
Abstract
Somatic embryogenesis (SE) is a method for producing embryos in vitro and is considered a highly promising approach for micropropagation and germplasm conservation. However, the application of SE for genetic breeding and ex situ conservation of certain species, such as Brazilian pine, faces several technical challenges, including the difficulty of inducing embryogenic cultures using tissues of mature trees, the loss of embryogenic competence of cell cultures and incomplete development of somatic embryos. In order to understand the genetic factors governing embryogenesis, a comparative transcriptome analysis was performed to elucidate differences between distinct cell cultures, early zygotic and somatic embryos and, unorthodox seed developmental stages. A total of 64 GB of sequence derived from high-throughput Illumina RNA-seq profiling was used for de novo transcriptome assembly. The reference transcriptome resulted in 112,772 predicted unigenes with an average length of 825 bp and an N50 of 1,638 bp. Sequence similarity searches using a public protein database revealed 19,947 unigenes that could be annotated with gene descriptions and gene ontology terms. Analysis of differential gene expression allowed pinpointing of genes whose products are predicted to be involved in cell line embryogenic potential, early somatic embryo formation and unorthodox seed development. The results expand our understanding of the complex molecular events that control embryogenesis suggesting that the regeneration impairment of Araucaria angustifolia cultures is consequence of the auxin signaling failure. The generated data lay the foundation for future functional genomic and evolutionary studies that will advance the understanding of conifer biology and unorthodox seed physiology.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Brazilian pine, Araucaria angustifolia (Bertol.) Kuntze, is the only native conifer species in Brazil with economic importance. The seeds have high nutritional value and are consumed by both humans and wild fauna, while the wood of adult trees is employed as both structural timber in the building industry and in the manufacturing of furniture (Steiner et al. 2008). Due to intense exploitation of wood resources and the lack of reforestation, A. angustifolia is classified as critically endangered, according to the International Union of Conservation of Nature Red List of Threatened Species (2013). Consequently, there is considerable interest in establishing biotechnology tools for germplasm conservation and genetic improvement in order to support reforestation and conservation programmes. Given that the seeds of A. angustifolia are unorthodox (i.e. they maintain high levels of water and active metabolic rates at the mature stage resulting in a rapid loss of viability), unlike those of most conifers, the need for effective technological approaches is even more pressing (Steiner et al. 2008). Somatic embryogenesis is a highly desirable in vitro propagation system because when optimized it yields high numbers of plants and can be coupled with cryopreservation, bioreactors, synthetic seed technologies and genetic transformation (Merkle and Dean 2000). However, despite attempts to develop a protocol for inducing somatic embryogenesis in A. angustifolia, to date only somatic embryos in early developmental stages have been obtained (Schlögl et al. 2012; Jo et al. 2014).
The difficulties in successfully establishing artificial conditions allowing continuous embryo development to maturity are associated with the fragmented knowledge concerning the genetic programmes that regulate zygotic embryogenesis. This in turn partly reflects the absence of genomic or transcriptomic data for this species. However, the development of next generation sequencing (NGS) technologies and the concomitant availability of powerful bioinformatic tools to process and analyze large sequence data sets (Van Verk et al. 2013) have enabled low-cost and high-throughput genome-scale DNA sequencing. For example, RNA-sequencing (RNA-Seq) platforms allow the assembly of short reads into genome-scale transcript profiles, providing a comprehensive and efficient means to assess transcriptome composition, obtain RNA expression patterns, and facilitate gene discovery. RNA-seq data can be used for de novo transcriptome assembly in the absence of a reference genome, which is particularly attractive for organisms whose genomic sequences have not yet been determined (Armengaud et al. 2014).
In this study, we used RNA-seq profiling to build a reference transcriptome and perform a comparative transcriptome profile analysis of A. angustifolia. Early somatic embryo formation and seed development. Transcriptome annotation revealed that Brazilian pine harbors a repertoire of expressed genes with annotated functional homologs in basal species of the Viridiplantae as well as Angiosperms. Analyses of differentially expressed genes provided insights into putative genetic determinants that contribute to cell line embryogenic potential, somatic embryo initiation, and differences in expression between the initial stages of somatic and zygotic embryogenesis as well as during seed development. Furthermore, the data provide a broad gene catalog that should be of general utility in the study of Brazilian pine biology.
Materials and methods
Plant material
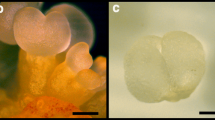
Seeds of A. angustifolia were collected in the Parque Estadual de Campos do Jordão, located in Campos do Jordão, São Paulo, Brazil. The seed samples were harvested from the same tree (22° 41.792′ south; 045° 29.393′ west, 1.529 m) on three different dates between December 2011 and June 2012, to obtain the three zygotic embryo developmental stages analysed: globular (GZE), late cotyledonal (CZE) and mature (MZE), as well as their respective megagametophyte (MG). For each stage, three pools of seeds from three different megastrobiles were made, allowing a random sampling of embryos. Each pool was considered to be a biological replicate. Two somatic embryogenic cultures were used, SE1 and SE6. These cell lines were previously selected by Jo et al. (2014), based on different responses under maturation conditions (MSG medium (Becwar et al.1989) supplemented with abscisic acid (ABA), maltose and PEG 4000). The SE1 cell culture produced precotyledonary embryos that eventually reached the early cotyledonary stage however, these somatic embryos did not develop further. The SE6 cell line is blocked and does not develop somatic embryos. For transcriptome analyses, SE1 and SE6 cultures were allowed to proliferate for 21 days on MSG medium before harvesting. The SE1 culture was additionally grown for 90 days on maturation medium (subcultured every 30 days) for development of globular somatic embryos (SE1M). The morphology of embryos, megagametophytes and cell cultures is shown in Fig. 1.

Araucaria angustifolia seeds at different developmental stages and morphology of somatic embryogenic cultures used for RNAseq. a Zygotic embryos and the corresponding megagametophytes at three different developmental stages were collected. Samples for transcriptome analyses are highlighted with a black box and named by an abbreviation. Stage 1: megagametophyte with globular zygotic embryo (GZE). Stage 2: zygotic embryo at the late cotyledonal stage (CZE) and the corresponding megagametophyte (MG2). Stage 3: mature zygotic embryo (MZE) and the corresponding megagametophyte (MG3). b Somatic embryogenic cultures were established from different immature zygotic embryos, and two were selected based on their differential response under maturation conditions (Jo et al. 2014). Samples for transcriptome analyses were: ABA-responsive embryogenic culture (SE1), ABA-non responsive embryogenic culture (SE6) and ABA-responsive embryogenic culture grown under maturation conditions showing a globular somatic embryo (SE1M)
RNA isolation, quality control and sequencing
RNA was extracted, using the PurelinkTM RNA Mini Kit (Life Technologies, Carlsbad, CA, USA) following the manufacturer’s protocol, from three biological replicates (1, 2 and 3) of each sample resulting in 24 samples. The RNA samples were quantified spectrophotometrically using a NanoDrop (Thermo Scientific, Wilmington, DE, USA). Samples with 260/280 nm and 260/230 nm ratios between 1.8–2.2 and 1.6–2.2, respectively, were considered to be of sufficient purity. The integrity of the total RNA (1 µg) was further confirmed by a 1 % (w/v) agarose gel electrophoresis and using the RNA 6000 Nano LabChip Kit and a Bioanalyzer 2100 (Agilent Technologies Inc., Santa Clara, CA, USA). RNA samples with RNA integrity number ≥6 were stored at −70 °C until further processing. For each sample, libraries with an insert size of approximately 200 bp were obtained with the TruSeq RNA Sample Preparation Kit v2, Set A (catalog # RS-122-2001, Illumina Inc., San Diego, CA, USA). Paired-end sequences of 100 bp were generated via the Illumina HiScanSQ platform at the Centro de Genômica Funcional Aplicada a Agropecuária e Agroenergia, ESALQ, USP, Piracicaba, São Paulo, Brazil.
Sequence trimming and de novo assembly
Raw sequences were filtered to remove adaptor sequences and low quality reads using SeqyClean (v.1.3.12) (https://bitbucket.org/izhbannikov/seqyclean/get/stable.zip) with the 28 and 26 Phred quality parameter for maximum average error and maximum error at end, respectively. Only high quality paired-end sequences were used for further analyses. Contaminant sequences were identified and removed with the Bowtie2 (v.2.1.0, 02-21-2013) program (Langmead and Salzberg 2012). The contaminant bank encompassed a total 29 GB of data including 2,150 microbial genomes; 999,366 sequences of arthropoda; 14,203,228 sequences of nematode; 20,747,849 sequences of flatworms, 25,479,398 ribosomal sequences and the complete genomes of Bos taurus (5,734,511,467 bp), Ixodes scapularis (411,892,114 bp) and Tribolium castaneum (190,173,473 bp). Sequence normalization was performed with the Trinity software package using the normalize_by_kmer utility (release 02-25-2013) with 30 defined as the maximum coverage (Grabherr et al. 2011). De novo transcriptome assembly was performed using Trinity and TransAbySS (assembly by short sequences) (v.1.3.4) (Robertson et al. 2011). For the latter, 13 values of k-mers between 52 and 64 were used. The complete sets of high quality reads are available at NCBI Sequence Read Archive (SRA) under accession numbers: Bioproject PRJNA240554; Project SRP039545; GZE: SRS567549, SRS567553 and SRS567554; CZE: SRS567550, SRS567551 and SRS567552; MG2: SRS567555, SRS567556 and SRS567557; MZE: SRS567561, SRS567562 and SRS567563; MG3: SRS567558, SRS567559 and SRS567560; SE1: SRS567564, SRS567565 and SRS567566; SE6: SRS567567, SRS567568 and SRS567569; SE1 M: SRS567570, SRS567571 and SRS567572.
Functional annotation and classification
The homology of the A. angustifolia assembled unigene sequences to sequences from three specific taxonomic groups: Viridiplantae (taxa ID 33090, VP), Monocotyledons (taxa ID 4447, MC) and two families of core Eudicotyledons, Brassicaceae (taxa ID 3700) and Solanaceae (taxa ID 4070) (BS) was evaluated using the BLASTX program of the BLAST suite (Camacho et al. 2013), searching against the Entrez protein system database (www.ncbi.nlm.nih.gov/protein). The e-value threshold was set as 1e−10 with a 100 bp as a minimum alignment. Functional annotation was performed based on BLASTX results using Blast2GO (www.blast2go.com) (Conesa et al. 2005). Transcription factor were annotated using PlantTFDB 3.0 (Jin et al. 2014).
Differential expression analysis
For sample-specific expression analysis, the reads obtained from each of the 24 sequenced samples were mapped to the 49,081 assembled contigs that displayed BLASTX results derived from the Viridiplantae protein database using Bowtie2 (v.2.1.0) (Langmead and Salzberg 2012). Before mapping, contigs were filtered using CD-HIT (v.4.6, 2012-04-25) (Li and Godzik, 2006) to eliminate redundant sequences with sequence similarity >95 %. To identify differentially expressed genes (DEGs), five pairwise comparisons were performed: SE1 versus SE6, SE1 versus SE1M, GZE versus (CZE+MG2), (CZE+MG2) versus (MZE+MG3) and SE1M versus GZE. The significance of differential gene expression was assessed with the edgeR (v.3.2.4) program (Robinson et al. 2010), according to standard protocols outlined in the package manual. These analyses were run in R/Bioconductor (v.3.0.2; Gentleman et al. 2004) and for each comparison analyses were conducted separately. To account for differences in sequencing effort and proportionality across libraries, count data were first normalized by TMM (Robinson and Oshlack 2010) using the calcNormFactors() function, while common dispersions were calculated with the estimateCommonDisp() function (Robinson and Smyth 2008). Gene wise exact tests for differences in the means between two groups of negative-binomially distributed counts were then computed. The threshold for evaluating significance was obtained by applying a p < 0.05, false discovery rate (FDR) ≤0.001 and log2 fold change ratio ≥2 as a threshold for determining significance levels. GO functional enrichment analysis of DEGs was performed by Fisher’s exact test (FDR < 0.05).
Results
Sequencing, quality trimming and de novo assembly
To obtain a global and comprehensive gene expression profile of A. angustifolia embryogenesis, RNA was extracted from three stages of seed development, as well as from embryogenic cell lines growing on proliferation and maturation culture medium. Twenty four mRNA libraries corresponding to three biological replicates for each of eight different samples (Fig. 1) were sequenced. In total, 642 million (64 GB) raw reads with a length of 100 bp were produced. The raw sequences were submitted to filtering processes to exclude low quality reads, adaptor sequences and contaminants, which together constituted about 40 % of the raw data.
Gymnosperm genomes are relatively large, highly heterozygous and abundant in repetitive elements (Nystedt et al. 2013). Therefore, to reduce redundancy and increase assembly efficiency, the sequences were normalized using the Trinity normalization utility, resulting in 56 million paired-end sequences that were further used for assembly. A schematic pipeline of the workflow for preprocessing raw data and the precise number of paired-end sequences per sample during the different steps is presented in Fig. 2 and Table 1, respectively. In order to compare assembly performance between programs, the transcriptome data were reconstructed by pooling the sequences of the eight samples and analyzing them with two different software packages: Trinity and TransABySS. The former program turned out to be more robust than TransABySS, as it produced fewer and larger contigs, as well as a higher N50 value (Table 2).

Schematic pipeline of the workflow for de novo assembly of the A. angustifolia transcriptome. Step 1: RNA extraction. Step 2: library construction. Step 3: paired-end sequencing using the Illumina platform HiScanSQ. Step 4: removal of low quality and adapter sequences. Step 5: removal of undesirable sequences. Step 6: normalization to eliminate redundancy. Step 7: assembly with two different platforms: Trinity (Grabherr et al. 2011) and TransABySS (Robertson et al. 2011)
In order to evaluate the diversity of transcripts in the different samples and developmental stages analyzed, separate assemblies were performed with Trinity. The algorithm of this program builds clusters that represent the full transcriptional complexity for a given gene or locus, thereby determining the total number of contigs. The most representative isoforms from each cluster, termed unigenes, were quantified to evaluate the abundance of different expressed genes. With a mean number of 7 million normalized paired-end sequences, a mean number of 68,968 contigs and 54,435 unigenes were obtained. No major differences were observed in the number of contigs or unigenes between zygotic and somatic tissues. However, it is worth noting that the cell line containing precotyledonary somatic embryos (SE1 M), even with the second lowest number of input sequences, displayed the highest number of unique unigenes (83,170). This could be the result of a low-quality assembly, as evidenced by the lowest N50, rather than a greater diversity in the transcript profile. A similar number of transcript variants (contigs) per locus (unigenes) were observed for all set of sequences, ranging from 1.1 to 1.4. In total, we identified 149,510 transcripts representing 112,772 expressed loci, which were used for further annotation and differential expression analyses (Table 3).
Functional annotation
The 149,510 transcripts associated with A. angustifolia embryogenesis were annotated using BLASTX based on three taxonomic groups: Liliopsida (monocots, MC), two families of core eudicots combined (Brassicaceae and Solanaceae, BS) and Viridiplantae (including MC and BS, VP). Of these, a total of 49,081 transcripts from the merged assembly had positive matches. Despite the pronounced difference in the number of protein entries between databases, the numbers of A. angustifolia sequences that showed BLASTX hits were similar but, as expected, a slight higher annotation rate was observed for VP database (Table 4). Further analysis revealed that 20,373 of the annotated unigenes are ubiquitous in VP as they have homologs in all three analyzed databases. In contrast, 667 and 813 A. angustifolia unigenes were annotated based on sequences from the VP and MC or VP and BS databases, respectively. Furthermore, 2,328 sequences showed homology to VP genes from species other than those included in the MC and BS data sets (Fig. 3). The 47 and 55 unigenes that were annotated exclusively based on the MC and BS databases, as well as 11 common sequences are missannotations that were deposited in the Entrez protein database (www.ncbi.nlm.nih.gov/protein) without a reference to the taxonomic origin within the Viridiplantae.

Venn diagram representing the number of A. angustifolia unigenes annotated using three different databases. Viridiplantae (VP), monocotyledons (MC) and Brassicaceae and Solanaceae (BS)
Of the 24,181 sequences that showed BLASTX hits against the VP database, 19,947 unigenes displayed associated gene anthology terms (GOs) that were subsequently separated in three main groups (‘biological process’, ‘molecular function’ and ‘cellular component’) (Table 5) and classified into 45 functional categories at level 2 using Blast2GO. The most highly represented categories, which comprised at least 4,000 genes, were ‘metabolic process’, ‘cellular process’, ‘single-organism process’ and ‘response to stimulus’ for the biological process category; ‘catalytic activity’ and ‘binding activity’ for the molecular function category; and ‘cell’, ‘organelle’ and ‘membrane’ for the cellular component category (Fig. 4).

Functional annotation of the embryogenesis reference transcriptome of A. angustifolia. The histograms represent the number of unigenes associated to level 2 GO categories
Differential expression analysis
In order to better understand the genetic bases of initiation and progression of embryogenesis, a differential expression analysis was performed. Five highly stringent (FDR < 0.001) pairwise comparisons were performed and the numbers of differentially expressed genes is shown in Table 6. DEGs were defined as those that were exclusively identified, or showed differences in transcript abundance in one of the samples in each pairwise comparison. Further annotation and GO functional enrichment analysis by Blast2GO allowed the identification of the putative function of the DEGs (Table S2 to Table S6) and the most highly represented GO categories within each comparison (Fig. 5).

GO functional enrichment within differentially expressed genes (DEGs). The histograms represent a multilevel chart of the most specific GO terms for biological process, cellular component or molecular function categories, which showed differential abundance according to Fisher’s exact test (FDR < 0.05). Values are expressed as percentage of the total upregulated genes in the sample. a Comparison between SE1 and SE6. b Comparison between SE1 and SE1M. c Comparison between CZE+MG2 and MZE+MG3. d Comparison between SE1 M and GZE. The comparisons between GZE and CZE+MG2 did not result in statistical differences. The list of the DEGs and the corresponding annotation are provided in Table S2, Table S3, Tables S5 and Table S6 for a, b, c and d, respectively
The comparative analysis of the two embryogenic cultures, responsive (SE1) and non-responsive (SE6) to maturation, showed the highest number of DEGs, which displayed a very broad spectra of functions. While SE6 expressed transcripts related to DNA replication, transcription, translation and cell division at high levels, the SE1 cell culture showed elevated expression of genes associated with defense, as well as cell wall and secondary metabolite biosynthesis (Fig. 5, Table S2). Additionally, a wide range of embryo development related genes and transcription factors (TFs) were differentially expressed (Fig. 5, Table S2). Within the 67 identified TFs, ERF (ethylene responsive factor) and NAC (NAM/ATAF/CUC) were the most represented families that showed distinct pattern of trancriptional regulation, in both cell culture, SE1 and SE6 (Table S7).
The expression of 1,242 genes was modified during maturation of the SE1 culture. A GO category enrichment analysis indicated active metabolism while growing in proliferation media, which is substantially reduced upon ABA, maltose and PEG 4000 treatment (see Materials and Methods). The abundance of ‘developmental process’ associated genes decreased following the treatment, while genes related to transposable element expression were exclusively present in the SE1M mature culture (Fig. 5, Table S3). Furthermore, the composition of expressed TFs differed between both growth conditions (Table S7). It is worth mentioning the upregulation of TFs belonging to ERF family in SE1M culture.
During the progression of zygotic development, the gene expression profile of the embryo and its corresponding megagametophyte showed little change: only 13 genes were differentially expressed from early embryogenesis (GZE) to the late cotyledonal (CZE+MG2) stage (Table 6). The GZE was characterized by genes involved in carbohydrate biosyntheses and storage, whereas embryos in the CZE+MG2 sample expressed genes associated with secondary metabolite production, as well as an ERF (Table S4, Table S7). No GO category was differentially represented at a statistically significant level. A comparison between CZE+MG2 and the mature stage (MZE+MG3) identified 78 DEGs (Table 6). At the cotyledonal stage, transcripts associated to ‘developmental process’ and ‘cell division’ categories were upregulated including an ERF TF (Table S5, Table S7). Interestingly, the MZE+MG3 expression profile was characterized by a high abundance of transcripts associated with photosystem and chlorophyll metabolism, suggesting the assembling of the photosynthetic machinery (Fig. 5, Table S5).
When comparing SE1M with its zygotic counterpart (GZE), 151 and 454 genes were upregulated, respectively. TFs were highly abundant in the SE1M transcriptome, being ERF and HD-ZIP (homeodomain leucine zipper motif) the most representative families (Table S7), while GZE showed elevated expression of mRNAs related to chloroplast and phosphorous metabolism (Fig. 5, Table S6).
Discussion
There are approximately 1,026 extant species of gymnosperms (conifers, cycads, genetophytes and ginkgo) (Christenhusz et al. 2011). Their genome sizes range from 18 to 35 GB, which is on average >200 times the size of the Arabidopsis thaliana genome (Mackay et al. 2012). Despite their economical and ecological importance, little is known about Gymnosperm genome structures (e.g. polyploidy) and molecular physiology compared to those of angiosperms. Indeed, the first whole-genome draft sequences of two Gymnosperms (Picea abies and Picea glauca) were only recently published, and these have genome sizes of approximately 20 GB (Nystedt et al. 2013; Birol et al. 2009). However, transcriptome analyses using a NGS approach provide a powerful tool for gene discovery and transcriptional regulation studies associated with specific organs or physiological/developmental programs in non-model species for which genome information is not available. This current study aimed to create the transcriptome profile of A. angustifolia embryogenesis to better understand the process and to help establish new strategies for Araucariaceae in vitro culture improvement. Eight transcriptomes from different developmental stages were constructed; specifically from in vitro embryogenic cell cultures, zygotic embryos and their corresponding megagametophytes (Fig. 1). Per sample, the mean number of input reads comprised 23.8 million cleaned high quality paired-end sequences, which resulted in an average of 68,968 transcripts representing 54,435 unigenes (Tables 1, 3). For comparison, the transcriptome reconstruction of the embryogenic cell culture of the Gymnosperm, Larix leptolepis, resulted in 65,115 assembled unigenes (Zhang et al. 2012). Using a similar approach to that used in this study (Illumina platform, paired-end protocol, 100 cycles and the Trinity assembler) Francis et al. (2013) analyzed the assembly performance of the mouse (Mus musculus) heart transcriptome with an increasing number of input sequences. They observed that this program reaches a performance plateau for contig and unigene counts when using >40 million reads. Similar results were obtained for the reconstruction of the Schizosaccharomyces pombe and Drosophila melanogaster transcriptomes, where the maximum number of assemblies by Trinity was reached with 22 and 25 millon paired-end sequences, respectively (Zhao et al. 2011). Based on these results the transcriptomes built here can be considered robust and deep-coverage. The observed differences in the numbers of assembled contigs and unigenes therefore probably reflect the actual diversity of loci being expressed in each sample and developmental stage analysed (Table 3). Our data indicate that the number of input sequences did not affect the number of transcript variants per locus assembled by Trinity, in accordance with the results of Francis et al. (2013).
The global transcriptome of A. angustifolia embryogenesis, constructed by merging the sequences obtained in all samples, produced 112,772 unigenes with high quality assembly parameters (N50 = 1,638 and mean contig length = 825) compared with previous reports of conifer transcriptomes (Zhang et al. 2012; Canales et al. 2013). Of these 24,181 (21 %) had BLASTX hits when searching VP database. Recent reports describing de novo assemblies of non-Gymnosperm species have shown that the annotation capability resulting from a BLAST search against the non-redundant protein database ranges from 43 to 68 % of assembled unigenes (Shi et al. 2011; Sangwan et al. 2013; Lai and Lin 2013; Xie et al. 2013). In contrast, for Gymnosperm transcriptomes the number was reported to be only 34 and 47 % for L. leptolepis (Zhang et al. 2012) and Pinus pinaster (Canales et al. 2013), respectively. The difficulty in annotating genes in Gymnosperm data sets is in large part due to insufficient genomic information (Nystedt et al. 2013; Birol et al. 2009) or information regarding expressed sequences, which is commonly not annotated (Zhang et al. 2012; Rigault et al. 2011; Ralph et al. 2008, Futamura et al. 2008). This problem is magnified by the scarcity of functional genomic data (Schlögl et al. 2012; Steiner et al. 2012; Cairney and Pullman, 2007; Hedman et al. 2013), which makes it difficult to experimentally confirm predicted gene functions or discover novel functions. It is also worth noting that most of the available sequence information regarding nuclear encoded mRNAs from Gymnosperm species was obtained from members of the Pinaceae, a family that is remotely related to the Araucaria clade (Burleigh et al. 2012). However, despite the relatively small amount of publically available data, some interesting results were obtained from the differential BLASTX analysis performed in this study. Most of the A. angustifolia unigenes represent common basal functions present throughout the Viridiplantae. However, 2,328 unigenes retrieved BLASTX hits only found in the VP database. Of these, 15, 32 and 39 % showed homology to genes from basal Viridiplantae (Bryophytes and Chlorophytes), Gymnosperms and Eudicotyledoneae (other than Solanaceae and Brassicaceae) species, respectively (Table S1). This suggests that these genes represent: ancestral functions that were lost or whose sequences changed substantially following divergence of the Gymnosperms (15 %), Gymnosperms novelties (32 %) or, ancestral functions lost or whose sequences diverged in the terminal Monocotyledoneae and Eudicotyledoneae species that are represented in the MC and BS databases (39 %). Interestingly, A. angustifolia also express unigenes that are exclusively shared with either Monocotyledoneae or Eudicotyledoneae species, and which might represent functions that arose between basal Viridiplantae and Gymnosperms (Fig. 3).
A total of 19,947 unigenes from the embryogenesis reference transcriptome of A. angustifolia had associated GO terms and could be assigned to a wide range of functions, indicating a broad functional diversity. Again, the functional transcriptome annotation of an embryogenic culture from Japanese larch (L. leptolepis) a basal Pinaceae genus (Burleigh et al. 2012), showed similar numbers to those presented in this study, comprising 20,324 unigenes that were classified into 51 functional categories. Moreover, the distribution profile of the categorized L. leptolepis transcripts (Zhang et al. 2012) closely resembles the profile obtained for A. angustifolia. A cotton (Gossypium hirsutum) embryogenesis transcriptome analysis has also been described and the entire reference transcript profile was reported to comprise 20,220 genes (Jin et al. 2013), which is a similar size to that of A. angustifolia and L. leptolepis.
The DEG analysis described here aimed to improve the knowledge of key genetic factors involved in A. angustifolia cell line embryogenic potential, somatic embryo formation and the developmental block observed during somatic embryo maturation. A comparison between SE1 and SE6 A. angustifolia embryogenic cell lines showed diverse transcriptional profiles, with 2,150 annotated DEGs (Table 6, Fig. 5, Table S2). Several TFs belonging to the NAC, WRKY, ERF, MYB, HD-ZIP and bZIP families, which have been associated with embryogenesis and stress responses induced by in vitro culture conditions (Xu et al. 2012; Jin et al. 2013), were differentially expressed, suggesting their involvement in embryogenic potential. A recent study of TF expression in cell cultures of Arabidopsis thaliana identified 141 genes that display differential expression patterns between highly embryogenic and non-embryogenic genotypes (Gliwicka et al. 2013). SE6 displayed high levels of DNA replication and cell division related genes, which correlates with the higher growth rate exhibited by this cell line compared to SE1 (Bueno and Floh, unpublished). This also correlates with data from arrested cell lines of P. abies that are deficient in embryo development, which showed a higher expression of metabolic process related genes than cell lines grown under normal conditions (Stasolla et al. 2004). We observed a conspicuous induction of defense-responsive genes in the SE1 line, which again is in agreement with the P. abies study, where cell defense related genes were also expressed at higher levels in a cell line with high embryogenic potential (Vestman et al. 2011). In addition, biosynthesis of secondary metabolites, in particular phenylpropanoids, has previously been reported in Larix embryogenic cultures (Zhang et al. 2012) and during embryogenesis in Oryza sativa (Xu et al. 2012). It is possible, that the activation of defense associated genes in cell lines with high embryogenic potential allows the adaptation to stressful conditions imposed by the in vitro environment (Rutledge et al. 2013). TFs belonging to the above mentioned families have been demonstrated to induce different phenylpropanoid pathways in Pinus sylvestris (Ueche 2012), Lotus corniculatus (Wang et al. 2013) and tomato (Solanum lycopersicum; Butelli et al. 2008). In agreement with the diverse transcriptional profile described here, Jo et al. (2014) observed noticeable differences in protein and biochemical (polyamines, ethylene and reactive oxygen species) profiles between SE1 and SE6 cultures. Interestingly, of the eight proteins identified as SE1-specific, by an analysis using two-dimensional electrophoresis coupled with mass spectrometry, upregulation of the corresponding mRNAs was observed for three: an ATPase (AAD03392), an S-adenosylmethionine synthase 3 (Q4LB22) and a hypothetical protein SELMODRAFT_404503 (XP_002962607). Additionally, of the two SE6-exclusive proteins the mRNA corresponding to a porin (AAD38145) was identified as up-regulated in the SE6 cell culture transcriptome. We note that SE1 and SE6 were established from different mother trees and so the biochemical, proteomic and transcriptional differences might be due to genetic diversity leading to a differential response to in vitro culture conditions.
When the culture of the responsive genotype SE1 was subjected to maturation conditions, most of the cell culture structures, which are incapable of going through embryogenesis, collapsed as reflected in the reduction in expression of genes related to cell maintenance (Filonova et al. 2000) (Fig. 5). In contrast, SE1 showed transcriptional induction of transposable elements (TEs), which have been widely demonstrated to be induced in response to stressful conditions (Chénais et al. 2012), but also to be associated with morphogenetic processes, as described in early zygotic embryogenesis of P. pinaster (Vega-Bartol et al. 2013).
The identity and reduced number of DEGs during the progression of zygotic embryogenesis suggests that few changes are present between GZE and CZE+MG2 trancriptional profiles (Table S4, Table S5, Table S7, Fig. 5). However, from the CZE+MG2 to the MZE+MG3 stage, when the primary plant body has already been established, the expression of several ‘developmental process’ related genes was down-regulated, while genes associated with photosynthetic machinery assembling increased their expression. In contrast, a microarray study comparing the transcriptional profile of zygotic embryos of P. pinaster identified several TFs as being differentially expressed from early embryogenesis to mature stages (Vega-Bartol et al. 2013). These apparently contradictory results can be explained by the different technical approaches that were used. The stringent cutoff used in the current study for the in silico DEG identification may have masked the fluctuation in expression of low abundance genes, such as those encoding TFs. However, it should be noted that A. angustifolia seed is unorthodox, which may involve different morphogenetical timing compared with orthodox seeds (dos Santos et al. 2006), and the major changes might therefore occur prior to the globular stage analyzed here.
A comparison of DEGs between early somatic (SE1M) and zygotic (GZE) embryos gave potential insights into the developmental block observed in the somatic embryos induced in SE1M. WUSCHEL, which belongs to the WOX family of TFs, was highly expressed in the SE1M transcriptome (9 fold change); whereas in GZE several genes that are predicted to encode ARFs (auxin responsive factors) and IAAs (indoleacetic acid-induced protein) (Table S7), genes associated with polar auxin transport (PAT) and leaf morphogenesis were up-regulated. PAT is crucial for normal embryo patterning in both Angiosperms and Gymnosperms (Larsson et al. 2012). During somatic embryo formation in P. abies, perturbations of PAT with auxin inhibitors was reported to promote procambium expansion, larger root apical meristem (Hakman et al. 2009), fused cotyledons and aborted shoot apical meristems (Larsson et al. 2008). Similarly altered phenotypes have been observed during somatic embryo maturation of A. angustifolia, although without auxin inhibitor supplementation (Steiner et al. 2008; Schlögl et al. 2012; Jo et al. 2014). Recently, a study of cotton embryogenic calli demonstrated that WUSCHEL over-expression led to a reduced mRNA level of ARF3, which alters polar auxin flux, resulting in abnormal morphology (Zheng et al. 2014). These data suggest that SE1M fails to establish the correct auxin distribution, consequently jeopardizing embryo development from early stages through maturity.
In addition to this hypothesis, the physiological differences between the samples may result from the actions of some of the unannotated DEGs; information that will eventually be revealed as functional genomics data is generated. In conclusion, the comprehensive repository of sequences produced here constitutes a valuable resource for improving our understanding of A. angustifolia physiology. Moreover, it enlarges the conifer gene catalog for further functional genomics, genetic diversity and evolutionary studies aiming to elucidate Gymnosperm biology.
References
Armengaud J, Trapp J, Pible O, Geffard O, Chaumot A, Hartmann EM (2014) Non-model organisms, a species endangered by proteogenomics. J Proteomics. doi:10.1016/j.jprot.2014.01.007
Becwar MR, Noland TL, Wyckoff JL (1989) Maturation, germination, and conversion of Norway spruce (Picea abies L.) somatic embryos to plant. In Vitro Cell Dev Biol Plant 26:575–580
Birol I, Jackman SD, Nielsen CB, Qian JQ, Varhol R, Stazyk G, Morin RD, Zhao Y, Hirst M, Schein JE, Horsman DE, Connors JM, Gascoyne RD, Marra MA, Jones SJM (2009) De novo transcriptome assembly with ABySS. Bioinformatics 25:2872–2877
Burleigh JG, Barbazuk WB, Davis JM, Morse AM, Soltis PS (2012) Exploring diversification and genome size evolution in extant gymnosperms through phylogenetic synthesis. J Bot. ID 292857
Butelli E, Titta L, Giorgio M, Mock HP, Matros A, Peterek S, Schijlen EGWM, Hall RD, Bovy AG, Luo J, Martin C (2008) Enrichment of tomato fruit with health-promoting anthocyanins by expression of select transcription factors. Nat Biotechnol 26:1301–1308
Cairney J, Pullman GS (2007) The cellular and molecular biology of conifer embryogenesis. New Phytol 176:511–536
Camacho C, Madden T, Ma N, Tao T, Agarwala R, Morgulis A (2013) BLAST command line applications user manual. National Center for Biotechnology Information (US). http://www.ncbi.nlm.nih.gov/books/NBK1763/. Accessed 09 Nov 2013
Canales J, Bautista R, Label P, Gómez MJ, Lesur I, Pozo NF, Rueda LM, Fernández D, Guerrero R, Vanessa C, Benzekri H, Cañas RA, Guevara MA, Andreia R (2013) De novo assembly of maritime pine transcriptome: implications for forest breeding and biotechnology. Plant Biotechnol J 1:1–14
Chénais B, Caruso A, Hiard S, Casse N (2012) The impact of transposable elements on eukaryotic genomes: from genome size increase to genetic adaptation to stressful environments. Gene 509:7–15
Christenhusz M, Zhang XC, Schneider H (2011) A linear sequence of extant families and genera of lycophytes and ferns. Phytotaxa 19:7–54
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
dos Santos ALW, Wiethölter N, El Gueddari NE, Moerschbacher BM (2006) Protein expression during seed development in Araucaria angustifolia: transient accumulation of class IV chitinases and arabinogalactan proteins. Physiol Plantarum 127:138–148
Filonova LH, Bozhkov PV, Brukhin VB, Daniel G, Zhivotovsky B, von Arnold S (2000) Two waves of programmed cell death occur during formation and development of somatic embryos in the gymnosperm, Norway spruce. J Cell Sci 113:4399–4411
Francis WR, Christianson LM, Kiko R, Powers ML, Shaner NC, Haddock SHD (2013) A comparison across non-model animals suggests an optimal sequencing depth for de novo transcriptome assembly. BMC Genom 14:167
Futamura N, Totok Y, Toyoda A, Igasaki T, Nanjo T, Seki M, Sakaki Y, Mari A, Shinozaki K, Shinohara K (2008) Characterization of expressed sequence tags from a full-length enriched cDNA library of Cryptomeria japonica male strobili. BMC Genom 9:383
Gentleman R, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5:R80
Gliwicka M, Nowak K, Balazadeh S, Mueller-Roeber B, Gaj MD (2013) Extensive modulation of the transcription factor transcriptome during somatic embryogenesis in Arabidopsis thaliana. PLoS ONE 8:e69261
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:644–652
Hakman I, Hallberg H, Palovaara J (2009) The effect of the polar auxin transport inhibitor NPA on embryo morphology and expression of an auxin efflux facilitator protein PIN during Picea abies somatic embryo development. Tree Physiol 29:483–496
Hedman H, Zhu T, von Arnold S, Sohlberg JJ (2013) Analysis of the WUSCHEL-RELATED HOMEOBOX gene family in the conifer Picea abies reveals extensive conservation as well as dynamic patterns. BMC Plant Biol 13:89
International Union of Conservation of Nature Red List of Threatened Species (2013) http://www.iucnredlist.org/details/32975/0. Accessed 03 March 2013
Jin F, Hu L, Yuan D, Xu J, Gao W, He L, Yang X, Zhang X (2013) Comparative transcriptome analysis between somatic embryos (SEs) and zygotic embryos in cotton: evidence for stress response functions in SE development. Plant Biotechnol J 2013:1–13
Jin JP, Zhang H, Kong L, Gao G, Luo JC (2014) PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res 42:D1182–D1187
Jo L, dos Santos ALW, Bueno CA, Barbosa HR, Floh EIS (2014) Proteomic analysis and polyamines, ethylene and reactive oxygen species levels of Araucaria angustifolia (Brazilian pine) embryogenic cultures with different embryogenic potential. Tree Physiol 34:94–104
Lai Z, Lin Y (2013) Analysis of the global transcriptome of longan (Dimocarpus longan Lour.) embryogenic callus using illumina paired-end sequencing. BMC Genom 14:561
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359
Larsson E, Sitbon F, Ljung K, von Arnold S (2008) Inhibited polar auxin transport results in aberrant embryo development in Norway spruce. New Phytol 177:356–366
Larsson E, Sundström JF, Sitbon F, von Arnold S (2012) Expression of PaNACO1, a Picea abies CUP-SHAPED COTYLEDON orthologue, is regulated by polar auxin transport and associated with differentiation of the shoot apical meristem and formation of separated cotyledons. Ann Bot Lond 110:923–934
Li W, Godzik A (2006) Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22:1658–1659
Mackay J, Dean JFD, Plomion C, Peterson DG, Cánovas FM, Pavy N, Ingvarsson PK, Savolainen O, Guevara MA, Fluch S (2012) Towards decoding the conifer giga-genome. Plant Mol Biol 80:555–569
Merkle SA, Dean JFD (2000) Forest biotechnology. Curr Opin Biotech 11:298–302
Nystedt B, Street NR, Wetterbom A, Zuccolo A, Lin YC, Scofield DG, Vezzi F, Delhomme N, Giacomello S, Alexeyenko A (2013) The Norway spruce genome sequence and conifer genome evolution. Nature 497:480–484
Ralph SG, Chun HJ, Kolosova N, Cooper D, Oddy C, Ritland CE, Kirkpatrick R, Moore R, Barber S, Holt RA (2008) A conifer genomics resource of 200,000 spruce (Picea spp.) ESTs and 6,464 high-quality, sequence-finished full-length cDNAs for Sitka spruce (Picea sitchensis). BMC Genom 14:484
Rigault P, Boyle B, Lepage P, Cooke JEK, Bousquet J, MacKay JJ (2011) A white spruce gene catalog for conifer genome analyses. Plant Physiol 157:14–28
Robertson G, Schein J, Chiu R, Corbett R, Field M, Jackman SD, Mungall K, Lee S, Okada HM, Qian JQ (2011) De novo assembly and analysis of RNA-seq data. Nat Methods 7:909–912
Robinson MD, Oshlack A (2010) A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol 11:R25
Robinson MD, Smyth GK (2008) Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9:321–332
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140
Rutledge RG, Stewart D, Caron S, Overton C, Boyle B, Mackay J, Klimaszewska K (2013) Potential link between biotic defense activation and recalcitrance to induction of somatic embryogenesis in shoot primordial from adult trees of with spruce (Picea glauca). BMC Plant Biol 13:116
Sangwan RS, Tripathi S, Singh J, Narnoliya LK, Sangwan NS (2013) De novo sequencing and assembly of Centella asiatica leaf transcriptome for mapping of structural, functional and regulatory genes with special reference to secondary metabolism. Gene 525:58–76
Schlögl PS, dos Santos ALW, do Nascimento VL, Floh EIS, Guerra MP (2012) Gene expression during early somatic embryogenesis in Brazilian pine (Araucaria angustifolia (Bert) O. Ktze). Plant Cell, Tissue Organ Cult 108:173–180
Shi CY, Yang H, Wei CL, Yu O, Zhang ZZ, Jiang CJ, Sun J, Li YY, Chen Q, Xia T (2011) Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genom 12:131
Stasolla C, Bozhkov PV, Chu TM, Zyl LV, Egertsdotter U, Suarez MF, Craig DC, Wolfinger RD, von Arnold S, Sederoff RR (2004) Variation in transcripts abundance during somatic embryogenesis in Gymnosperms. Tree Physiol 24:1073–1085
Steiner N, Santa-Catarina C, Andrade JBR, Balbuena TS, Guerra MP, Handro W, Floh EIS, Silveira V (2008) Araucaria angustifolia biotechnology. Funct Plant Sci Biotech 2:20–28
Steiner N, Santa-Catarina C, Guerra MP, Cutri L, Dornelas MC, Floh EIS (2012) A gymnosperm homolog of somatic embryogenesis receptor-like kinase-1 (SERK1) is expressed during somatic embryogenesis. Plant Cell, Tissue Organ Cult 109:41–50
Ueche OG (2012) Pine transcriptomics—RNA-Seq data analysis of Scots pine (Pinus sylvestris) seedlings subjected to a wounding experiment. Dissertation, University of Helsinki
Van Verk MC, Hickman R, Pieterse CMJ, Van Wees S (2013) RNA-Seq: revelation of the messengers. Trends Plant Sci 18:175–179
Vega-Bartol JJ, Simões M, Lorenz WW, Rodrigues AS, Alba R, Dean JFD, Miguel CLM (2013) Transcriptomic analysis highlights epigenetic and transcriptional regulation during zygotic embryo development of Pinus pinaster. BMC Plant Biol 13:123
Vestman D, Larsson E, Uddenberg D, Cairney J, Claphan D, Sundberg E, von Arnold S (2011) Important process during differentiation and early development of somatic embryos of Norway spruce as revealed by changes in global gene expression. Tree Genet Genomes 7:347–362
Wang Y, Hua W, Wang J, Hannoufa A, Xu Z, Wang Z (2013) Deep sequencing of Lotus corniculatus L. reveals key enzymes and potential transcription factors related to the flavonoid biosynthesis pathway. Mol Genet Genomics 288:131–139
Xie C, Li B, Xu Y, Ji D, Chen C (2013) Characterization of the global transcriptome for Pyropia haitanensis (Bangiales, Rhodophyta) and development of cSSR markers. BMC Genom 14:107
Xu H, Zhang W, Gao Y, Zhao Y, Guo L, Wang J (2012) Proteomic analysis of embryo development in rice (Oryza sativa). Planta 235:687–701
Zhang Y, Zhang S, Han S, Li X, Qi L (2012) Transcriptome profiling and in silico analysis of somatic embryos in Japanese larch (Larix leptolepis). Plant Cell Rep 31:1637–1657
Zhao QY, Wang Y, Kong YM, Luo D, Li X, Hao P (2011) Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinformatics 12:2–12
Zheng W, Zhang X, Yang Z, Wu J, Li F, Duan L, Li F (2014) AtWuschel promotes formation of the embryogenic callus in Gossypium hirsutum. PLoS ONE 9:e87502
Acknowledgments
P.E. was a recipient of a CAPES fellowship. B.L., S.C.S.A. and A.L.W.S. were recipients of FAPESP fellowships. M.R. and E.F. hold fellowships from CNPq. This work was partially supported by grants from FAPESP (Brazil), CNPq (Brazil), USP (Brazil) and Petrobras. The authors thank Roberta Alvares Campos for technical assistance during sample preparation, Horácio Montenegro and Marcelo Brandão for help and access to the Computer Cluster Thunder (Bioinformatics Group from the Laboratório de Biologia Molecular de Plantas, ESALQ, USP) and PlantScribe (www.plantscribe.com) for carefully editing this paper.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Eny Iochevet Segal Floh and Magdalena Rossi have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Elbl, P., Lira, B.S., Andrade, S.C.S. et al. Comparative transcriptome analysis of early somatic embryo formation and seed development in Brazilian pine, Araucaria angustifolia (Bertol.) Kuntze. Plant Cell Tiss Organ Cult 120, 903–915 (2015). https://doi.org/10.1007/s11240-014-0523-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-014-0523-3