanyushin and Ashapkin 2011). The process of methylation is executed by methylases, counting up to three subfamilies in higher plants: the methyltransferases (Met), chromo methyltransferases (CMT), and domain rearranged methyltransferases (DRM). Thus, being responsible for DNA methylation in methylation contexts “CG” and “CHG”, respectively Met and CMT methylases executing maintenance methylation of structure genes. While DRM methylases able to methylate assymetrical “CHH” sites are mainly targeted for transposons inactivation via mechanism of de novo methylation (Law and Jacobsen 2010). In contrast to DNA methylation, DNA demethylation can be passive and/or active (Ikeda and Kinoshita 2009). Whereas passive DNA demethylation may take place due to lack of maintenance methylation during DNA replication, active demethylation occurs enzymatically by removing methylated cytosines. In plants several enzymes assigned as DNA glycosylases were shown to exhibit a DNA demethylation activity in combination with base excision repair process. DEMETER (DME) and REPRESSOR OF SILENCING 1 (ROS1) are the founding members of a family of DNA glycosylases in plants that also includes DEMETER-LIKE 2 (DML2) and DML3 (Zhu 2009).
Recent study on changes in the methylation pattern of transcribed and satellite DNA in plant meristematic tissue under UV-C treatment showed a significant difference suggesting DNA methylation to be a key factor in the process of plant UV response (Kravets et al. 2013). Therefore, the primary goal of the current study is to analyze the level of cytosine methylation within STS genes at normal conditions and under UV-C treatment in connection to their expression and the level of stilbenes content in the cultured cells of Vitis amurensis Rupr. The current paper focuses on the differences in the DNA methylation pattern of certain VaSTS genes and the putative mechanism of VaSTS genes regulation is described.
Materials and methods
Vitis amurensis cell culture
As described previously, the V2 callus culture of wild-growing grape Vitis amurensis Rupr. (Vitaceae) was established in 2002 (Kiselev et al. 2009). The V2 callus culture were cultivated in 35 day subculture intervals in the dark at 24 ± 1 °C in 15 ml of WB/A solid medium (Kiselev et al. 2009). Reagents for cell culture were purchased from Sigma Chemical Co. (MO, USA) and Serva Feinbiochemica GmbH & Co. (Heidelberg, Germany). The samples were harvested from the 35 day cultures during their linear growth phase and when the highest t-resveratrol content was observed. These samples were then aseptically transferred into polystyrol Petri dishes (diameter 40 mm), obtained from Medpolimer (Saint Petersburg, Russia) and subjected for UV-C irradiation.
Ultraviolet treatment
In vitro grown V. amurensis calli (35 days old) were exposed to UV-C irradiation in the same controlled growth conditions described above being transferred into plastic Petri dishes with open lids. UV-C treatment of 254 nm wave length were conducted in the dark using UV lamp VL-215.MC provided by Vilber Lourmat company (Marne-la-Vallée, France). During the treatment period of 60 min mentioned UV-C emitter was suspended 13 cm above opened Petri dishes containing calli within. Control calli group after being transferred into plastic Petri dishes were contained in the same growth conditions in the dark for 60 min. Time of 60 min for UV-C treatment was chosen according to the data of preliminary experiments. Though, time-point of 60 min corresponded to the highest level of resveratrol production in UV-C treated calli. Throughout the ultraviolet treatment, calli were exposed to 230 µW cm−2 of 254 nm UV-C. Three independently grown, harvested, and extracted sets of samples corresponding to experimental UV-C treatment were prepared as biological replicates. After irradiation samples were collected for nucleic acids extraction and cDNA preparation. The calli were dried using hot air (50 °C for 2 h), and their levels of t-resveratrol were measured by HPLC as described previously (Dubrovina and Kiselev 2012; Kiselev et al. 2013b).
Total RNA isolation and quantitative real-time PCR
Total RNA was isolated using the CTAB (hexadecyltrimethylammonium bromide)-based extraction protocol (Bekesiova et al. 1999). The cDNA was synthesized as described previously (Aleynova et al. 2015). The gene-specific primer pairs and TaqMan-probes for VaSTS1–VaSTS10, VaPAL1–VaPAL5, VaDem1–VaDem3, Actin1, GAPDH genes were presented in previously published papers (Kiselev et al. 2013a; Shumakova et al. 2011). The efficiency of primer annealing was evaluated using the Primer Premier 5.0 program. Each TaqMan probe for the Actin1 and GAPDH genes were labeled with an FAM reporter dye at the 5′-end and a RTQ-1 quencher dye at the 3′-end, and each TaqMan probe for the VaPAL and VaSTS genes was labeled with an ROX reporter dye at the 5′-end and a BHQ-2 quencher dye at the 3′-end (Syntol, Russia).
For TaqMan real-time PCR of cDNA transcripts corresponding to the VaPAL and VaSTS genes, cDNAs were amplified (Aleynova et al. 2015) in 20-µl reaction mixtures containing 1× TaqMan Buffer B, 2.5-mM MgCl2, 250-µM dNTP, 1-U Taq DNA polymerase, 0.5-µl (15 ng) cDNA and 0.25 µM of each primer and probe (Real-time PCR Kit, Syntol, Russia). PCR amplification consisted of one cycle of 2 min at 95 °C, followed by 50 cycles of 10 s at 95 °C and 25 s at 62 °C.
For real-time PCR of cDNA transcripts corresponding to the VaDem1–VaDem3 genes, cDNAs were amplified (Shumakova et al. 2011) in 20-μl reaction mixtures containing 1× TaqMan Buffer B, 2.5 mM MgCl2, 250 μM dNTP, 1 U Taq DNA polymerase, 0.5 μl (15 ng) cDNA, 0.25 μM of each primer (Real-time PCR Kit, Syntol, Russia) and 1× EvaGreen (Biotium, Hayward, USA). PCR amplification consisted of one cycle of 2 min at 95 °C, followed by 50 cycles of 10 s at 95 °C and 25 s at 62 °C. Both types of real-time PCR assays were performed in an iCycler thermocycler that was equipped with the iQ5 Multicolor Real-time PCR detection system (BioRad, Hercules, CA), and data were analyzed using the iQ5 Optical System software v.2.0 according to the manufacturer’s instructions.
Vitis amurensis Actin 1 and GAPDH genes (GenBank Acc. Nos. AY907207; GU585870) were used as endogenous controls to normalize variance in the quality and the amount of cDNA used in each real-time RT-PCR experiment. Expression was calculated by the 2−ΔΔCT method (Livak and Schmittgen 2001). Scaling options are the highest (the highest expressing sample accrued the value 1 in the relative mRNA calculation). The data were summarized from three quantitative real-time PCR reactions for each biological replicate with V. amurensis Actin 1 as the endogenous control and three independent PCR reactions with GAPDH as the endogenous control (Kiselev et al. 2013a; Shumakova et al. 2011).
Bisulfite sequencing
The DNA methylation status of the VaSTS1, VaSTS2, VaSTS6, VaSTS10 5′-upstream region, 5′, and 3′ end of protein coding region (GeneBank Acc. Nos. GQ167204; EU659863; EU659867; JQ780328) was investigated using the bisulfite sequencing method as described previously (Kiselev et al. 2013b, 2015; Tyunin et al. 2013). A total of 1.5 μg of V. amurensis genomic DNA was subjected to bisulfite modification using the EZ DNA Methylation-Gold Kit (Zymo Research, USA), as per the manufacturer’s instructions. Before DNA samples were subjected to bisulfite sequencing restriction digestion with PalA I enzyme (50 µ/µL, SibEnzyme, Novosibirsk, Russia) having no restriction site within sequences of interest. The DNA was converted using the follow conditions: 95 °C for 5 m and 50 °C for 1.5 h. The cloned PCR products from the 5′-upstream region and parts of the coding region were used as a positive control for the bisulfite chemical reactions. The level of C to T transitions in the converted PCR products was greater than 95 %.
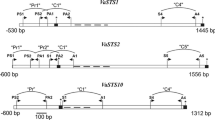
The primer sets for bisulfite sequencing of 5′-upstream region, 5′, and 3′ end of protein coding region of VaSTS1, VaSTS2, and VaSTS10 were described in articles published earlier (Kiselev et al. 2013b, 2015; Tyunin et al. 2013). For amplifying of VaSTS6 fragment named Cod1 corresponding to 5′ upstream and 5′ end of protein coding sequences primers S1: 5′AAA GTG ATG YTA YYA AYT TGG AGT TA and A1: 5′TAA ACT TCT TCT TRA RCT CRA TCA T were used (Fig. 1). The area of the 3′ end of protein coding region of the VaSTS6 (Cod5) was amplified using primers S5: 5′TAA TTY AGY AGG TGY TAT TGY AGG TA and A5: 5′TTA RTT TRA ATC TRT ACC AAC RCT AT (Fig. 1).

Schematic diagram of the VaSTS1, VaSTS2, VaSTS6 and VaSTS10 promoter and protein coding regions. PS1, PS2, PA1, S1, S4, S5, A1, A4, A5 P1Ms, P1Ma, P10Ms, P10Ma are primers. 5′CACCTACA3′, 5′CACCTACC3′, 5′CACCAACC3′—shows positions of hypothetical sites for MYB transcription factors binding. The asterisks indicate the start codons, black colored squares at the terminal part of the genes indicate stop codons and the dashes indicate the intron position. For further explanation, see the “Materials and methods” section
For searching of hypothetical MYB transcription factors binding sites within promoters of studied VaSTS genes on-line database of plant cis-acting regulatory DNA elements PLACE (http://www.dna.affrc.go.jp/PLACE) was used. Thus, nucleotide sequences of a 600 b.p. prior to the start codon were scanned for MYB transcription factors binding sites. By following this method one hypothetical MYB transcription factor binding site was described on “sense” DNA strand for promoter of VaSTS1 and two more were described for VaSTS1 and VaSTS10 being located on “antisense” DNA strand, one for the each of the genes (Fig. 1).
For bisulfite sequencing of the hypothetical MYB transcription factor binding site 5′CACCTACA3′ located on the “sense” strand of VaSTS1 promoter region we used the same pair of primers as we used to amplify promoter region (Fig. 1). For bisulfite sequencing of the hypothetical MYB transcription factor binding site 5′CACCTACC3′ located on the “antisense” strand we used primers P1Ms: 5′AAT RTT TCT CTT TAR CCG TRA CCR RAT T and P1 Ma: 5′TYT YAT YTA TTG GYG GTY AAT AAA TAA T (Fig. 1). For bisulfite sequencing of the hypothetical MYB transcription factor binding site 5′CACCAACC3′ located on the “antisense” strand we used primers P10Ms: 5′TTC ACA TAT CAT CAA RTA TCC ATT RAC T and P10Ma: 5′TGA TGA YTA YTG AAA TYG AAG YTT AA (Fig. 1). Figure 1 schematically demonstrates positions of the primers within nucleotide sequences of VaSTS1, VaSTS2, VaSTS6, VaSTS10 and their promoters that were used in current study.
Amplification reactions were performed in volumes of 25 µl containing 10 mM Tris–HCl (pH 8.5), 50 mM KCl, 2.5 mM MgCl2, 0.01 % gelatin, 0.1 mM Triton X-100, 0.25 mM of each dNTP, 0.25 µM of primer, 1 unit of Taq polymerases (Silex M, Moscow, Russia). Analysis was performed in an thermocycler (Bis-N Novosibirsk, Russia) programmed for an initial denaturation step of 2 min at 95 °C, followed by 35 cycles of 15 s at 95 °C, 10 s at 52 °C, 40 s at 72 °C, and a last cycle of 72 °C for 20 min, using the fastest available transitions between each temperature.
The PCR products were isolated from agarose gels using a Cleanup Mini Kit (Evrogene, Moscow, Russia) and were subcloned into the pTZ57R/T plasmid using the InsT/Aclone PCR Product Cloning Kit (Fermentas, Vilnius, Lithuania) and sequenced using a Big Dye Terminator Cycle Sequencing Kit (Perkin–Elmer Biosystems, Forster City, CA), following the manufacturer’s protocol and recommendations with an ABI 3130 Genetic Analyzer (Applied Biosystems, Foster City, USA). A total of 8–10 individual clones were sequenced for each region of used genes. The BLAST search program was used for sequence analysis. Multiple sequence alignments were performed using the ClustalX program.
Statistical analysis
Statistical analysis was performed using Statistica 10.0 and statistical significance was determined using the paired Student’s t test. The data are presented as mean ± standard error (SEM). We required that P < 0.05 for statistical significance.
Results
VaPAL and VaSTS expression and stilbene content in cultured cells of V. amurensis
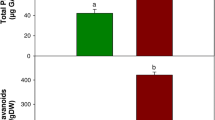
Being very suitable model for investigation of stilbene biosynthesis calli culture of V. amurensis V2 used in this experiment normally contains up to 0.002 % of a dry weight (DW) of t-resveratrol as one of the major stilbene compound (Kiselev et al. 2009). In the course of our experiment V2 calli were exposed to 230 µW cm−2 of 254 nm UV-C for 60 min and the content of all stilbene compounds was measured. As the results extension of t-resveratrol content in 7.5-fold was observed being statistically significant (Table 1). However, not only t-resveratrol content increased in response to UV-C in V2 calli. According to our data t-piceid, ε-vininferin, and δ-viniferin being derivatives of t-resveratrol also increased their content in 3.8-, 6.6-, and 5.4-fold upon UV-C exposure of V2 calli.
Biosynthesis of stilbenes occurs via the phenylpropanoid pathway where PAL enzymes are working at the initial step and STS enzymes are directly catalyzing the reaction of t-resveratrol formation. Thus, up-regulation of the t-resveratrol biosynthesis as the main precursor, and increasing of the PAL and STS gene expression are the reliable indicators of the activation of the stilbene biosynthesis. According to our previous findings expression of 5 VaPAL (Genbank Acc. Nos. EU659859; EU659860; EU659861; GQ443744; GQ443745) and 10 VaSTS (Genbank Acc. Nos. GQ167204; EU659863; EU659864; EU659865; EU659866; EU659867; EU659868; GQ443746; GU266256; JQ780328) isoforms is typical for calli culture of V. amurensis V2 (Kiselev et al. 2015). Real-time PCR analysis on the VaPAL and VaSTS gene expression revealed activation of VaSTS2–VaSTS7, VaSTS10, VaPAL1, VaPAL4 gene expression in response to UV-C exposure (Fig. 2). Moreover, our data showed that expression of VaPAL2 and VaPAL3 decreased under UV-C treatment, while VaSTS1, VaSTS8 and VaSTS9 expression remained to be the same (Fig. 2). Thus, UV-C exposure mostly increased VaSTS2, VaSTS6 and VaSTS10 expression (in 6.4-, 2.2- and 3.7-times, respectively, compared with untreated calli group); therefore, those genes were selected for DNA methylation pattern analysis. For correct data interpretation methylation pattern of VaSTS1 gene was also analyzed, because VaSTS1 gene expression was predominant in different grape cell cultures (Kiselev et al. 2009; Tyunin et al. 2013). Thus, the DNA methylation level of VaSTS1, VaSTS2, VaSTS6 and VaSTS10 genes in the V2 callus culture treated by UV-C was subject of further analysis.

STS and PAL expression levels detected by real-time PCR in V. amurensis callus culture V2 at normal conditions (control) and after UV-C exposure for 60 min (UV-C exposure). The data are presented as the mean ± SEM obtained from three independent experiments, **P < 0.01; *P < 0.05 versus the values measured from the untreated V2k calli. r.u. relative units
Analysis on the DNA methylation patterns within 5′ and 3′ end of protein coding sequences of VaSTS1, VaSTS2, VaSTS6 and VaSTS10 in cultured cells of V. amurensis under UV-C exposure
Being the “golden standard” for determination of cytosine methylation status bisulfite sequencing technique allows to analyze fragments of ~400 b.p. (Fig. 1). All the mentioned fragments were analyzed individually to determine the cytosine methylation levels in V2 calli at normal conditions and after UV-C exposure. We found that the methylation profiles corresponding to the 5′ end protein-coding sequences of the VaSTS1 (Fig. 4a), VaSTS2 (Fig. 4b), VaSTS6 (Fig. 4c) and VaSTS10 (Fig. 4d) were considerably different. Moreover, alternations in methylation profiles in response to UV-C treatment also have different amplitude depending from the rate of gene induction (Fig. 4). According to the data obtained the total level of cytosine methylation within 5′ end protein-coding sequences of the VaSTS1 did not significantly changed, while the same parameter decreased in 1.8-, 1.5- and 2.5-fold within 5′ end protein-coding sequences of the VaSTS2, VaSTS6, and VaSTS10 respectively upon UV-C induction of mentioned genes (Fig. 3). Furthermore, alternation in methylation profiles within 5′ end protein-coding sequences of the VaSTS2, VaSTS6, and VaSTS10 can be noted by the eye from the graphics presented (Fig. 4b–d) in contrary to VaSTS1 5′ end protein-coding sequence (Fig. 4a). Analysis on the level of cytosine methylation within different contexts (“CG”, “CHG”, “CHH”, where H=A, T or G), of VaSTS1 5′ end of protein-coding sequences in V2 calli at normal conditions and after UV-C exposure revealed no significant alternation (Electronic supplementary material, Table S1). In contrary to VaSTS1, VaSTS1 5′ end protein-coding sequences of VaSTS2, VaSTS6, and VaSTS10 demonstrated significant reduction in the level of methylation within all mentioned methylation contexts connected to gene activation, with less pronounced effect on “CG” and “CHG” contexts of VaSTS6, and “CHH” context of VaSTS2 5′ end protein-coding sequences (Electronic supplementary material, Table S1).

Analysis of the total DNA methylation level within the different regions of the VaSTS1 (a), VaSTS2 (b), VaSTS6 (c) and VaSTS10 (d) genes under normal conditions in V2 calli (V2-k) and V2 cells exposed to the UV-C for 60 min (V2-UV). Pr: methylation levels withinin the promoter region of VaSTS1, VaSTS2, VaSTS6 and VaSTS10 genes; 5′-methylation level within the 5′ end of protein-coding sequences of VaSTS1, VaSTS2, VaSTS6 and VaSTS10 genes; 3′-methylation level within the 3′ end of protein-coding sequences of VaSTS1, VaSTS2, VaSTS6 and VaSTS10 genes. The data are presented as the mean (n = 14) ± SE obtained from two independent experiments. *P < 0.05; **P < 0.01 versus the values measured from the untreated V2 cells

The DNA methylation profiles corresponding to the 5′ end protein-coding sequences of the VaSTS1 (a), VaSTS2 (b), VaSTS6 (c) and VaSTS10 (d). The each graph represents two methylation profiles: at normal conditions (dotted line) and after UV-C exposure (firm line) of the V2 cells. The horizontal axis represents every cytosine position within analyzed region and the vertical axis represents the percentage of methylated cytosines detected by bisulfite sequencing
Analysis on the level of cytosine methylation within 3′ end protein-coding sequences of the VaSTS1 (Fig. 5a), VaSTS2 (Fig. 5b), VaSTS6 (Fig. 5c), and VaSTS10 (Fig. 5d) revealed differences in the level and distribution of methylated cytosines at normal conditions. Moreover, alternations in the methylation profiles in response to UV-C exposure connected to activation of VaSTS2, VaSTS6, and VaSTS10 were expressed differently. The data obtained showed that the total level of cytosine methylation within 3′ end protein-coding sequences of the VaSTS1 did not considerable varied, while the same parameter decreased in 1.6-, 2.8- and 3.2-fold within 3′ end protein-coding sequences of the VaSTS2, VaSTS6, and VaSTS10 respectively upon UV-C induction of mentioned genes (Fig. 3). Interestingly, the level of cytosine methylation detected within 3′ end protein-coding sequences of constantly expressing gene VaSTS1 averaged 12 % at normal conditions, and the same percentage or lesser were detected for 3′ end protein-coding sequences of the VaSTS2, VaSTS6, and VaSTS10 upon UV-C induction (Fig. 3). Comparative analysis on the level of methylation at different methylation contexts within 3′ end protein-coding sequences revealed significant reduction of the methylation level at “CG” and “CHH” contexts for VaSTS6 and VaSTS10. Furthermore, significant reduction was observed at “CHG” context for VaSTS2 and VaSTS6 within their 3′ end protein-coding sequences (Electronic supplementary material, Table S1).

The DNA methylation profiles corresponding to the 3′ end protein-coding sequences of the VaSTS1 (a), VaSTS2 (b), VaSTS6 (c), and VaSTS10 (d). The each graph represents two methylation profiles: at normal conditions (dotted line) and after UV-C exposure (firm line) of the V2 cells. The horizontal axis represents every cytosine position within analyzed region and the vertical axis represents the percentage of methylated cytosines detected by bisulfite sequencing
Analysis on the DNA methylation patterns within gene promoters of VaSTS1, VaSTS2, VaSTS6, and VaSTS10 in cultured cells of V. amurensis under UV-C exposure
Comparative analysis on the methylation profiles corresponding to the promoter sequences of VaSTS1, VaSTS2, VaSTS6, and VaSTS10 in V2 cells at normal conditions and under UV-C exposure revealed no considerable alternations for promoters of VaSTS1 and VaSTS2 in response to UV-C exposure (Fig. 6), while the average level of cytosine methylation decreased in 2.2- and 3.3-fold within promoters of VaSTS6 and VaSTS10 in compare to the control calli group (Fig. 3). The data obtained on the level of cytosine methylation within different methylation contexts showed significant reduction in all methylation contexts for VaSTS6 and VaSTS10 gene promoters and no significant alternations for VaSTS1 and VaSTS2 gene promoters (Electronic supplementary material, Table S1). However, additional analysis on the level of cytosine methylation within hypothetical sites for MYB transcription factors binding found in promoters of VaSTS1 and VaSTS10, revealed this sites to be slightly methylated up to 22.2 % detected within 5′CACCAACC3′ site of VaSTS10 in V2 cells at normal conditions (Table 2). Further investigation demonstrated redistribution of cytosine methylation in VaSTS10 5′CACCAACC3′ site, and increase in the level of cytosine methylation within both 5′CACCTACA3′ and 5′CACCTACC3′ sites described for promoter of VaSTS1 up to 50 % (Table 2) being statistically significant.

The DNA methylation profiles corresponding to the gene promoter sequences of the VaSTS1 (a), VaSTS2 (b), VaSTS6 (c), and VaSTS10 (d). The each graph represents two methylation profiles: at normal conditions (dotted line) and after UV-C exposure (firm line) of the V2 cells. The horizontal axis represents every cytosine position up-stream the start codon within analyzed region and the vertical axis represents the percentage of methylated cytosines detected by bisulfite sequencing
Expression of demethylases VaDem1, VaDem2 and VaDem3 in cultured cells of V. amurensis at normal conditions and upon the UV-C exposure
To determine the mechanism of the DNA demethylation observed within certain nucleotide sequences of VaSTS genes upon the UV-C exposure the expression level of VaDem1 (Genbank Acc. No. KC491211), VaDem2 (Genbank Acc. No. KC491212) and VaDem3 (Genbank bankit 1592679) were measured by real-time PCR. The data on demethylases gene expression earlier detected in V. amurensis cells (Kiselev et al. 2013a) namely: VaROS1-like (VaDem1), VaDML3-like (VaDem2), and VaDemeter-like (VaDem3) genes revealed no significant alternation in gene expression in response to UV-C treatment (Electronic supplementary material, Figure S1). Nevertheless, the obtained data revealed a tendency to decrease in gene expression for all mentioned demethylases genes, but these alternations were statistically insignificant (Electronic supplementary material, Figure S1).
Discussion
Being one of the most damage-inducing source of stress for plant cells, UV irradiation has numerous regulatory effects on plant morphology, physiology and can be used as an elicitor for induction of plant secondary metabolites production. Several studies conducted on plant species from Vitaceae family revealed UV of B and C types to be powerful triggers for induction of stilbene compounds biosynthesis (Table 3). Thus, the data on the enhancement of stilbenes content obtained in current study are consistent with the literature data (Table 3). According to our study UV-C exposure for 1 h mostly increased t-resveratrol, ε- and δ-viniferin content. This result indicates that chosen parameters for UV-C exposure were sufficient as we observed accumulation of stilbene precursor (t-resveratrol) and stilbene compound corresponding to the stressed grape cells (δ-viniferin) in response to UV-C treatment (Pezet et al. 2003; Xu et al. 2015). Moreover, our data revealed induction upon UV-C exposure of VaSTS2–VaSTS7, VaSTS10 genes encoding key enzymes in resveratrol biosynthesis and VaPAL1, VaPAL4 genes encoding enzymes working up-stream compared with STS in phenylpropanoid pathway.
According to the literature data induction of certain gene expression in response to UV exposure are governed by transcription factors activating their expression (Höll et al. 2013; Yuan et al. 2015). However, this observation cannot explain the differential nature of certain gene activation, leading us to investigation of epigenetic mechanisms regulating these processes. According to the data obtained by Lister et al. (2008) cytosine methylation suppresses gene expression, especially being concentrated within 5′ and 3′ end of protein coding sequences. Our data revealed reduction in the cytosine methylation level within 5′ and 3′ end of protein coding regions of the VaSTS2, VaSTS6, and VaSTS10 genes, activated upon UV-C exposure. Moreover, we detected no significant alternation in the level of cytosine methylation for VaSTS1 gene and this observation correlated with no significant alternation in expression of mentioned gene upon UV-C exposure. In addition, the data obtained demonstrated no correlation between the total level of cytosine methylation within promoter region of analyzed VaSTS genes and their expression and these data were consistent with our previous findings (Kiselev et al. 2013b, 2015; Tyunin et al. 2013).
As DNA methylation was shown to play a pivotal role in regulation of plant development and stress response, methylases are involved in the regulation of a great number of different genes and basing on their expression it is hard to determinate their function in certain process (Tyunin et al. 2012). Nevertheless, basing on the data obtained from comparative analysis on the level of cytosine methylation within different methylation contexts we propose that maintenance methylases of Met and CMT subfamilies play leading part in regulation of VaSTS genes. This hypothesis based on the fact that “CG” and “CHG” methylation contexts within 5′ and 3′ end of protein coding regions of the VaSTS2, VaSTS6, and VaSTS10 genes were methylated to the greater extension than “CHH” methylation context and in the most cases alternation in the level of methylation there were the most significant in response to UV-C treatment.
Recent studies of Höll et al. (2013) assigned VvMyb14 and VvMyb15 transcription factors as positive regulators of VvSTS expression in V. vinifera cells in response to UV-C treatment. However, authors provided no data on the location of sites for VvMyb14 and VvMyb15 binding within promoters of VvSTS genes. Thus, by taking advantage of utilizing PLACE database, two putative MYB R2R3 transcription factors binding sites for promoter of VaSTS1 and one for promoter of VaSTS10 were assigned and the level of cytosine methylation within was measured. According to our data, cytosine methylation within analyzed sites for MYB R2R3 transcription factor binding in promoters of studied VaSTS genes are slightly methylated at normal conditions. Moreover, our data suggests little or no effect of cytosine methylation within hypothetical transcription factor binding sites on gene expression, as we observed no correlation between mentioned parameters. Thus, we hypothesize that DNA methylation and transcription factors are working in parallel on regulation of VaSTS genes in V. amurensis cells in response to UV-C treatment. Mentioned data altogether reveals DNA methylation as the main regulator for VaSTS gene expression and stilbene production consequently, in response to UV-C elicitation of V. amurensis calli. However, we were unable to detect induction of demethylases gene expression in response to UV-C exposure, since C type of UV were intentionally chosen as the less devastating for DNA and less inductive for enzymes involved in reparation processes including demethylases. Nevertheless, the last point is out of scope of the current study leading us to further investigations.
Abbreviations
- STS:
-
Stilbene synthase
- PAL:
-
Phenylalanine ammonia-lyase
- UV-C:
-
Ultraviolet C (100–280 nm wave length)
- t-resveratrol:
-
3,5,4′-trihydroxy-trans-resveratrol
References
Aggarwal BB, Bhardwaj A, Aggarwal RS, Seeram NP, Shishodia S, Takada Y (2004) Role of resveratrol in prevention and therapy of cancer: preclinical and clinical studies. Anticancer Res 24:2783–2840
Aleynova OA, Dubrovina AS, Manyakhin AY, Karetin YA, Kiselev KV (2015) Regulation of resveratrol production in Vitis amurensis cell cultures by calcium-dependent protein kinases. Appl Biochem Biotechnol 175:1460–1476
Alscher RG, Donahue JL, Cramer CL (1997) Reactive oxygen species and antioxidants: relationships in green cells. Physiol Plant 100(2):224–233
Bekesiova I, Nap JP, Mlynarova L (1999) Isolation of high quality DNA and RNA from leaves of the carnivorous plant Drosera Rotundifolia. Plant Mol Biol Rep 17:269–277
Belchi-Navarro S, Almagro L, Lijavetzky D, Bru R, Pedreno M (2012) Enhanced extracellular production of trans-resveratrol in Vitis vinifera suspension cultured cells by using cyclodextrins and methyljasmonate. Plant Cell Rep 31:81–89
Bidel LP, Chomicki G, Bonini F, Mondolot L, Soulé J, Coumans M, La Fisca P, Baissac Y, Petit V, Loiseau A, Cerovic ZG, Gould KS, Jay-Allemand C (2015) Dynamics of flavonol accumulation in leaf tissues under different UV-B regimes in Centella asiatica (Apiaceae). Planta. doi:10.1007/s00425-015-2291-7
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21
Brosché M, Strid Å (2003) Molecular events following perception of ultraviolet-B radiation by plants. Physiol Plant 117(1):1–10
Casati P, Walbot V (2003) Rapid transcriptome responses of maize (Zea mays) to UV-B in irradiated and shielded tissues. Genome Biol 5(3):R16
Dáder B, Gwynn-Jones D, Moreno A, Winters A, Fereres A (2014) Impact of UV-A radiation on the performance of aphids and whiteflies and on the leaf chemistry of their host plants. J Photochem Photobiol B 138:307–316
Dubrovina AS, Kiselev KV (2012) Effect of long-term cultivation on resveratrol accumulation in a high-producing cell culture of Vitis amurensis. Acta Physiol Plant 34:1101–1106
Finnegan EJ, Genger RK, Peacock WJ, Dennis ES (1998) DNA methylation in plants. Annu Rev Plant Physiol Plant Mol Biol 49:223–247
Frankel EN, Waterhouse AL (1993) Inhibition of human LDL oxidation by resveratrol. Lancet 341:1103–1104
Frohnmeyer H, Staiger D (2003) Ultraviolet-B radiation-mediated responses in plants. Balancing damage and protection. Plant Physiol 133:1420–1428
Gill SS, Anjum NA, Gill R, Jha M, Tuteja N (2015) DNA damage and repair in plants under ultraviolet and ionizing radiations. Sci World J 2015:250158. doi:10.1155/2015/250158
Höll J, Vannozzi A, Czemmel S, D’Onofrio C, Walker AR, Rausch T, Lucchin M, Boss PK, Dry IB, Bogs J (2013) The R2R3-MYB transcription factors MYB14 and MYB15 regulate stilbene biosynthesis in Vitis vinifera. Plant Cell 25(10):4135–4149
Ikeda Y, Kinoshita T (2009) DNA demethylation: a lesson from the garden. Chromosoma 118:37–41
Jansen MAK, Gaba V, Greenberg BM (1998) Higher plants and UV-B radiation: balancing damage, repair and acclimation. Trends Plant Sci 3(4):131–135
Keskin N, Kunter B (2008) Production of trans-resveratrol in ‘Cabernet Sauvignon’ (Vitis vinifera L.) callus culture in response to ultraviolet-C irradiation. Vitis 47:193–196
Kiselev KV (2011) Perspectives for production and application of resveratrol. Appl Microbiol Biotechnol 90:417–425
Kiselev KV, Dubrovina AS, Bulgakov VP (2009) Phenylalanine ammonia–lyase and stilbene synthase gene expression in rolB transgenic cell cultures of Vitis amurensis. Appl Microbiol Biotechnol 82:647–655
Kiselev KV, Tyunin AP, Karetin YA (2013a) Influence of 5-azacytidine and salicylic acid on demethylase gene expression in cell cultures of Vitis amurensis Rupr. Acta Physiol Plant 35:1843–1851
Kiselev KV, Tyunin AP, Zhuravlev YN (2013b) Involvement of DNA methylation in the regulation of STS10 gene expression in Vitis amurensis. Planta 237:933–941
Kiselev KV, Tyunin AP, Karetin YA (2015) Salicylic acid induces alterations in the methylation pattern of the VaSTS1, VaSTS2, and VaSTS10 genes in Vitis amurensis Rupr. cell cultures. Plant Cell Rep 34(2):311–320
Kostyuk V, Potapovich A, Suhan T, de Luca C, Pressi G, Dal Toso R, Korkina L (2008) Plant polyphenols against UV-C-induced cellular death. Planta Med 74:509–514
Kravets AP, Sokolova DA, Vengzhen GS, Grodzinskii DM (2013) Fractionated UV-C irradiation effects on the changes of transcribed and satellite DNA methylation profile and unstable chromosomal aberration yield. Radiats Biol Radioecol 53(6):583–591
Kuo-Lung KU, Chang PS, Cheng YC, Lien CY (2005) Production of stilbenoids from the callus of Arachis hypogaea: a novel source of the anticancer compound piceatannol. J Agric Food Chem 53:3877–3881
Langcake P, Pryce RJ (1977) A new class of phytoalexins from grapevines. Experientia 33:151–152
Law JA, Jacobsen SE (2010) Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 11:204–220
Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR (2008) Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 3:523–536
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408
Lu Y, Zhang M, Meng X, Wan H, Zhang J, Tian J, Hao S, Jin K, Yao Y (2015) Photoperiod and shading regulate coloration and anthocyanin accumulation in the leaves of malus crabapples. Plant Cell Tissue Organ Cult 121:619–632
Marti G, Schnee S, Andrey Y, Simoes-Pires C, Carrupt PA, Wolfender JL, Gindro K (2014) Study of leaf metabolome modifications induced by UV-C radiations in representative Vitis, Cissus and Cannabis species by LC-MS based metabolomics and antioxidant assays. Molecules 19:14004–14021
Messeguer R, Ganal MW, Steffens JC, Tanksley SD (1991) Characterization of the level, target sites and inheritance of cytosine methylation in tomato nuclear DNA. Plant Mol Biol 16:753–770
Migicovsky Z, Kovalchuk I (2014) Transgenerational changes in plant physiology and in transposon expression in response to UV-C stress in Arabidopsis thaliana. Plant Signal Behav 9(11):e976490
Pandey N, Pandey-Rai S (2014) Modulations of physiological responses and possible involvement of defense-related secondary metabolites in acclimation of Artemisia annua L. against short-term UV-B radiation. Planta 240:611–627
Parage C, Tavares R, Rety S, Baltenweck-Guyot R, Poutaraud A, Renault L, Heintz D, Lugan R, Marais GAB, Aubourg S, Hugueney P (2012) Structural, functional, and evolutionary analysis of the unusually large stilbene synthase gene family in grapevine. Plant Physiol 160:1407–1419
Pezet R, Perret C, Jean-Denis JB, Tabacchi R, Gindro K, Viret O (2003) Delta-viniferin, a resveratrol dehydrodimer: one of the major stilbenes synthesized by stressed grapevine leaves. J Agric Food Chem 51:5488–5492
Pontin MA, Piccoli PN, Francisco R, Bottini R, Martinez-Zapater JM, Lijavetzky D (2010) Transcriptome changes in grapevine (Vitis vinifera L.) cv. Malbec leaves induced by ultraviolet-B radiation. BMC Plant Biol 10:224–237
Rastogi RP, Kumar A, Tyagi MB, Sinha RP (2010) Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J Nucleic Acids 2010:592980
Rozema J, van de Staaij J, Björn LO, Caldwell M (1997) UV-B as an environmental factor in plant life: stress and regulation. Trends Ecol Evol 12(1):22–28
Shumakova OA, Manyakhin AY, Kiselev KV (2011) Resveratrol content and expression of phenylalanine ammonia-lyase and stilbene synthase genes in cell cultures of Vitis amurensis treated with coumaric acid. Appl Biochem Biotechnol 165:1427–1436
Siipola SM, Kotilainen T, Sipari N, Morales LO, Lindfors AV, Robson TM, Aphalo PJ (2015) Epidermal UV-A absorbance and whole-leaf flavonoid composition in pea respond more to solar blue light than to solar UV radiation. Plant Cell Environ 38(5):941–952
Sullivan JH, Muhammad D, Warpeha KM (2014) Phenylalanine is required to promote specific developmental responses and prevents cellular damage in response to ultraviolet light in Soybean (Glycine max) during the seed-to-seedling transition. PLoS One 9(12):e112301
Suzuki M, Nakabayashi R, Ogata Y, Sakurai N, Tokimatsu T, Goto S, Suzuki M, Jasinski M, Martinoia E, Otagaki S, Matsumoto S, Saito K, Shiratake K (2015) Multiomics in grape berry skin revealed specific induction of the stilbene synthetic pathway by ultraviolet-C irradiation. Plant Physiol 168(1):47–59
Takahashi M, Teranishi M, Ishida H, Kawasaki J, Takeuchi A, Yamaya T, Watanabe M, Makino A, Hidema J (2011) Cyclobutane pyrimidine dimer (CPD) photolyase repairs ultraviolet-B induced CPDs in rice chloroplast and mitochondrial DNA. Plant J 66(3):433–442
Tuteja N, Singh MB, Misra MK, Bhalla PL, Tuteja R (2001) Molecular mechanisms of DNA damage and repair: progress in plants. Crit Rev Biochem Mol Biol 36(4):337–397
Tuteja N, Ahmad P, Panda BB, Tuteja R (2009) Genotoxic stress in plants: shedding light on DNA damage, repair and DNA repair helicases. Mutat Res Rev Mutat Res 681(2–3):134–149
Tyunin AP, Kiselev KV, Zhuravlev YN (2012) Effects of total DNA demethylation on methyltransferase gene expression and resveratrol production in cell cultures of Vitis amurensis. Plant Cell Tissue Organ Cult 111:91–100
Tyunin AP, Kiselev KV, Karetin YA (2013) Differences in the methylation patterns of the VaSTS1 and VaSTS10 genes of Vitis amurensis Rupr. Biotechnol Lett 35:1525–1532
Vannozzi A, Dry I, Fasoli M, Zenoni S, Lucchin M (2012) Genome-wide analysis of the grapevine stilbene synthase multigenic family: genomic organization and expression profiles upon biotic and abiotic stresses. BMC Plant Biol 12:130
Vanyushin BF, Ashapkin VV (2011) DNA methylation in higher plants: past, present and future. Biochim Biophys Acta 8:360–368
Xu A, Zhan JC, Huang WD (2015) Effects of ultraviolet C, methyl jasmonate and salicylic acid, alone or in combination, on stilbene biosynthesis in cell suspension cultures of Vitis vinifera L. cv. Cabernet Sauvignon. Plant Cell Tissue Organ Cult 122:197–211
Yuan Y, Qi L, Yang J, Wu C, Liu Y, Huang L (2015) A Scutellaria baicalensis R2R3-MYB gene, SbMYB8, regulates flavonoid biosynthesis and improves drought stress tolerance in transgenic tobacco. Plant Cell Tissue Organ Cult 120:961–972
Zhu JK (2009) Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet 43:143–166
Acknowledgments
This work was supported by Grants from the following institutions and foundations: the Russian Foundation for Basic Research (14-04-31122) and the Russian Scientific Foundation (14-14-00366). The Russian Foundation for Basic Research supported all work related to grape cell culture transformations, selection of the transgenic calli, sequencing, and real-time PCR analysis; the Russian Scientific Foundation supported binary vector construction and HPLC analysis.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tyunin, A.P., Kiselev, K.V. Alternations in VaSTS gene cytosine methylation and t-resveratrol production in response to UV-C irradiation in Vitis amurensis Rupr. cells. Plant Cell Tiss Organ Cult 124, 33–45 (2016). https://doi.org/10.1007/s11240-015-0872-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-015-0872-6