
Abstract
Genetic engineering of secondary metabolic pathways is an emerging area of research for production and improvement of natural products in plant biotechnology. Here, we describe a systematic approach to manipulate a key regulatory step of isoprenoid biosynthetic pathway in Withania somnifera to study its effect on withanolide production. We generated T0 W. somnifera plants overexpressing squalene synthase (WsSQS) by Agrobacterium tumefaciens mediated transformation, which were analyzed by Gus biochemical assay and PCR of hygromycin phosphotransferase (hptII) and WsSQS. qRT-PCR analyses of various transformed tissues indicated 2–5 fold increase in WsSQS transcripts in both T0 and T1 generations. The tissue specific protein expression studies revealed 2–3 fold increase in WsSQS, which was further confirmed by enzyme activity. These observations were corroborated with the 1.5–2 fold increase in total withanolide content of the transformed tissues. However, in leaf tissue, the levels of Withaferin A and Withanolide A increased significantly up to 4–4.5 fold. These findings demonstrate genetic engineering of isoprenoid pathway in W. somnifera resulting in enhanced production of withanolides, and also provide insights into such metabolic pathways for their manipulation to improve the pharmacological content of different medicinally important plants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Isoprenoids are one of the largest groups of natural products with structurally diverse carbon skeleton present in all forms of life. They include several biologically active and commercially important compounds like sterols, steroidal sapogenins, alkaloids and lactones. Withanolides are one such class of naturally occurring steroidal lactones, commonly found in members of Solanaceae. A majority of these secondary metabolites (SMs) are concentrated in Withania somnifera, characterized by C28 ergostane backbone with a side chain of C9 having six-membered lactone ring which is a distinctive feature of these metabolites responsible for most of their biological activities.
W. somnifera (L.) Dunal, commonly known as Indian ginseng, is principally recognized for its medicinal value in Ayurveda and has been well investigated for its antiserotogenic, adaptogenic and anticancer activity (Winters 2006). These pharmacological efficacies of the plant are attributed to different withanolides which are actively involved in various plant processes such as defense, signaling, flavor and fragrance, and also accounts for hormonal, antibiotic, insecticidal and therapeutic activities of the plant. Withanolides are widely known for their antioxidant, anti-inflammatory, immunomodulating, antistress and rejuvenating effects (Bhattacharya et al. 2001; Gupta et al. 2003).
For large scale cultivation, in vitro regeneration has been widely studied in W. somnifera using various explants like axillary meristems (Roja et al. 1991), mature leaf (Joshi and Padhya 2010), shoot tips (Ray and Jha 2001), hypocotyl (Rani et al. 2003), and others (Ghimire et al. 2010; Logesh et al. 2010; Sharma et al. 2010). But there are few reports where nodal explants have been used for the regeneration and shoot multiplication in W. somnifera (Kumar et al. 2011; Sivanandhan et al. 2011). Agrobacterium tumefaciens mediated genetic transformation of W. somnifera using nodal explants have been shown to produce more number of transgenic shoots directly from nodal explants within a short period of time (Udayakumar et al. 2014). Apical and nodal explants have resulted in higher transformation efficiency in W. somnifera with proper concentration of phytohormones (Patel et al. 2014). In a present study, apical and nodal segments were used as explants for transformation.
W. somnifera is a slow growing shrub producing minute quantity of withanolides which depends on the physiological and developmental stage of the plant (Oksman-Caldentey and Inze 2004). Such a low abundance is incapable of meeting the present economic demand for medicinal formulations. In this regard, chemical synthesis of these plant natural products were attempted, however, sheer structural complexity and the specific stereochemical requirements of the compounds resulted in their low yield (Jana et al. 2011). Plant cell cultures had been set up as an alternative for extraction of SMs which ended up in limited commercial success and technological issues such as maintenance of large bioreactors and cultivation conditions (Oksman-Caldentey and Inze 2004).
This limitation, however, may be overcome by looking into the biosynthetic pathway of withanolides (Fig. 1) and using genetic engineering as a tool to manipulate the crucial steps of this metabolic network to increase their yield. The first committed step which diverts the carbon flux away from the central isoprenoid pathway and towards withanolide biosynthesis is the formation of squalene; catalyzed by a 47 kDa membrane associated enzyme, squalene synthase (SQS; EC 2.5.1.21) (Gupta et al. 2012). The substrate for this enzyme, farnesyl diphosphate (FPP), originates from isoprenoid biosynthetic pathway and can be channeled towards squalene accumulation which is supposed to be the first precursor of triterpenoids. SQS catalyses head to head condensation of two FPP molecules to form presqualene diphosphate which undergo rearrangements and cyclopropyl ring opening to form squalene in the presence of NADPH and Mg2+ (Pandit et al. 2000). Squalene oxidizes in presence of NADPH to afford squalene 2, 3-epoxide, subsequently cyclizing into lanosterol which acts as a backbone structure for various steroidal triterpenoids (Mirjalili et al. 2009). The complete biosynthetic pathway of withanolides is still unclear but they are supposed to be derived from 24-methylenecholesterol (Lockley et al. 1976).

Simplified scheme of withanolide biosynthetic pathway. Abbreviations: IPP Isopentenyl-5-pyrophosphate, DMAPP Dimethylallyl pyrophosphate, GPP Geranyl pyrophosphate, FPP Farnesyl pyrophosphate, and SQS Squalene synthase. Dashed arrows show that multiple steps are involved in the reaction
Since SQS acts at a regulatory branch point, it has attracted considerable interest as a possible genetic engineering target for diverting the metabolic flux to promote SM biosynthesis in plants. Many approaches have been investigated to understand the regulatory role of SQS in sterol biosynthesis using SQS mutants (Tozawa et al. 1999), fungal elicitors (Vogeli and Chappell 1988) and specific inhibitors of SQS (Wentzinger et al. 2002). Genetically improved Withania with increased levels of withanolides would have a profound impact on pharmaceutical industries. We show here that upregulation of squalene synthase in triterpenoid pathway leads to generation of transformed Withania plants by Agrobacterium mediated transformation, having normal phenotype and enhanced withanolides content as compared to the controls. These modified lines and the approach used are valuable for developing improved cultivars of beneficial crops.
Materials and methods
Plant material and propagation
The seeds of W. somnifera (L.) Dunal were procured from Vindhya herbals, Bhopal, MP, India. Seeds were surface sterilized, germinated, and micropropagation was achieved according to the protocol described already (Patel et al. 2014). In brief, the sterilized seeds were inoculated on half-strength solidified MS (Murashige and Skoog) medium (Murashige and Skoog 1962) and incubated in dark for 15 days. The germinated seed were transferred to half-strength liquid MS medium for further development. The grown seedlings were multiplied in proliferation medium (MS supplemented with 0.1 mg l−1 kinetin and 0.2 mg l−1 6-BAP) and rooted in rooting medium (half strength MS liquid medium containing 2 mg l−1 IBA). The apical and nodal segments of 1 cm length containing a single node were excised from in vitro grown shoots and propagated separately to observe their response in the medium. Both the tissues resulted in shoot multiplication and regeneration, and there was no significant difference noticed with respect to the time of regeneration in the medium, number of multiple shoot formation, rooting time and morphology of root (data not shown). Therefore, both apical and nodal segments of 1 cm length were used as explants for transformation irrespective of their type.
Vector construction
Previously cloned full length squalene synthase from W. somnifera (WsSQS; GenBank GU732820) with an open reading frame of 1242 bp (Gupta et al. 2012) was amplified using primers having sites for the restriction enzymes KpnI and SacI. The resulting fragment was positioned between cauliflower mosaic virus (CaMV) 35S promoter and a nopaline synthase (Nos) terminator in modified T-DNA region of pCAMBIA 1301 vector to ensure high level of gene expression (Patel et al. 2014). This vector contains a reporter gene namely gus (β–Glucuronidase) and a selectable marker hptII (hygromycin phosphotransferase) gene imparting resistance to hygromycin B under the control of the constitutive CaMV 35S promoter to confirm transgene expression The construct was transformed into A. tumefaciens GV2260 for subsequent genetic transformation. W. somnifera explants transformed with Agrobacterium harboring the empty vector were used as control.
Genetic transformation of W. somnifera
Genetic transformation in W. somnifera was achieved by A. tumefaciens GV2260 carrying WsSQS in three sets of 30 explants each, according to the already described protocol (Patel et al. 2014). Essentially, the preconditioned explants were wounded and immersed in a freshly prepared bacterial suspension for 15 min, blotted dry to remove excess of bacterial culture and transferred to the proliferation medium. Cocultivation was carried out for 48 h in dark at 26 ± 2 °C. After 48 h, the cocultivated explants were washed with sterile distilled water containing 250 mg l−1 cefotaxime, blotted dry and shifted to proliferation medium supplemented with 250 mg l−1 cefotaxime. After 10 days, the explants were transferred to the selection medium (proliferation medium containing 10 mg l−1 hygromycin B) and shoot development was monitored. Expression of the inserted genes in the transformed tissues was regularly observed by visualizing the β–Glucuronidase activity in different sectors of treated explants and growing shoots under a stereomicroscope (Leica MZ 125, Switzerland). The hygromycin B-resistant Gus-positive shoots were continuously maintained on selection pressure while the concentration of cefotaxime was reduced to 100 mg l−1 and then completely eliminated. To produce independent transformed lines, the shoots were detached from the transformed explant, cultured on proliferation medium and rooted onto the rooting medium.
Molecular identification of transformants
PCR (C1000 BIO-RAD thermal cycler, BIO-RAD, USA) was carried out using primers specific to the hptII and WsSQS. Total genomic DNA was extracted from tissues of wild-type and hygromycin B-resistant transformed shoots by using plant DNA extraction kit (Hipura Plant Genomic Purification kit, Himedia, India). The hptII gene specific forward and reverse primer sequences used were 5′-TCCTGCAAGCTCCGGATGCCTC-3′ and 5′-CGTGCACAGGGTGTCACGTTGC-3′, respectively. For WsSQS gene specific PCR, the forward primer was designed from the sequence of CaMV 35S promoter (GeneBank GQ336528.1; 5′-ACAGTCTCAGAAGACCAAAGGGCA-3′) to avoid endogenous SQS amplification, and reverse primer was designed from the 3′ terminal sequence of WsSQS (5′-GAGCTCCTAAGATCGGTTGCCAG-3′). The components of PCR reaction mixture were: 15 ng template DNA, 150 µM dNTPs, hptII/WsSQS gene specific forward and reverse primers (0.66 pmol each), 0.5 U of Taq DNA polymerase in a total volume of 15 µl with 1X reaction buffer. The PCR reaction was carried out as follows: an initial denaturation at 94 °C for 5 min followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s and extension at 72 °C for 1 min (for hptII) and 1.6 min (for WsSQS) and a final 5 min extension at 72 °C. The amplified products were subjected to 1 % (w/v) agarose gel electrophoresis and visualized by ethidium bromide staining under UV.
qRT-PCR analysis
Total RNA was isolated from different tissues of WsSQS transformed lines and controls using Plant RNA Isolation Kit (Invitrogen, USA) as per manual instructions and treated with DNase using DNase I Digest kit (Sigma, USA). Total RNA (2 µg) was reverse transcribed into cDNA using AMV reverse transcription system (Promega, USA) with oligo dT primers in a 20 μL reaction volume. The reaction mixture was incubated for 1 h at 42 °C. For normalization of the relative expression data, ubiquitin gene (UBC) was employed as an internal standard using primer mix from Eurogentec (Belgium). To quantify the WsSQS transcripts, first-strand DNA was PCR amplified using gene-specific primers: SQS-F (5′-TTTATGATCGTGAATGGCACTTTTC-3′) and SQS-R (5′-AGCGGTTGAAACATGATGGAAC-3′) synthesized from WsSQS. All qRT-PCR reactions were performed with SYBR Green Brilliant® II QPCR Master Mix (2× with low ROX, Stratagene, USA) on Mx 3000P instrument (Stratagene, USA) according to the manufacturer’s instructions. PCR cycling conditions included a DNA denaturing stage of 95 °C for 10 min, followed by 40 cycles of 95 °C for 30 s, 55 °C for 45 s and 72 °C for 30 s. The amplified products were analyzed with MxPro software provided with the machine. Data were analyzed by comparative Ct method (Pfaffl 2001).
Indirect ELISA and western blot
Fresh tissues (500 mg each) of WsSQS transformed and control plants were ground and resuspended in 1 ml of phosphate buffered saline (PBS; 136 mM NaCl, 2 mM KCl, 8 mM Na2HPO4, 1 mM KH2PO4, pH 7.5) containing CHAPS (5 mM) to solubilize membrane proteins. Total protein quantity was estimated in the supernatant by Bradford assay using bovine serum albumin as standard. The polyclonal antibodies against purified WsSQS were raised in rabbits (New Zealand White) at National Toxicology Centre, Pune, India, and the antibody titer of the anti-SQS serum was determined. For detection of WsSQS in total plant protein, equal concentration of extracted protein (100 μL/well) was coated on 96 well polystyrene microtiter plate (Costar, USA) overnight at 4 °C followed by washings with PBST (PBS + 0.05 % Tween 20). Non-specific sites were blocked with blocking buffer (PBS + 1 % BSA) and incubated for 2 h at 37 °C. After washing thrice with PBST, primary antibody (rabbit polyclonal IgG against WsSQS) was added and incubated for 2 h at 37 °C. The unbound primary antibody was washed thrice with PBST and the plate was exposed to secondary antibody (goat anti-rabbit IgG-alkaline phosphatase conjugate, 1:20000) followed by incubation for 1–2 h at 37 °C. The presence of antigen was determined by the addition of enzyme specific substrate pNPP (p-Nitro Phenyl Phosphate; 1 mg ml−1) and incubating for 45 min in dark for the color development. The reaction was stopped by adding 10 mM EDTA and the absorbance was measured at 405 nm using an xMark ELISA plate reader (BIO-RAD, USA). Detection limit of ELISA was determined by plotting a standard curve using the purified recombinant truncated WsSQS protein. The concentration of the WsSQS present in total soluble protein extracted from control and WsSQS transformed plants were analyzed in each case of three replications from the standard curve.
For western blot, total crude protein (50 μg) from WsSQS transformed and control tissues was electrophoresed on 10 % SDS-PAGE and electro-transferred on to PVDF membrane using iBlot gel transfer system (Invitrogen) as per manufacturer’s instructions, with recombinant truncated WsSQS used as a size marker. WesternBreeze kit (Invitrogen, USA) was used for further processing of the blot. Blot was placed in blocking solution and incubated at room temperature for 30 min on rotatary shaker. The membrane was rinsed and incubated with primary antibody solution for 1 h. The membrane was washed thrice and incubated in secondary antibody for 30 min. Signals were detected with ready to use 5-bromo-4-chloro-3-indolyl phosphate and nitrobluetetrazolium (BCIP/NBT) solution (Calbiochem, Germany).
WsSQS enzyme acitivity
The microsomal protein fractions were prepared from different tissues as already described (Vogeli and Chappell 1988). Essentially, 1 g of frozen tissue was homogenized in 10 mL of protein extraction buffer, filtered and centrifuged at 10,000 g for 25 min at 4 °C. The supernatant was again centrifuged at 100,000 g for 60 min to obtain the microsomal pellet. The pellet was resuspended in 200 μL of 100 mM Tris·Cl (pH 8), 1.5 mM DTT and 20 % glycerol, and the protein concentration were determined by Bradford method.
Assay for WsSQS enzyme was carried out with 10 µg microsomal protein according to the method described previously (Gupta et al. 2012). The enzyme activity was determined fluorimetrically by measuring NADPH depletion during the reaction on an LS 55 spectrofluorimeter (Perkin Elmer). The assay mixture was excited at 340 nm and emission was recorded in the range 400–500 nm with characteristic maxima around 460 nm corresponding to NADPH fluorescence. Excitation and emission slits were kept at 7.5 and 2.5 nm, respectively, with a scan speed of 100 nm min−1. The reaction was carried out at 30 °C for 1 h and averaged fluorescence of 5 accumulated scans were recorded at regular time intervals. A standard curve was prepared by plotting fluorescence of commercially available NADPH (dissolved in 50 mM Tris–Cl; pH 8) at 460 nm against concentration.
In order to validate the enzyme reactions, squalene formed in each reaction was checked on GC–MS. Replicates of the above mentioned reactions, after 2 h of incubation, were extracted using tert-butyl methyl ether, and concentrated to 100 µL by bubbling dry nitrogen. The concentrate (1 µL) was injected on GC–MS (Agilent 5975C mass selective detector interfaced with an Agilent 7890A gas chromatograph) fitted with a capillary column HP-5 (25 m × 0.25 mm, film thickness 0.33 µm 5 % methylpolysiloxane cross-linked capillary column, Hewlett-Packard, USA) with a split ratio of 10:1. The injector temperature was set at 290 °C with helium as the carrier gas (10 ml min−1). The oven temperature was programmed from 150 to 250 °C at 10 °C min−1 and from 250 to 310 °C at the rate of 5 °C min−1, and maintained at final temperature for 5 min. The chromatogram obtained was compared with the authentic squalene (Sigma, USA) for its retention time and mass fragmentation pattern (see Supplementary Fig. 1). The squalene content was calculated from the standard curve plotted from the peak area versus concentrations of authentic squalene, and expressed as nmol mg−1 protein.
Withanolides extraction and LC–MS analysis
Dried tissues (100 mg each) were separately crushed to fine powder and percolated thrice with 5 ml methanol for 1 h under shaking conditions at room temperature. The extracts were pooled, filtered, concentrated under reduced pressure at 45 °C and thoroughly washed with double volume of n-hexane. The methanolic fraction was dried completely and further partitioned twice with water:chloroform (1:1). The chloroform fractions were pooled, concentrated and finally dissolved in 150 µl methanol. The samples were filtered and subjected to liquid chromatography. All the solvents used in the study were HPLC grade purchased from Fischer Scientific, USA.
LC–MS was performed on Waters Acquity UPLC system (Milford, MA, USA) with an Acquity UPLC® BEH C18 column (2.1 × 100 mm, 1.7 µm) attached to a positive ion elecrospray ionization-mass spectrometer (Waters) for the identification and quantification of withanolides in W. somnifera extracts. Separations were achieved using a binary gradient elution of water (solvent A) and acetonitrile (solvent B) with the following program carried out at 25 °C: 10 % B for 2 min; 45 % B for 8 min; 75 % B for 10 min; and 95 % B for 5 min, at a flow-rate of 0.4 ml min−1, with a total run time of 25 min. External standards of different withanolides (Chromadex, USA) were used to construct calibrated graph of peak area versus withanolide concentration, being linear over 10 measurements at different concentrations.
Statistical analysis
All the experiments were repeated thrice and the data of each experiment were obtained in three replicates. Data were analyzed using Graphpad Prism 5 software (San Diego, California). Values are represented as mean ± SE. Statistical analysis was performed using Student’s t test to evaluate difference between the control and transformed plants. P values <0.05 are considered significant.
Results and discussion
Genetic transformation of W. somnifera
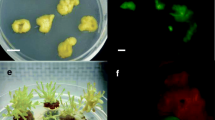
The apical and nodal explants derived from tissue culture raised W. somnifera were transformed with WsSQS with the aim of improving withanolide accumulation in the plant. Gus assay enabled the monitoring of expression of the introduced genes in planta, thus identifying the Gus-positive shoots (Fig. 2e–g). This was the first level of screening where the chimeric and untransformed shoots were identified and separated, while the Gus-positive transformants were allowed to flourish in presence of hygromycin B. Shoot elongation and multiplication were observed in some explants while many shoots turned necrotic indicating elimination of the untransformed tissues in the growth process. Green and living shoots were separated and cultured in fresh selection medium (Fig. 2b). The hormonal combination of 0.1 mg l−1 Kinetin and 0.2 mg l−1 6-BAP supported quick and maximum shoot proliferation and multiplication of the transformed explants (Patel et al. 2014). Elimination of any remaining chimeric shoots having untransformed branches was carried out by performing Gus assay regularly with different sectors and tissues of the growing shoots. Green shoots were subsequently transferred onto the rooting medium (Fig. 2c). In this process, total 10 hygromycin B resistant transformed lines were recovered. The rooted T0 transformed plants were successfully hardened and shifted to green house (Fig. 2d). These plants were normal in growth and appearance with no phenotypic aberrations.

Stages of genetic transformation of W. somnifera. a Nodal explant in proliferation media after 2 days of transformation; b shoot elongation and proliferation; c rooted plantlet; d successfully hardened transformed plant in green house; e Gus stained leaf tissue of T0 plant; f Gus stained apical bud of T0 plant; and g regenerating shoot of T0 plantlet showing origin of Gus positive tissue
The genomic DNA isolated from wild-type and hygromycin B-resistant T0 plants were analyzed by PCR for the presence of hptII and WsSQS genes which showed an amplification of ~600 bp (Fig. 3a) and ~1.6 kb (Fig. 3b) respectively. Out of 10 explants that survived on selection medium, only 6 transformed lines tested positive for PCR analysis, thus resulting in an overall transformation frequency of 6.66 %. The empty vector control and WsSQS transformed T0 plants of same age, size and similar growth characteristics were taken into consideration for further analyses, and the mean values of three control plants were used for each experiments.

Molecular analyses of T0 and T1 lines. a hptII specific PCR showing ~600 bp amplified products; and b WsSQS specific PCR showing ~1.6 kb amplified products. M: Low range molecular weight ladder (Banglore Genei, India); P: Positive control (plasmid pCAMBIA 1301 carrying WsSQS insert); N: Negative control (wild-type plant); 1–10: putative T0 transformed lines; and 11–13: T1 individuals
Although, regeneration has been reported frequently in W. somnifera (Kanungo and Sahoo 2011; Sivanesan 2007), transformation studies were not carried out in any one of them. Genetic transformation of W. somnifera for increased production of withanolides has been confined to hairy roots through A. rhizogenes (Bandyopadhyay et al. 2007; Murthy et al. 2008), however, hairy root cultures normally produce chemicals which are synthesized in the roots of intact plants. None of previous reports on transformation in W. somnifera resulted in stable and successful regeneration of plants from the transgenic tissue. There are only two reports on A. tumefaciens-mediated transformation in W. somnifera where in one study, the wild type strain of A. tumefaciens resulted in the formation of shooty teratomas (Ray and Jha 1999) while another report demonstrated very low transformation efficiency using leaf explants from green house grown plants (Pandey et al. 2010). Obtaining good transformation efficiency has always been a major concern and prerequisite for manipulating the SM biosynthetic pathways. The higher transformation efficiency in the present study can be attributed to the efficient axillary proliferation method employed which utilized apical and nodal segments derived from in vitro raised Withania plants. Rapid proliferation system provides large number of genetically identical explants for transformation. Explant selection is another factor that is important for successful transformation and plant regeneration. The present study used apical and nodal segments as explants. The use of organized meristematic tissues maintains cultivar integrity, and has the benefit of rapid shoot elongation and proliferation, thus reducing the chances of somaclonal variations (Sivanesan 2007). Apical and nodal segments are considered to be the ideal for achieving higher transformation efficiency with the proper concentrations of growth regulators (Patel et al. 2014). Nodal explants have also shown to form multiple shoots both from pre-existing and de novo buds on MS medium containing 0.1–0.5 mg l−1 BAP (Kulkarni et al. 2000). The greatest number of shoots were obtained from the nodal explants in MS medium supplemented with phytohormones (Kumar et al. 2011; Manickam et al. 2000; Sivanesan and Murugesan 2008). The age and type of donor tissue also play crucial role in biochemistry of subsequent cultures lines. In vitro cultures have the differential capacity to synthesize SMs depending on the morphological nature of type of explant utilized. The plantlets regenerated from apical and nodal explants have been shown to possess higher content of Withanolide A and Withanone (Sharada et al. 2007).
Accumulation of WsSQS mRNA in transformed tissues
The accumulation of WsSQS mRNAs in control and WsSQS transformed lines was analyzed by qRT-PCR. In control plants, the maximum expression of WsSQS was observed in root followed by leaf and stem (Fig. 4). Root and leaf tissues accumulated 4 and 2 fold higher WsSQS mRNA than stem, respectively. Of all the WsSQS transformed T0 plants analyzed by qRT-PCR, three best performing lines (T020, T058 and T079) are shown where the mRNA levels increased up to 2–5 fold (Fig. 4). Similar results were obtained with Eleutherococcus senticosus (Seo et al. 2005) and Panax ginseng (Lee et al. 2004), where transformation with SQS, resulted in accumulation of SQS mRNA in the leaves and adventitious roots of the transgenic plants, respectively. This was later observed in Bupleurum falcatum also, where SQS mRNA expression was enhanced in sense transgenic roots but suppressed in antisense roots as compared to non-transgenic roots (Kim et al. 2011). In the present study, almost 3.5 and 2.5 fold increase in WsSQS mRNA was observed in leaf and root tissue of WsSQS transformed lines, respectively. Although, stem showed the maximum fold increase (about 5 fold), the maximum expression of WsSQS was observed in root than rest of the tissues (Fig. 4). These results demonstrate that WsSQS, catalyzing the regulatory step leading to withanolide biosynthesis, was up-regulated in all the transformed tissues.

Tissue specific WsSQS transcript analysis in transformed W. somnifera T0 and T1 lines by qRT-PCR. Ubiquitin gene was used as an internal control. Three best performing T0 transformed line (T020, T058 and T079) and one T1 progeny (T179) were chosen to study mRNA accumulation against the control plant. Values are the means of three replicate measurements and error bars show the SE of the mean. Asterisk indicates significant differences, (*) for P < 0.05; (**) for P < 0.001; (***) for P < 0.0001, based on Student’s t test
WsSQS protein levels and enzyme activity in transformed plants
Indirect ELISA was performed to investigate the relative increase of WsSQS protein levels in WsSQS overexpressing T0 transformed tissues. The antisera dilution 1:5000 was found to contain optimum antibody titer for carrying out the immunological studies. The control tissues showed almost equal expression of WsSQS in root and leaf, which was 2 fold greater than stem. It was found different from the transcript expression pattern in control tissues where root showed highest mRNA levels followed by leaf and stem. There was no direct co-relation found between mRNA expression and protein accumulation patterns, however, consistent with the transcript expression results, ELISA showed increased protein levels in all the WsSQS overexpressing transformed tissues, with maximum accumulation in leaf followed by root and stem. Absolute quantification of protein was carried out by plotting the standard curve. In WsSQS transformed leaf, WsSQS increased from 1.3 to 3.6 µg g−1 fr wt, in stem from 0.51 to 1.2 µg g−1 fr wt, and in root from 1.42 to 3.6 µg g−1 fr wt (Fig. 5a). This overexpression was further witnessed by western blotting (Fig. 5b). The recombinant truncated WsSQS protein (44 kDa) was used as size marker. Different tissues of T079 showed clear immune-precipitated protein bands with the higher intensity corresponding to the expected size (~47 kDa).

WsSQS protein expression analyses. a WsSQS protein quantification in total soluble protein extracted from different tissues of control and WsSQS transformed lines by ELISA, determined from the standard curve plotted between purified recombinant WsSQS protein concentration and absorbance at 405 nm. Values are the means of three replicates from each biological sample and error bars show the SE of the mean. Asterisk indicates significant differences, (*) for P < 0.05; (**) for P < 0.001; (***) for P < 0.0001, based on Student’s t test; and b Western blot analysis of W. somnifera transformed with pCAMBIA 1301 harboring WsSQS gene. Total soluble protein (50 μg) from different tissues of T079 and control plant was immunodetected with antisera against recombinant WsSQS (1:5000 dilution). RP recombinant truncated WsSQS protein as size marker, CL control leaf, TL WsSQS transformed leaf, CS control stem, TS WsSQS transformed stem, CR control root; and TR WsSQS transformed root
SQS activity was measured flourimetrically by calculating the amount of NADPH consumed in the reaction, as well as detecting squalene formation in these reactions by GC–MS (see Supplementary Fig. 1). The maximum increase in enzyme activity was found to be 3.3 fold in root tissue followed by 2.7 fold in leaf and 2.1 fold in stem (Table 1). The results obtained clearly indicated the increased SQS content in the T0 WsSQS transformed lines (Table 1), which is a consequence of the increased expression of WsSQS. Likewise, the enzyme activity was 1.5–3 fold higher and 1.2–3 fold higher in transgenic embryos and transgenic plants of E. senticosus, respectively (Seo et al. 2005). Similar results were obtained with P. ginseng (Lee et al. 2004) and B. falcatum (Kim et al. 2011).
Tissue specific accumulation of withanolides in transformed W. somnifera
The present study offers a simple analytical LC–MS system providing well-resolved and symmetrical peaks required for accurate quantitative analysis of withanolides. This allowed identification of four withanolides in the plant extract which were confirmed by retention time (RT) and mass spectrum of their respective standards facilitating their quantification: Withaferin A (11.31 min), Withanolide A (12.35 min), Withanolide B (15.03 min) and Withanone (18.35 min) (see Supplementary Fig. 2). The metabolite profiles indicated variability between the different tissues but their level was found to be higher than control (see Supplementary Fig. 2). The overexpression of WsSQS resulted in up to 2 fold increase in total withanolide content of WsSQS T0 transformed plants. The total withanolide content of control leaf, stem and root was 1.84, 2.24 and 2.25 mg g−1 dry wt of the tissue, respectively, which increased to 3.55, 3.37 and 3.98 mg g−1 dry wt of their respective tissue of T079 (Fig. 6). The total withanolide increase of about 1.9 fold was achieved in leaf tissue followed by 1.8 in root and 1.5 in stem. The leaf tissue showed maximum fold increase but the overall concentration of total withanolides was highest in WsSQS transformed root (Fig. 6). The major change in concentration of withanolides was found in leaf where Withaferin A and Withanolide A increased up to 4–4.5 folds while there was negligible change in Withanolide B and Withanone. This pattern also reflected in root where Withaferin A and Withanolide A increased up to 2–2.5 fold whereas the concentration of Withanolide B and Withanone reached only 1.2 and 1.3 fold, respectively. The metabolite profile of stem was completely different from leaf and root where although Withaferin A and Withanolide B increased to about 2 and 1.5 fold, respectively, the quantity of Withanolide A was found to be almost same, while the concentration of Withanone increased to 3.7 fold. There are previous reports on tissue specific accumulation of SMs in W. somnifera where in one study, root tissue accumulated maximum Withanolide A and Withaferin A (Manwar et al. 2012), which is in sharp contrast to another study where the plant was found to accumulate these withanolides more in leaf tissue (Sharma et al. 2007).

Improved production of withanolides in T0 and T1 transformed tissues overexpressing WsSQS. Four different withanolides are shown in leaf, stem and root tissue of control (C), three transformed lines (T020, T058 and T079) and one T1 progeny (T179) where T079 is the best performing line. The control line (C) is a tissue transformed with Agrobacterium harboring the empty vector. Vertical bars indicate the mean values ± SE from three independent experiments. Asterisk indicates significant differences, (*) for P < 0.05; (**) for P < 0.001; (***) for P < 0.0001, based on Student’s t test
Analysis of W. somnifera T1 plants
The primary transformants showing increased withanolides content were raised to advance to the T1 progeny by selfing the seeds under green house conditions. DNA was extracted from T1 progeny plants, and subjected to hptII and WsSQS specific PCR, as described in Materials and Methods section, which showed an amplification of ~600 bp (Fig. 3a) and ~1.6 Kb (Fig. 3b), respectively; thus confirming the inheritance of inserted genes in the progeny. It was further verified by monitoring T1 individuals for the increased expression of mRNA and protein, and withanolides production. The data of T179 is presented here which is the progeny of maximum withanolides producing T0 plant. qRT-PCR of T1 progeny showed WsSQS mRNA expression levels similar to T0 lines, suggesting a stable expression of gene in T1 line (Fig. 4). ELISA analysis of T179 indicated that the protein expression remained at par with that of T0 plants. The protein quantity ranged from 0.75 to 3.25 µg g−1 fr wt of the tissue (Fig. 5a). The slight variation in the expression is acceptable which may be arising due to the developmental stage of the plant and season of growth. LC–MS of T1 plants was conducted to identify the plants that had retained increased withanolide content which was found more or less consistent with T0 plants (Fig. 6).
There are few reports on plant cell and hairy root cultures initiated for the increased production of SM in Withania. Murthy et al. (2008) developed transformed hairy root cultures of W. somnifera which produced 2.7 fold higher Withanolide A. In another study, W. somnifera transformation with A. tumefaciens resulted in formation of shooty teratomas which accumulated Withaferin A and Withanolide D, and rooty teratomas with Withanolide D, but the levels of these withanolides was low (Ray and Jha 1999). The overexpression of Arabidopsis thaliana SS1 gene (encoding SQS) in W. coagulans using A. rhizogenes resulted in increased phytosterol and withanolide production in engineered hairy roots, thus indicating the potentiality of SQS to promote triterpenoid biosynthesis (Mirjalili et al. 2011). None of the previous studies on hairy root and plant cell cultures have resulted in significant levels of the metabolites required for their economic exploitation. Moreover, an important constraint in the commercial utilization of hairy root culture is development and up-scaling of appropriate vessels for the delicate and sensitive hairy roots.
Metabolic engineering of withanolide biosynthetic pathway is an alternative approach for their improvement. SQS operates at a branch point of the pathway regulating the metabolic flux and catalyzes the first committed step leading to the synthesis of different withanolides (Mirjalili et al. 2009). Thus, manipulating the expression of SQS could be a tool for modifying the SMs of this plant. This paper describes the metabolic engineering of isoprenoid pathway by overexpressing SQS which resulted in enhanced production of withanolides in W. somnifera. Similar results were obtained with P. ginseng (Lee et al. 2004) and E. senticosus (Seo et al. 2005) where the transformed plants accumulated 1.6–3 fold higher total ginsenoside content in adventitious roots and 2–2.5 times higher triterpene saponin, respectively. Likewise, transgenic roots of B. falcatum overexpressing BfSS1 in the sense orientation resulted in SQS mRNA accumulation and enhanced production of both phytosterol and saikosaponins (Kim et al. 2011). Overexpression of SQS in Glycyrrhiza uralensis led to 2.6 fold higher glycyrrhizin content as compared to control hairy roots (Lu et al. 2008). There is only one report of SQS overexpression in W. somnifera which resulted in 2.5 fold increase in Withanolide A and production of Withaferin A from the transformed callus and cell suspension cultures (Grover et al. 2013). Cell suspension cultures are more prone to contamination and are difficult to maintain, and callus is a chimeric tissue, thus reducing the complete effect of total number of transformed cells actually contributing in improved withanolide production. The fate of developing cells in callus is unknown, thus providing no idea about the specific tissue involved in the production of SMs. It has also been reported previously that transformed undifferentiated callus and cell suspension cultures of W. somnifera are generally unable to synthesize withanolides (Bandyopadhyay et al. 2007; Roja et al. 1991). The major drawback of callus culture is their tendency to undergo frequent genetic erosions (D’Amato 1977), and therefore, pure clones cannot be maintained. So callus culture could be limited source of metabolites while transformed plant production is suitable option for conservation of germplasm. Development of whole transformed plants presents a one step ahead and convenient technology for steady supply of bioactive compounds.
In summary, the present study establishes significant involvement of SQS in withanolide biosynthesis. Previously, we reported the isolation and cloning of WsSQS from W. somnifera (Gupta et al. 2012), and here we demonstrate enhanced withanolides in W. somnifera by overexpressing squalene synthase. Such plant metabolic engineering studies can be used as a tool to improve the production of Withania bioactive compounds, as well as obtain greater understanding of secondary metabolism, in general.
References
Bandyopadhyay M, Jha S, Tepfer D (2007) Changes in morphological phenotypes and withanolide composition of Ri-transformed roots of Withania somnifera. Plant Cell Rep 26(5):599–609
Bhattacharya A, Ghosal S, Bhattacharya SK (2001) Anti-oxidant effect of Withania somnifera glycowithanolides in chronic footshock stress-induced perturbations of oxidative free radical scavenging enzymes and lipid peroxidation in rat frontal cortex and striatum. J Ethnopharmacol 74(1):1–6
D’Amato F (1977) Cytogenetics of differentiation in tissue and cell cultures. In: Applied and fundamental aspects of plant cell, tissue, and organ culture. Springer, Berlin, pp 343–357
Ghimire BK, Seong ES, Kim EH, Lamsal K, Yu CY, Chung IM (2010) Direct shoot organogenesis from petiole and leaf discs of Withania somnifera (L) Dunal. Afr J Biotechnol 9:7453–7461
Grover A, Samuel G, Bisaria VS, Sundar D (2013) Enhanced withanolide production by overexpression of squalene synthase in Withania somnifera. J Biosci Bioeng 115(6):680–685
Gupta SK, Dua A, Vohra BP (2003) Withania somnifera (Ashwagandha) attenuates antioxidant defense in aged spinal cord and inhibits copper induced lipid peroxidation and protein oxidative modifications. Drug Metabol Drug Interact 19(3):211–222
Gupta N, Sharma P, Santosh Kumar RJ, Vishwakarma RK, Khan BM (2012) Functional characterization and differential expression studies of squalene synthase from Withania somnifera. Mol Biol Rep 39(9):8803–8812
Jana CK, Hoecker J, Woods TM, Jessen HJ, Neuburger M, Gademann K (2011) Synthesis of withanolide A, biological evaluation of its neuritogenic properties, and studies on secretase inhibition. Angew Chem Int Ed Engl 50(36):8407–8411
Joshi AG, Padhya MA (2010) Shoot regeneration from leaf explants of Withania somnifera (L.) Dunal. Not Sci Biol 2:63–65
Kanungo S, Sahoo SL (2011) Direct organogenesis of Withania somnifera L. from apical bud. International Research. J Biotechnol 2(3):058–061
Kim YS, Cho JH, Park S, Han JY, Back K, Choi YE (2011) Gene regulation patterns in triterpene biosynthetic pathway driven by overexpression of squalene synthase and methyl jasmonate elicitation in Bupleurum falcatum. Planta 233(2):343–355
Kulkarni AA, Thengane SR, Krishnamurthy KV (2000) Direct shoot regeneration from node, internode, hypocotyl and embryo explants of Withania somnifera. Plant Cell Tissue Organ Cult 62:203–209
Kumar OA, Jyothirmayee G, Tata SS (2011) Multiple shoot regeneration from nodal explants of Ashwagandha (Withania somnifera) (L.) Dunal. Asian J Exp Biol Sci 2(4):636-640
Lee MH, Jeong JH, Seo JW, Shin CG, Kim YS, In JG, Yang DC, Yi JS, Choi YE (2004) Enhanced triterpene and phytosterol biosynthesis in Panax ginseng overexpressing squalene synthase gene. Plant Cell Physiol 45(8):976–984
Lockley WJS, Rees HH, Goodwin TW (1976) Biosynthesis of steroidal withanolides in Withania somnifera. Phytochemistry 15:937–939
Logesh P, Settu A, Thangavel K, Ganapathi A (2010) Direct in vitro regeneration of Withania somnifera (L.) Dunal through leaf disc culture. Int J Biol Technol 1:1–4
Lu H-Y, Liu J-M, Zhang H-C, Yin T, Gao S-L (2008) Ri-mediated Transformation of Glycyrrhiza uralensis with a Squalene Synthase Gene (GuSQS1) for Production of Glycyrrhizin. Plant Mol Biol Rep 26:1–11
Manickam VS, Mathavan RE, Antonisamy R (2000) Regeneration of Indian ginseng plantlets from stem callus. Plant Cell Tissue Organ Cult 62:181–185
Manwar JV, Mahadik KR, Paradkar AR, Takle SP, Sathiyanarayanan L, Patil SV (2012) Determination of withanolides from the roots and herbal formulation of Withania somnifera by HPLC using DAD and ELSD detector. Der Pharmacia Sinica 3(1):41–46
Mirjalili MH, Moyano E, Bonfill M, Cusido RM, Palazon J (2009) Steroidal lactones from Withania somnifera, an ancient plant for novel medicine. Molecules 14(7):2373–2393
Mirjalili MH, Moyano E, Bonfill M, Cusido RM, Palazon J (2011) Overexpression of the Arabidopsis thaliana squalene synthase gene in Withania coagulans hairy root cultures. Biol Plantarum 55(2):357–360
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassay with tobacco tissue cultures. Physiol Plant 15(3):473–497
Murthy HN, Dijkstra C, Anthony P, White DA, Davey MR, Power JB, Hahn EJ, Paek KY (2008) Establishment of Withania somnifera hairy root cultures for the production of withanolide A. J Integr Plant Biol 50(8):975–981
Oksman-Caldentey KM, Inze D (2004) Plant cell factories in the post-genomic era: new ways to produce designer secondary metabolites. Trends Plant Sci 9(9):433–440
Pandey V, Misra P, Chaturvedi P, Mishra MK, Trivedi PK, Tuli R (2010) Agrobacterium tumefaciens-mediated transformation of Withania somnifera (L.) Dunal: an important medicinal plant. Plant Cell Rep 29(2):133–141
Pandit J, Danley DE, Schulte GK, Mazzalupo S, Pauly TA, Hayward CM, Hamanaka ES, Thompson JF, Harwood HJ Jr (2000) Crystal structure of human squalene synthase. A key enzyme in cholesterol biosynthesis. J Biol Chem 275(39):30610–30617
Patel N, Patel P, Kumari U, Kendurkar SV, Khan BM (2014) Microprojectile bombardment assisted agroinfection increases transformation efficiency of Withania somnifera (L.). Res Biotechnol 5(4):13–24
Pfaffl MW (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucl Acids Res 29(9):e45
Rani G, Virk GS, Nagpal A (2003) Callus induction and plantlet regeneration in Withania somnifera (L.) Dunal. In Vitro Cell Dev Biol Plant 39:468–474
Ray S, Jha S (1999) Withanolide synthesis in cultures of Withania somnifera transformed with Agrobacterium tumefaciens. Plant Sci 146:1–7
Ray S, Jha S (2001) Production of withaferin A in shoot cultures of Withania somnifera. Planta Med 67:432–436
Roja G, Heble MR, Sipahimalani AT (1991) Tissue cultures of Withania somnifera: morphogenesis and withanolide synthesis. Phytother Res 5:185–187
Seo JW, Jeong JH, Shin CG, Lo SC, Han SS, Yu KW, Harada E, Han JY, Choi YE (2005) Overexpression of squalene synthase in Eleutherococcus senticosus increases phytosterol and triterpene accumulation. Phytochemistry 66(8):869–877. doi:10.1016/j.phytochem.2005.02.016
Sharada M, Ahuja A, Suri KA, Vij SP, Khajuria RK, Verma V, Kumar A (2007) Withanolide production by in vitro cultures of Withania somnifera and its association with differentiation. Biol Plantarum 51(1):161–164
Sharma V, Gupta AP, Bhandari P, Gupta RC, Singh B (2007) A validated and densitometric HPTLC method for the quantification of Withaferin-A and Withanolide-A in different plant parts of two morphotypes of Withania somnifera. Chromatographia 66:801–804
Sharma MM, Ali DJ, Batra A (2010) Plant regeneration through in vitro somatic embryogenesis in ashwagandha (Withania somnifera L. Dunal). Researcher 2:1–6
Sivanandhan G, Mariashibu TS, Arun M, Rajesh M, Kasthurirengan S, Selvaraj N, Ganapathi A (2011) The effect of polyamines on the efficiency of multiplication and rooting of Withania somnifera (L.) Dunal and content of some withanolides in obtained plants. Acta Physiol Plant 33:2279–2288
Sivanesan I (2007) Direct regeneration from apical bud explants of Withania somnifera Dunal. Indian J Biotechnol 6:125–127
Sivanesan I, Murugesan K (2008) An efficient regeneration from nodal explants of Withania somnifera Dunal. Asian J Plant Sci 7:551–556
Tozawa R, Ishibashi S, Osuga J, Yagyu H, Oka T, Chen Z, Ohashi K, Perrey S, Shionoiri F, Yahagi N, Harada K, Gotoda T, Yazaki Y, Yamada N (1999) Embryonic lethality and defective neural tube closure in mice lacking squalene synthase. J Biol Chem 274(43):30843–30848
Udayakumar R, Kasthurirengan S, Mariashibu TS, Rayan JJS, Ganapathi A, Kim SC, Kim JJ, Choi CW (2014) Agrobacterium-mediated genetic transformation of Withania somnifera using nodal explants. Acta Physiol Plant 36:1969–1980
Vogeli U, Chappell J (1988) Induction of sesquiterpene cyclase and suppression of squalene synthetase activities in plant cell cultures treated with fungal elicitor. Plant Physiol 88(4):1291–1296
Wentzinger LF, Bach TJ, Hartmann MA (2002) Inhibition of squalene synthase and squalene epoxidase in tobacco cells triggers an up-regulation of 3-hydroxy-3-methylglutaryl coenzyme a reductase. Plant Physiol 130(1):334–346
Winters M (2006) Ancient medicine, modern use: Withania somnifera and its potential role in integrative oncology. Altern Med Rev 11:269–277
Acknowledgments
The authors are thankful to S. Haldar and A. Shukla for their help in LC–MS. The authors thank Council of Scientific and Industrial Research (CSIR), New Delhi, India for financial support and University Grants Commission (UGC), New Delhi, India for providing fellowship.
Conflict of interest
The authors declare that they have no conflict of interest and the publication of the work has been approved by all co-authors.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Patel, N., Patel, P., Kendurkar, S.V. et al. Overexpression of squalene synthase in Withania somnifera leads to enhanced withanolide biosynthesis. Plant Cell Tiss Organ Cult 122, 409–420 (2015). https://doi.org/10.1007/s11240-015-0778-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-015-0778-3