
Abstract
The desert shrub Leptadenia pyrotechnica (Forssk.) Decne (Asclepiadaceae) is an important multipurpose woody species of tropical and sub-tropical arid regions. The shrub’s excellent pharmacological properties, importance in desert afforestation and role in sand dune fixation has been elaborately studied in recent years which make it a potential candidate for genetic manipulation in the global warming scenario. We have developed an Agrobacterium-mediated transformation protocol for L. pyrotechnica using hypocotyl explants from 5 days old seedlings. The reliability of the protocol has been tested by transforming the species with gus and gfp reporter genes separately. Hypocotyl explants were sonicated for 40 s, infected with Agrobacterium suspension of OD600 0.5, co-cultivated in the dark for 2 days at 26 °C in presence of 200 μM acetosyringone. Transgenic plants were obtained after 20–22 weeks at a frequency of 14 and 11 % for gus and gfp reporter genes respectively. Transgenic plants were confirmed by PCR and Southern blots. Expression of the two reporter genes has been tested in different stages of transgenic plant development. No phenotypic differences between the wild-type and transgenic plants were noted. This method will be very helpful to introduce alien genes-of-interest for various biotechnological applications.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Leptadenia pyrotechnica (Forssk.) Decne is a multipurpose woody shrub of the Asclepiadaceae family. It is widespread in the tropical and sub-tropical arid regions across Africa to western India including Pakistan, Iran, Saudi Arabia, Egypt, Sudan, Somalia, Chad, Libya and Algeria (Dhawan and Singh 1976). The plant is an important species of the United Arab Emirates (UAE) and other countries of the Middle East and has high economic and environmental values. As a xerophyte, L. pyrotechnica is well adapted to grow in extremely severe climatic conditions (−0.4 to 49.5 °C) of arid regions where the rainfall is very scanty, sporadic and variable (Migahid et al. 1972). The strong soil binding property of L. pyrotechnica, due to its extensive and long root system, makes the species as a prime choice in sand dune fixation and desert afforestation programs (Alyemeni 2000). A successful restoration of a threatened lake “Budha Puskar,” Rajasthan, India, was recently achieved by growing L. pyrotechnica on the slope of the surrounding sand dunes, preventing erosion of soil into the water body (Sharma and Chouhan 2008).
The plant species has great, nutritional, economic and medicinal importance (Singh et al. 2007) and is judiciously used by the people living in desert areas, which are otherwise unsuitable for farming because of very harsh climatic conditions. The green parts of this shrub, used as vegetable and fodder, are a very good source of protein, dietary fiber, calcium, phosphorus, iron and vitamin C (Goyal and Sharma 2009). The plant is a source of good quality fiber which is useful for making ropes, bags, mats, carpets, and even textiles by blending with cotton and polyester fiber. The fiber is also suitable as a raw material in pulp and paper industries (Goyal and Sharma 2006). L. pyrotechnica makes excellent firewood and is commonly used for fuel and for making long-burning matches, from where it derives its Latin name, pyrotechnica. In addition, L. pyrotechnica is a source of many pharmacologically active constituents such as cardenolides, alkaloids, flavonoids, sterols, triterpenes and polyoxypregnane derivatives and their anti-tumor, anti-inflammatory, analgesic and diuretic actions has been intensively studied (Moustafa et al. 2007, 2009; Chaudhary et al. 2011; Khasawneh et al. 2011; Soliman et al. 2012).
Leptadenia pyrotechnica possesses a long reproductive cycle. The improvement of this important multipurpose shrub seems difficult through conventional breeding strategies. Transfer of any gene of biotechnological interest in this plant species depends on an efficient and reproducible genetic transformation system. Many plant species are now being routinely transformed through Agrobacterium-mediated transformation techniques (Choi et al. 2012; Liu et al. 2012; Sujatha et al. 2012). Nevertheless, Asclepiadacean plant species are recalcitrant to genetic transformation methodologies. There are only few successful reports of Agrobacterium-mediated transformation among Asclepiadacean members. Transgenic plant regeneration through Agrobacterium rhizogenes-mediated transformation was accomplished in Tylophora indica (Chaudhuri et al. 2006), while Periploca sepium (Miyabashira et al. 2003; Chen et al. 2010) was transformed by Agrobacterium tumefaciens.
Sonication of tissue during infection with Agrobacterium-mediated transformation has been reported to enhance the efficiency of transformation by producing small and uniform fissures and channels throughout the plant tissue, allowing the access of Agrobacterium to internal plant tissue (Trick and Finer 1997; Bakshi et al. 2011; Subramanyam et al. 2011). In the present study an efficient Agrobacterium-mediated genetic transformation method expressing gus and gfp reporter genes separately has been developed for L. pyrotechnica. The method provides an efficient tool for the introduction of genes of biotechnological interest in this species.
Materials and methods
Plant material and seed surface sterilization
Seeds of L. pyrotechnica were collected from different areas in United Arab Emirates during period of 2009–2010. A voucher specimen of this species has been deposited in the herbarium of the Biology Department, Faculty of Science, UAE University. Seeds (stored at 4 °C in dark) were surface sterilized in a 250 ml sterile flask by rinsing in 50 ml 20 % (v/v) commercial bleach solution with 0.1 % (v/v) Tween-20 for 15 min. The seeds were rinsed five times with sterile water. Surface sterilized seeds were kept at 4 °C for 2 days and transferred to a growth chamber maintained at 25 ± 1 °C and 16 h light: 8 h dark photoperiod under white fluorescent tubes (40 μmol m−2 s−1). After emergence of radicles, the seeds were transferred to half-strength MS (Murashige and Skoog 1962; Phytotechnology laboratory, USA) medium M1 in 150 ml transparent sterile plastic sundae cups (Solo, Urbana, IL, USA). The cups were kept in the growth chamber (16 h light: 8 h dark photoperiod at 25 ± 1 °C) for further seedling growth. Approximately 3 mm hypocotyl segments were prepared from the 5 days old seedlings immediately beneath the cotyledonary node.
Medium and culture conditions
The development of transgenic plants was accomplished by adopting the in vitro plant regeneration protocol established in our laboratory using 5 days old seedling-derived hypocotyl (3 mm) explants. The protocol consists of plant regeneration through callus mediated somatic embryogenesis. The pH of all plant tissue culture media was adjusted to 5.8 before addition of agar (Sigma-Aldrich, St. Louis, USA) for solid media and was autoclaved at 121 °C for 20 min. Plant growth regulators and antibiotics from sterile stock solutions were added to the media after autoclaving. The cultures were incubated in a growth chamber (Percival Scientific, series 101) maintained at 16 h light: 8 h dark photoperiod; 25 ± 1 °C throughout the study. MS media supplemented with different plant growth regulator combinations were used to determine the morphogenetic response (data not shown) of the hypocotyl explants. The best media optimized for morphogenesis and transformation has been summarized in Table 1 for ready reference. The hypocotyl explants were cultured on M3 medium (callus induction) for 4 weeks and then transferred to M4 medium. The tissues were cultured 6–8 weeks on M4 medium for regeneration and elongation of shoots with one change of medium after 3–4 weeks. Tips (1 cm) of the elongated shoots were excised and cultured on M5 medium for 2 days. The shoots tips were subsequently transferred to M6 medium for root formation, further growth and maintenance. Regenerated transgenic plants were clonally propagated by cutting shoot tips of 1 cm and its subsequent culture on M5 and M6 media respectively at an interval of 2–3 weeks. This strategy enabled the clonal propagation of transgenic plants without any callus phase, which in turn circumvent the possible chances of variation through callus mediated regeneration. The regenerated plants were used as controls in the reporter gene assays and molecular analysis later. In transformation experiments, the above mentioned plant culture media were supplemented with different doses of timentin and hygromycin for suppression of Agrobacterium tumefaciens and selection of transgenic plants (Table 1) respectively. Petri plates (9 cm) were covered with aluminum foil and incubated in growth chamber for the dark treatments throughout the study.
Plasmid vectors and A. tumefaciens strain
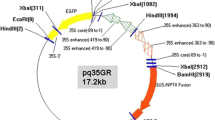
Binary vectors pCAMBIA1201 (pC1201) and pH7m24-35S:mGFP were used. The T-DNA of binary vector pCAMBIA1201 includes β-glucuronidase gene (gus) with a catalase intron and hygromycin phosphotransferase gene (hpt), both driven by the cauliflower mosaic virus (CaMV) 35S promoter (Fig. 1a). The binary vector pH7m24-35S:mGFP includes gfp and hpt genes, driven by CaMV35S promoter and nos terminator respectively (Fig. 1b). The 35S:mGFP construct was generated using a multisite gateway method (Invitrogen, NY, USA). A 1,333 bp of an enhanced CaMV35S promoter was cloned from pEarleyGate101 (Earley et al. 2006) using B4-35Sp-F (GGGGACAACTTTGTATAGAAAAGTTGTCCAATCCCACAAAAATCTGAGC) and B1r-35Sp-R (GGGGACTGCTTTTTTGTACAAACTTGCGTGTCCTCTCCAAATGAAATG) by BP recombination reaction with pDONRP4P1r (Invitrogen, New York, USA) to generate pDONRL4R1-35S as the first fragment entry vector. For the second fragment, mgfp was cloned from pEarleyGate103 (Earley et al. 2006) using B1-GFP-HIS pEG103 (GGGGACAAGTTTGTACAAAAAAGCAGGCTATGGTAGATCTGACTAGTAAAGGAG) and B2-GFP-HIS pEG103 (GGGGACCACTTTGTACAAGAAAGCTGGG TTCACACGTGGTGGTGGTGGTGGTGG) by BP recombination reaction with pDONR207 (Invitrogen, NY, USA) to generate pDONR207-mGFP. Those two entry vectors were recombined with pH7m24GW destination vector (Karimi et al. 2007) by LR reaction to create pH7m24-35S:mGFP expression vector (Fig. 1b).

Schematic diagram of gene constructs used for L. pyrotechnica transformation. a The T-DNA region of construct pCAMBIA1201 with gus and hpt genes. b The T-DNA region of construct pH7m24-35S:mGFP with gfp and hpt genes. MCS multiple cloning site, LB T-DNA left border, RB T-DNA right border. Restriction enzyme sites for Southern blots are shown. The bars illustrate the location of probes used for Southern blot analysis. The lines below the constructs show the expected minimum band size in Southern blots
The binary vectors were introduced into the disarmed hyper virulent A. tumefaciens strain EHA105 (Hood et al. 1993) by electroporation and streaked on solid Luria–Bertani (LB) medium (tryptone 10 g l−1; yeast extract 5 g l−1; NaCl 5 g l−1; agar, bacteriological grade 15 g l−1) containing plates with 25 mg l−1 rifampicin and the appropriate antibiotics for the corresponding binary vectors. The plates were incubated at 28 °C for 48 h in dark.
Determination of hygromycin dose for the selection of transformed L. pyrotechnica cells
The hygromycin sensitivity to select the transformed cells of L. pyrotechnica was determined. Hypocotyl explants were cultured on solid M3 medium (Table 1) containing 0, 10, 20, 30 and 40 mg l−1 hygromycin (Fig. 2) in the growth chamber. The explants were subcultured twice at 2 weeks interval. Thirty explants were used for each dose of hygromycin and the experiments were repeated twice.

Effects of different concentrations of hygromycin on callus induction response in L. pyrotechnica from 5 days old hypocotyl explants after 4 weeks of culture. Each value represents mean ± SD. At 20 mg l−1 hygromycin the callus induction was significantly (P < 0.05) reduced compared to no hygromycin treatment. Statistical analysis performed using Microsoft Excel 2010, n = 3
SAAT treatment of hypocotyl explants
To determine the optimum sonication time, hypocotyl explants were immersed in 50 ml screw capped corning tubes containing 5 ml liquid M3 medium (Table 1) and the tubes were placed at the center of a bath type sonicator (Branson 1025). The explants were subjected to sonication at a frequency of 60 kHz for 0, 20, 40, 60 and 80 s. The explants were then infected with Agrobacterium strain containing the plasmid pC1201 as described in the following section. The explants were co-cultivated with Agrobacterium on M2 medium for 2 days at 25 ± 1 °C in dark. After co-cultivation, the explants were washed 3 times in sterile water in sterile 50 ml tubes and cultured on M2 solid medium supplemented with 300 mg l−1 timentin and 20 μM silver nitrate for subsequent transient GUS assays. Frequency of transient gus expression was calculated as the percentage of explants showing at least one locus of GUS staining on the explant out of the total number of Agrobacterium infected explants.
Agrobacterium infection and plant transformation
Bacterial growth on solid LB plates with appropriate antibiotics was started 2 days before inoculation. On the day of infection, two loops of Agrobacterium culture were scraped off the petri plate and added to 20 ml liquid LB medium containing appropriate antibiotics in a 150 ml sterile flask. The flask was incubated at 28 °C with shaking (250 rpm) for 4 h. The cells were pelleted by centrifugation at 20 °C, 9,000g for 5 min after 4 h when the OD600 of the culture reached 0.5–0.6. The cells were resuspended in Agrobacterium vir gene induction medium as described by Gelvin (2006) which consisted of 1× AB salts, 2 mM NaH2PO4, 50 mM 2-(N-morpholino)ethanesulfonic acid, 0.5 % glucose, pH 5.6 (20× AB salts: per liter: 20 g NH4Cl, 6 g MgSO4·7H2O, 3 g KCl, 0.2 g CaCl2, 50 mg FeSO4·7H2O). Acetosyringone (Arcos Organics, Geel, Belgium) was added to the culture at a final concentration of 200 μM. The cultures were kept on gentle shaking (50 rpm) for 2 h at room temperature (21–23 °C). The culture was centrifuged at 20 °C; 9,000g for 5 min and the cells were resuspended in 15 ml M2 medium (Table 1). The explants were immediately infected with the prepared Agrobacterium culture (Table 2).
Approximately 200–300 hypocotyl explants were prepared from 5 days old seedlings 1 day before the Agrobacterium infection and incubated on M2 medium containing 200 μM acetosyringone at 26 °C in dark (Table 1) for each sonication treatment. While inducing the vir genes, the explants were collected in a 50 ml corning tube containing 5 ml liquid M2 medium. The tube was placed at the center of the bath type sonicator and subjected to sonication for either 40 or 60 s. Following sonication, the final Agrobacterium suspension (15 ml) was added to the tube and acetosyringone at a final concentration of 200 μM was added to the 20 ml final culture. The explants were infected with Agrobacterium for 30 min with gentle shaking (50 rpm) at room temperature in the dark. The explants in Agrobacterium suspension were poured in a sterile Petri plate and the bacterial suspension was removed as much as possible using a sterile 1 ml pipette. Using a sterile spatula, the explants were spread on to M2 solid medium containing 200 μM acetosyringone. The explants were co-cultivated with Agrobacterium for 2 days at 25 ± 1 °C in dark. Explants inoculated in bacterial suspension without prior sonication were considered as control for micro-wounding. After co-cultivation, the explants were washed 3 times in sterile water in a sterile 50 ml tube and cultured biweekly on M3 medium supplemented with 300 mg l−1 timentin, 20 μM silver nitrate, and 15 mg l−1 hygromycin for selection of transgenic calli for 4–6 weeks. Control treatments consisted of either non-infected explants or explants infected without sonication.
Selection and transgenic plant regeneration
Explants were biweekly transferred to fresh M3 selection medium up to 4–6 weeks. Emerged calli were then transferred to clear 150 ml sterile sundae cups each containing 50 ml M4 selection medium (shoot regeneration/elongation) supplemented with 300 mg l−1 timentin, 20 μM silver nitrate and 5 mg l−1 hygromycin. The tissue at this stage was subcultured twice in a period of 6–8 weeks. Tips (1 cm) of the elongated shoots were cut and cultured on M5 medium with 150 mg l−1 timentin for 2 days in the growth chamber. The shoots were then transferred to M6 medium supplemented with 100 mg l−1 timentin, 10 μM silver nitrate and 5 mg l−1 hygromycin for rooting and further growth.
Acclimatization of transgenic plants in greenhouse
Plants were propagated clonally through cutting of stems and rooting as described above in presence of hygromycin. The plantlets with approximately 1 cm long roots were taken out of the agar medium and transferred to soil mix (Sunshine Mix 4, Sun Gro Horticulture) in clear plastic 500 ml solo cups. The cups were covered by another cup to retain moisture while acclimatizing the plants in greenhouse. The covering cup was gradually removed as the roots spread to the bottom of the soil and new shoots emerged out of the stem nodes. The plantlets with roots spread in soil mix were then transferred to soil (4 sand: 1 clay) in 10 L pots and grown in greenhouse.
gus and gfp expression
The transient and stable gus expression assay was based on the protocol described by Jefferson et al. (1987). Transient histochemical GUS assay was performed on Agrobacterium infected explants cultured on M2 medium supplemented with 300 mg l−1 timentin for 3 days after co-cultivation and stable expression of gus was monitored in hygromycin resistant calli, germinated somatic embryos, and mature plant parts such as shoot tips, leaves and flowers. Samples were incubated in 100 mM sodium phosphate buffer pH 6.8 with 0.1 % (v/v, Tween-20) at 37 °C for 1 h. Samples were then washed two times with 100 mM sodium phosphate buffer pH 6.8. The assay buffer (1 mg ml−1 X-Gluc, 100 mM sodium phosphate buffer pH 6.8, 0.5 mM potassium ferrocyanide, 0.5 mM potassium ferricyanide, 100 μM Na2EDTA) was then added and vacuum applied for 5 min. Plant parts were incubated in the assay buffer overnight at 37 °C. After development of GUS stain, the chlorophyll was removed by repeated washing with 3:1 absolute alcohol: acetic acid (v/v).
The gfp expression was visualized under a fluorescence stereomicroscope. Hygromycin resistant calli, matured shoot tip and roots were visualized under either visible reflected light or UV fluorescent light and non-transformed and transformed tissues were compared to eliminate the possibility of autofluorescence from chlorophyll.
Genomic DNA extraction, PCR and Southern blot analysis of transgenic plants
Genomic DNA was extracted from soft young green leaves and shoot tips using the cetyltrimethyl ammonium bromide (CTAB) method as follows. The tissues were ground in liquid nitrogen and genomic DNA was extracted from 1 g tissue powder. The tissues were incubated in extraction buffer [0.1 M Tris pH 7.5; 1 M NaCl; 0.01 M EDTA; 1 % (w/v) CTAB; 2 % (w/v) polyvinylpyrrolidone and 5 % (v/v) β-mercaptoethanol] in 50 ml tubes. β-Mercaptoethanol was added to the buffer just before use. Extraction buffer (20 ml) was added to 1 g powdered tissue and mixed by gentle inversion. The tissue powder in buffer was kept at 65 °C for 30 min with occasional gentle shaking. The samples were brought to room temperature and 100 μl RNase (10 mg ml−1) was added. The tubes were incubated at 37 °C for 20 min. An equal volume of Chloroform: Isoamyl alcohol 24:1 was added and the tubes were kept at room temperature with gentle shaking for 20 min. The samples were centrifuged at 9,000g for 10 min. The supernatant was carefully removed into another tube without disturbing the pellet at the junction of aqueous and organic phase. The Chloroform: Isoamyl alcohol 24:1 treatment was repeated. An equal volume of chilled isopropanol was added to the supernatant collected and incubated at −20 °C for 1 h. The precipitate was collected by centrifugation at 9,000g for 10 min. The precipitated DNA was washed with 70 % ethanol, air-dried and dissolved in 200 μl 10 mM Tris–HCl buffer (pH 8.0).
PCR analysis was performed with 100 ng of genomic DNA in a 20 μl reaction volume to confirm the presence of transgenes in the putatively transformed plants. The presence of gus (353 bp amplified band) was detected using the primers GUS1F: ATGAACATGGCATCGTGGTGATTG and GUS1R: GAGATCGCTGATGGTATCGGTGTG. The reaction conditions were 94 °C for 5 min, 30 cycles of 94 °C for 30 s, 60 °C for 15 s, 72 °C for 30 s and 5 min extension at 72 °C. The presence of gfp (753 bp amplified band) was detected using the primers B1-GFP-HIS pEG103 and B2-GFP-HIS pEG103 mentioned before. The reaction conditions were 94 °C for 5 min, 30 cycles of 94 °C for 30 s, 60 °C for 20 s, 72 °C for 30 s and 5 min extension at 72 °C. The PCR reaction mixtures were analyzed in 1.2 % (w/v) agarose gels.
Genomic DNA (20 μg) of transgenic and non-transgenic (control) plants were digested with 3 U μg−1 EcoRI and HindIII restriction enzyme (New England Biolabs Inc. MA, USA) respectively. The digested DNA was concentrated and purified by ethanol precipitation and was electrophoresed for 8 h. The gels were treated with 0.25 N of HCl to depurinate briefly the DNA and then denatured with an alkaline solution for 30 min and neutralized for 30 min. The denatured DNA was then transferred to a nitrocellulose membrane following the protocol of Amersham Hybond-N+. (GE Health-Care Life Sciences, NJ, USA).
Southern blot analysis was based on the methods of Sambrook et al. (1989). The 353 and 753 bp amplified DNA from pCAMBIA 1201 and pH7m24-35S:mGFP constructs for gus and gfp genes, respectively were used to synthesize probes for Southern blot. The amplified products were gel-purified using the Qiagen gel extraction kit (Qiagen CA, USA). The probes were radio-labeled with α-dCTP32 (MP Biomedicals, CA, USA) using the Random primer labeling kit (Stratagene, USA). The membranes were pre-hybridized with SSC buffer (6× SSC, 1 % SDS) and Sperm DNA (10 mg ml−1 as blocking reagent) at 65 °C for 2 h. The membranes were hybridized with radiolabeled probes at 65 °C overnight. The membranes were rinsed once and washed twice with preheated 20 ml 2× SSC with 0.5 % SDS at 60 °C. The membranes were further washed with 0.2× SSC with 0.1 % SDS at 60 °C. The washed membranes were blotted dry on a filter paper and the radioactivity was assessed. The membranes were exposed to a phospho-screen prior to detection with a Phosphoimager (Typhoon 8600 Variable Mode Imager).
Statistical analysis
The frequency of transient and stable transformation is a mean of three independent experiments ±SE. The statistical analysis was done in Microsoft Excel 2010 and the means were compared at P < 0.05 using Duncan multiple range test.
Results
Sensitivity of hypocotyl explants to hygromycin
Different concentrations of hygromycin were tested in non-transformed L. pyrotechnica hypocotyl explants to determine the appropriate selection dose. All the explants died at 30 mg l−1 hygromycin within 4 weeks of hygromycin treatment. The ability of the explants to produce calli at the cut surface was significantly (P < 0.05) reduced in the presence of 20 mg l−1 hygromycin as compared to no hygromycin (11.7 vs. 100 %; Fig. 2). About 90 % of hypocotyl explants turned brown and died after 4 weeks on 20 mg l−1 hygromycin without any callus formation at the cut ends. At this hygromycin concentration, the calli formed from cut ends of the remaining 10 % explants were very small compared to no hygromycin control and the calli eventually turned brown after one more week of exposure to hygromycin. Callus formation at the cut ends of hypocotyl explants was also significantly reduced at 10 mg l−1 hygromycin (P < 0.05) as compared to the no hygromycin treatment and the callus turned brown after 4 weeks. Therefore, 15 mg l−1 hygromycin was used in subsequent transformation experiments to select transgenic tissue without compromising the stringency after 4–6 weeks of initial selection. At this hygromycin concentration, the transgenic tissues were visibly distinct from the dead explants grown on selection medium.
Effect of sonication on transient expression of the gus gene
There was no Agrobacterium growth around the explants 3 days after culturing on M2 medium (Table 1) supplemented with 300 mg l−1 timentin. The cut ends of the explants swelled by this time indicating rapid cell division. Infected explants were incubated in X-Gluc solution to test tissue susceptibility to Agrobacterium infection, and transient GUS assays were performed to test the effects of sonication on Agrobacterium infection. Sonication significantly (P < 0.05) enhanced gus transient expression (Fig. 3) and T-DNA delivery efficiency (79 vs. 26 % in 40 s sonicated and non-sonicated explants, respectively) at the cut surface of the explants. Up to 40 s of sonication, the GUS foci were clustered into small spots on the cut surface of the explants. Increase of sonication time up to 80 s increased the GUS foci on the overall surface of the explants. The increase in transient gus expression after 40, 60 and 80 s sonication remained insignificant (Fig. 3). The high transient gus expression and the stringent selection on hygromycin-containing medium indicated the feasibility of obtaining stable transformation of L. pyrotechnica.

Effect of SAAT treatment duration on transient gus expression frequency. Each value represents mean ± standard error. Means followed by same letters are not significantly different (P < 0.05)
Production of regenerated plants
The non-infected hypocotyl explants cultured on M3 medium formed friable greenish embryogenic calli in 2–3 weeks of culture. Callus induction from hypocotyl explants on M3 medium was 100 %. Embryos appeared on the callus in 4–5 weeks of callus induction with distinct root poles emerging out of the callus and the shoot pole partly buried into the callus. The calli at this stage were transferred to M4 medium in sundae cups for another 6–8 weeks with one change of medium. During this time, the shoots emerged out from 90 % of the calli and they elongated with distinct nodes and internodes. The excised shoot tips were rooted and clonally propagated as described in materials and methods.
Production of transgenic plants
Transgenic plant regeneration followed the order of culture media and procedure as described for plant regeneration from non-infected hypocotyl explants with exception of adding timentin and hygromycin to the media for controlling the Agrobacterium growth and selection of transgenic tissue (Table 1). Following sonication (40, 60 s) and co-cultivation with A. tumefaciens, the hypocotyl explants (Fig. 4a) were incubated on M3 medium supplemented with timentin and hygromycin for a stringent selection (Table 1). Distinct green calli appeared at the cut ends of the selected explants after 5–6 weeks of selection. The non-infected explants turned brown and were visibly distinct from the green transgenic calli (Fig. 4b). At this stage hygromycin resistant calli were further subcultured in sterile sundae cups containing 50 ml M4 medium with 300 mg l−1 timentin, 5 mg l−1 hygromycin to maintain the independent transgenic plants from each treatment. Shoot development on the surface of the hygromycin resistant green calli was observed in another 6–8 weeks on M4 selection medium (Fig. 4c). An average of 92 % shoots rooted with an average of 7 roots from the cut ends of each shoot within 10–20 days of transfer to M6 medium as described in materials and methods (Fig. 4d). Plantlets were grown in covered cups in the greenhouse for 3–4 weeks and in 10 L pots afterwards (Fig. 4e). The transformation frequency, based on the number of hygromycin-resistant T0 plants out of total number of co-cultivated hypocotyl explants was determined. Stable transformation frequency was determined for three sonication treatments (0, 40 and 60 s) because the transient transformation frequency was not significantly different between 40, 60 and 80 s sonication, but they were significantly different from no sonication treatment (Fig. 3). The average stable transformation frequencies were 4, 14 and 11 % for 0, 40 and 60 s sonication treatments respectively. The average stable transformation frequency at 40 s was higher compared to 60 s, but both were significantly (P < 0.05) different from no sonication treatment. Therefore, the transformation was repeated with the gfp construct following 40 s of sonication in order to check the reproducibility of the protocol. The stable transformation frequency for gfp gene construct was 11 % at 40 s sonication treatment. The plants acclimatized and grown in greenhouse showed 95 % survival with no morphological difference from the non-transformed control plants. Twelve independent transgenic lines expressing gus and nine independent transgenic lines expressing gfp were grown in the greenhouse.

Different stages of transgenic plant development in L. pyrotechnica. a Hypocotyl explant from 5 days old seedlings (bar, 3 mm). b Selection of Agrobacterium infected hypocotyl explant on M3 medium with 15 mg l−1 hygromycin (bar, 1.3 cm); green hygromycin resistant calli with distinct shoot buds and root poles (circled in green) is visually distinguishable from the non-transformed dead brown explants (circled in red) after 6 weeks of selection. c Shoots from hygromycin resistant calli elongated on M4 medium with 5 mg l−1 hygromycin (bar, 4 mm). d Root induction from transgenic shoots and maintenance of transgenic plants on M6 medium with 5 mg l−1 hygromycin (bar, 0.8 cm). e Transgenic plants acclimatized and grown in greenhouse. (Color figure online)
Reporter gene expression
In order to confirm the progress of the transformation process, GUS activity and GFP fluorescence were assessed at different developmental stages. Distinct blue foci along the cut edges of the explants, 3 days after co-cultivation was the first step confirmation of Agrobacterium infection on the plant tissues (data not shown). No gus expression was observed in regenerated non-transformed control plantlets (Fig. 5a). GUS activity was observed in roots and shoots of the hygromycin-resistant plantlets (Fig. 5b). GUS staining was also observed in the shoot tip, stem, petiole, leaves and flowers of mature transgenic plants (Fig. 5d, f, g). No endogenous GUS activity was detected in the non-transformed control plants (Fig. 5c, e).

Expression of gus gene in different plant parts. a No GUS activity in the germinated somatic embryo from non-transformed hypocotyl callus (bar, 3 mm). b Constitutive expression of gus gene in germinated somatic embryo from hygromycin resistant callus; root and shoots are GUS stained (bar, 3 mm). c No gus expression in shoot tip of non- transformed plant (bar, 3 mm). d GUS staining in shoot tip of transgenic plant (bar, 3 mm). e No gus expression in mature leaf of non-transformed plant (bar, 5 mm). f GUS staining in mature leaf, leaf petiole and stem of transgenic plant (bar, 5 mm). g GUS staining in flowers of transgenic L. pyrotechnica (bar, 4 mm)
Expression of gfp at different stages of plant transformation showed a similar pattern to that of GUS activity in the transgenic plants as both the reporter genes were driven by the CaMV35S promoter. Non-transformed plants did not display fluorescence (Fig. 6b, f, j), while a bright green fluorescence was observed in the hygromycin-selected calli (Fig. 6d), shoot tip, stem, leaves and roots of the transgenic plants (Fig. 6h, l).

GFP fluorescence in different stages of transgenic plant development in L. pyrotechnica. The left panel shows different parts of non-transgenic control plants viewed under bright light (left side, a, e, i) and UV light using GFP filter (right side, b, f, j). a, b Callus from non-transformed hypocotyl explant (bar, 2 mm). e, f Shoot of non-transformed plant (bar, 5 mm). i, j Root of non-transformed control plant (bar, 5 mm). The right panel shows different parts of transgenic plants viewed under bright light (left side, c, g, k) and UV light using GFP filter (right side, d, h, l). c, d Four week old hygromycin resistant callus (bar, 2 mm). g, h Stem and emerging shoot of transgenic plant (bar, 5 mm). k, l Root of transgenic plant under bright light and GFP expression in the same roots under UV light (bar, 5 mm)
Molecular analysis of transgenic L. pyrotechnica
The detection of 353 and 753 bp amplified bands corresponding to gus and gfp genes in PCR analysis confirmed the presence of the transgenes in the transformed T0 plants (Fig. 7a, b). Six 35S:GUS and five 35S:mGFP PCR positive transgenic plants were further analyzed by Southern blot hybridization to verify the number of insertions in their genome. Hybridizations of radiolabeled probes to total genomic DNA digested with EcoRI and HindIII for gus and gfp gene blots were expected to identify DNA fragments unique to individual integration events greater than 3.0 and 4.6 kb respectively (Fig. 1). The transgenic plants showed differential integration events and simple integration patterns that ranged from single to two loci. All the hybridized fragments were more than 3.0 and 4.6 kb as expected (Fig. 8a, b). No hybridization signal was detected in the untransformed plants (Fig. 8a, b, lanes NC). One out of six 35S:GUS plants and one out of five 35S:mGFP transgenic plants showed double T-DNA insertion (Fig. 8a, b). The integrity of the T-DNA was indicated by hygromycin selection and display of GUS/GFP activity in mature plant parts.

a PCR detection of gus gene in transgenic plants. Lane M molecular weight DNA marker (50 bp). Lane B blank, PCR mix without template DNA, Lane C DNA from non-transformed control plant (negative control). Lane P plasmid pCAMBIA1201 DNA (positive control). Lanes 1–6 are six transgenic L. pyrotechnica plants from independent transformation events; the arrow indicates the fragment corresponding to the gus gene (353 bp). b PCR detection of gfp gene in transgenic plants. Lane M molecular weight DNA marker (100 bp). Lane B blank, PCR mix without template DNA. Lane C DNA from non-transformed control plant (negative control). Lane P plasmid pH7m24-35S:mGFP DNA (positive control). Lanes 1–5 are five transgenic L. pyrotechnica plants from independent transformation events; the arrow indicates the fragment corresponding to the gfp gene (753 bp)

a Southern blot analysis of gus gene in transgenic plants. DNA from plant material was digested with EcoRI, electrophoresed and probed with a α-P32 labeled gus gene fragment (353 bp). The number of bands corresponds to number of transgene insertion. Lanes 1–6 transgenic plants from independent transformation events. Lane NC DNA from non-transformed control plant (negative control). Lane PC 353 bp PCR amplified fragment (positive control). The size of the markers is indicated on the left. b Southern blot analyses of gfp gene in transgenic plants. DNA from plant material was digested with HindIII, electrophoresed and probed with a α-P32 labeled gfp gene fragment (753 bp). The number of bands corresponds to number of transgene insertion. Lanes 1–5 transgenic plants from independent transformation events. Lane NC DNA from non-transformed control plant (negative control). Lane PC 753 bp PCR amplified fragment (positive control). The size of the markers is indicated on the left
Discussion
Woody plants, shrubs as well as trees are recalcitrant to transformation as compared to herbaceous plants. Here, we describe an efficient and reproducible Agrobacterium-mediated genetic transformation method for the multipurpose woody desert shrub L. pyrotechnica.
Asclepiadacean species are considered as difficult-to-transform plants due to the presence of latex and other plant specific secondary metabolites. The non-amenability of Agrobacterium infection of Asclepiadacean members has been emphasized during the studies on P. sepium (Miyabashira et al. 2003). A scrutiny of successful transgenic plant regeneration among Asclepiadacean members accounts only two species: T. indica (Chaudhuri et al. 2006) and P. sepium (Chen et al. 2010). Chen et al. (2010) have studied various factors affecting the efficiency of Agrobacterium-mediated transformation using stem (from seedling derived shoots) explants of P. sepium. They accomplished an efficacy of 50–60 % transformation using the A. tumefaciens EHA105 after analyzing various parameters including other Agrobacterium strains. In T. indica, the induction of hairy roots and transgenic plants was dependent on A. rhizogenes strains and explant type (Chaudhuri et al. 2005, 2006). In their study, the A4 strain inoculated only at the nodes of intact shoots grown in vitro induced hairy roots (60 %) and was followed by spontaneous formation of transgenic plants. The suitability and high amenability of hypocotyl explants to Agrobacterium-mediated transformation as in the present study has been demonstrated in several woody plant species (Vengadesan et al. 2006; Du and Pijut 2009; Prakash and Gurumurthi 2009).
Here, the transformation efficiency of L. pyrotechnica hypocotyl explants was significantly increased by sonication treatments. Sonication has been shown to augment transformation efficiency, particularly in species recalcitrant to transformation (Pathak and Hamzah 2008; Bakshi et al. 2011; Subramanyam et al. 2011). Sonication of L. pyrotechnica explants, before the Agrobacterium infection, contributed to create micro-wounds on the plant surface, providing better access of the Agrobacterium cells to the explants. Similar results have been reported during the infection of other plant species (Pathak and Hamzah 2008; Bakshi et al. 2011; Subramanyam et al. 2011). In the present study, an increase of sonication time increased the frequency of transient gus expression, but there were no significant difference in increase of transient gus expression observed after 40 s of sonication. This was indicative of significant difference in stable transformation frequencies between no sonication treatment and sonication for more than 40 s. In spite of no significant difference in transient gus expression, the stable transformation frequency at 60 s was lower compared to 40 s sonication which might indicate tissue damage due to extended sonication.
Both M2 and M3 media produced callus at the cut ends with 100 % efficiency. The calli proliferated faster on M2 medium compared to M3 medium. This fast cell division probably contributed to the efficient T-DNA transfer during the co-cultivation step. Infected explants after 40 s sonication and co cultivation on M2 medium (Table 1) showed 79 % of transient GUS infection compared to 64 % on M3 medium. Nevertheless, there was no plant regeneration on M2 medium even after prolonged culture. Therefore the M3 selection medium was used to obtain hygromycin resistant calli and shoots subsequently.
Maintaining 300 mg l−1 timentin up to M4 selection medium was important in order to prevent tissue necrosis because of Agrobacterium overgrowth. Both the 35S promoter and the nos promoter for hygromycin resistance were able to select transgenic plants at 15 mg l−1 hygromycin. The stringency of hygromycin selection made the protocol false positive-free and highly reliable without compromising the growth of transgenic plants.
Similar transformation efficiencies with two vectors harboring different reporter genes, their expression in all stages of transgenic plant development and a low copy integration of T-DNA demonstrate the reliability and reproducibility of the transformation protocol. The transformation procedure was completed within 20–22 weeks (starting from seed surface sterilization to planting putative transgenic plants in the greenhouse) and can be used to generate transgenic plants with genes of biotechnological interest.
Abbreviations
- SAAT:
-
Sonication-assisted Agrobacterium-mediated transformation
- GUS:
-
β-Glucuronidase
- GFP:
-
Green fluorescent protein
- hpt :
-
Hygromycin phosphotransferase
- X-Gluc:
-
5-bromo-4-chloro-3-indolyl glucuronide
- BA:
-
N6-Benzyladenine
- NAA:
-
Naphthaleneacetic acid
- ABA:
-
Abscisic acid
- IBA:
-
Indole-3-butyric acid
References
Alyemeni NM (2000) Ecological studies on sand dunes vegetation in AI-Kharj region, Saudi Arabia. Saudi J Bio Sci 7:64–87
Bakshi S, Sadhukhan A, Mishra S, Sahoo L (2011) Improved Agrobacterium-mediated transformation of cowpea via sonication and vacuum infiltration. Plant Cell Rep 30:2281–2292
Chaudhary S, Khosa RL, Jha KK, Kumar S (2011) Evaluation of antidiabetic activity of whole plant of Leptadenia pyrotechnica (Forssk.) Decne against streptozotocin induced diabetes in rats. Pharmacol Online 2:1196–1204
Chaudhuri KN, Ghosh B, Tepfer D, Jha S (2005) Genetic transformation of Tylophora indica with Agrobacterium rhizogenes A4: growth and tylophorine productivity in different transformed root clones. Plant Cell Rep 24:25–35
Chaudhuri KN, Ghosh B, Tepfer D, Jha S (2006) Spontaneous plant regeneration in transformed roots and calli from Tylophora indica: changes in morphological phenotype and tylophorine accumulation associated with transformation by Agrobacterium rhizogenes. Plant Cell Rep 25:1059–1066
Chen R, Gyokusen M, Nakazawa Y, Su Y, Gyokusen K (2010) Establishment of an Agrobacterium-mediated transformation system for Periploca sepium Bunge. Plant Biotechnol 27:173–181
Choi J, Shin J, Chung Y, Hyung N-I (2012) An efficient selection and regeneration protocol for Agrobacterium-mediated transformation of oriental melon (Cucumis melo L. var. makuwa). Plant Cell Tissue Organ Cult 110:133–140
Dhawan AK, Singh H (1976) Properties of acid phosphatase from Leptadenia pyrotechnica Forsk. Indian J Exp Biol 14:344–345
Du N, Pijut P (2009) Agrobacterium-mediated transformation of Fraxinus pennsylvanica hypocotyls and plant regeneration. Plant Cell Rep 28:915–923
Earley KW, Haag JR, Pontes O, Opper K, Juehne T, Song K, Pikaard CS (2006) Gateway-compatible vectors for plant functional genomics and proteomics. Plant J 45:616–629
Gelvin SB (2006) Agrobacterium virulence gene induction. In: Wang K (ed) Agrobacterium protocols. Humana Press, Totowa, pp 77–84
Goyal M, Sharma SK (2006) Prospects and dimensions for utilization arid foods Yash publishing house. Bikaner, India
Goyal M, Sharma SK (2009) Traditional wisdom and value addition prospects of and foods of desert region of North West India. Indian J Tradl Knowl 8:581–585
Hood EE, Gelvin SB, Melchers LS, Hoekema A (1993) New Agrobacterium helper plasmids for gene transfer to plants. Transgenic Res 2:208–218
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions beta glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO 6:3901–3908
Karimi M, Bleys A, Vanderhaeghen R, Hilson P (2007) Building blocks for plant gene assembly. Plant Physiol 145:1183–1191
Khasawneh MA, Elwy HM, Hamza AA, Fawzi NM, Hassan AH (2011) Antioxidant, anti-lipoxygenase and cytotoxic activity of Leptadenia pyrotechnica (Forssk.) Decne polyphenolic constituents. Molecules 16:7510–7521
Liu M, Lu S, Liu L, Tan J, Guo Z (2012) Agrobacterium-mediated transformation of centipede grass (Eremochloa ophiuroides [Munro] Hack.). Plant Cell Tissue Organ Cult 109:557–563
Migahid AM, Batanoun K, Abdelwah A (1972) Eco-physiological studies on desert plants. 7. Water relations of Leptadenia pyrotechnica (Forsk.) Decne. Growing in Egyptian desert. Oecologia 10:79–91
Miyabashira A, Gyokusen K, Sando T, Fukusaki E, Kobayashi A, Nakazawa Y, Bamba T, Su YQ (2003) Genetic transformation of Periploca (Periploca sepium Bunge). Kyushu J For Res 56:178–179
Moustafa AMY, Khodair AI, Saleh MA (2007) Phytochemical investigation and toxicological studies of lipid constituents isolated from Leptadenia pyrotechnica. J Pharmacol Toxicol 2:681–697
Moustafa AMY, Khodair AI, Saleh MA (2009) Isolation, structural elucidation of flavonoid constituents from Leptadenia pyrotechnica and evaluation of their toxicity and antitumor activity. Pharm Biol 47:539–552
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 5:473–497
Pathak M, Hamzah R (2008) An effective method of sonication-assisted Agrobacterium-mediated transformation of chickpeas. Plant Cell Tissue Organ Cult 93:65–71
Prakash M, Gurumurthi K (2009) Genetic transformation and regeneration of transgenic plants from precultured cotyledon and hypocotyl explants of Eucalyptus tereticornis Sm. using Agrobacterium tumefaciens. In Vitro Cell Dev Biol Plant 45:429–434
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning a laboratory manual second edition, vols 1, 2 and 3
Sharma KC, Chouhan CS (2008) Ecology and restoration of lake Budha Pushkar—a threatened water body of Ajmer, Rajasthan. In: Sengupta M, Dalwani R (eds) The 12th world lake conference, pp 1757–1764
Singh JP, Rathore VS, Beniwal RK (2007) Kheep (Leptadenia pyrotechnica): potential rangeland shrub of Western Rajasthan, India. Indian J Plant Genet Resour 20:199–203
Soliman GA, Donia AERM, Awaad AS, Alqasoumi SI, Yusufoglu H (2012) Effect of Emex spinosa, Leptadenia pyrotechnica, Haloxylon salicornicum and Ochradenus baccatus extracts on the reproductive organs of adult male rats. Pharm Biol 50:105–112
Subramanyam K, Subramanyam K, Sailaja K, Srinivasulu M, Lakshmidevi K (2011) Highly efficient Agrobacterium-mediated transformation of banana cv. Rasthali (AAB) via sonication and vacuum infiltration. Plant Cell Rep 30:425–436
Sujatha M, Vijay S, Vasavi S, Reddy PV, Rao SC (2012) Agrobacterium-mediated transformation of cotyledons of mature seeds of multiple genotypes of sunflower (Helianthus annuus L.). Plant Cell Tissue Organ Cult 110:275–287
Trick HN, Finer JJ (1997) SAAT: sonication-assisted Agrobacterium-mediated transformation. Transgenic Res 6:329–336
Vengadesan G, Amutha S, Muruganantham M, Anand R, Ganapathi A (2006) Transgenic Acacia sinuata from Agrobacterium tumefaciens-mediated transformation of hypocotyls. Plant Cell Rep 25:1174–1180
Acknowledgments
The authors are thankful to Ms Allison Hutmacher for her technical support. This work was supported by a grant from the UAE Ministry of Presidential Affairs.
Conflict of interest
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dutta, I., Kottackal, M., Tumimbang, E. et al. Sonication-assisted efficient Agrobacterium-mediated genetic transformation of the multipurpose woody desert shrub Leptadenia pyrotechnica . Plant Cell Tiss Organ Cult 112, 289–301 (2013). https://doi.org/10.1007/s11240-012-0236-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-012-0236-4