
Abstract
Double-stranded RNA interference can be used to silence gene expression in various organisms. Sustained RNAi-mediated gene silencing is typically triggered by hairpin RNAs (hpRNAs) generated by the transcription of inverted repeat (IR) DNA constructs. In this paper, we describe a transient expression assay that uses a green fluorescent protein marker gene (GFP) fused to the coat protein gene (CP) of papaya ring spot virus. This yields a nuclear and cytomembrane localized protein expression vector that can be used to monitor the silencing efficiency of inverted repeats. An ihpRNA was derived from a 215 nt IR segment of the 3′ end of the CP gene and recombined into the pHellsgate12 vector. We also recombined the 861 nt CP into the destination vectors pHellsgate12 and pWatergate using the GatewayTM technology. Nicotiana benthamiana protoplasts were co-transfected with (1) pGA, (2) pGA + pHellsgate12-CP IR, (3) pGA + pWatergate-CP IR, and (4) pGA + pHellsgate12-CP 215IR. Expression of the GFP fusion protein decreased by 77.7, 75.4, and 65.6% for the three constructs. Transfection with (2) and (3) yielded a silencing efficiency significantly higher than that of the (4) (P < 0.05), as measured by fluorescence intensity upon co-transfection, using the vector pGA-DsRed as an internal control. Depletions of 90.7, 91.5, and 87.5% of the CP transcript were observed by RT-PCR in protoplasts co-transfected with the (1) pGA, (2) pGA + pHellsgate12-CPIR, and (3) pGA + pWatergate-CPIR, (4) pGA + pHellsgate12-CP 215IR. Transient RNAi depletion of the target polypeptide was also detected by western blot analysis. Short RNA fragments complementary to the p6a and p7a antisense probes were detected by Ribonuclease Protection Assays.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The molecular biology of papaya has been considerably advanced by the completion of the papaya genome sequencing project in 2008 (Ming et al. 2008). The current challenges are now to determine the functions of all of the annotated papaya genes. In papaya, a total of 21,784 (76.1%) of the predicted papaya genes, with average length of 1,057 base pairs, have similarity to proteins in the non-redundant database from the National Center for Biotechnology Information of USA, with 9,760 (44.8%) of these supported by papaya unigenes. Functional analyses of these genes have not yet been initiated.
RNA interference (RNAi) is a powerful tool for studying gene function through gene silencing. In plants, there are three natural pathways for RNA silencing. Cytoplasmic small interfering RNA (siRNA) silencing is most likely a defence mechanism against plant viruses, endogenous messages can be silenced by microRNA (miRNA) genes and DNA methylation can lead to silencing of transcription (Baulcombe 2004). RNA can also be artificially silenced by the introduction of dsRNA into cells to initiate RNAi silencing. The introduced dsRNA is detected as aberrant and is processed by type III RNase Dicers to short interfering RNAs (siRNAs, 20–26 nucleotides long) (Bernstein et al. 2001; Brodersen and Voinnet 2006). One of the two siRNA strands is then incorporated into the RNA-induced-silencing complex (RISC), which uses siRNAs to recognize complementary motifs in target nucleic acids. The result is sequence specific inhibition of gene expression at either the transcription, the mRNA stability, or the translational levels (Baulcombe 2004; Brodersen and Voinnet 2006; Brodersen et al. 2008).
RNAi can be introduced into plant cells by constructing DNA expression vectors that encode self-complementary hpRNAs and homologues that target the gene sequence of interest. For instance, constructs have been specifically designed to express dsRNA for viral coat protein genes (Zrachya et al. 2007; Tougou et al. 2006), replicase genes (Lennefors et al. 2006), etc. in the form of hairpin RNAs (hpRNAs). These hpRNAs can elicit a high frequency of post-transcriptional gene silencing (PTGS) of an invading virus. Constructs manipulating the expression of inverted repeats act as endogenous sources of sustained dsRNA supply. These can be used to generate stably transformed organisms that show heritable gene silencing. This strategy of expressing an ihpRNA gene to silence a virus can induce PTGS with ~100% efficiency (Smith et al. 2000). However, the production and analysis of transgenic plants is time-consuming and labour intensive, and requires maintenance of multiple plant lines. A rapid, genome-wide reverse-genetic screen for initial selection of multiple genes of interest would greatly expedite functional genetic studies.
Plant protoplasts, which lack cell walls, are versatile cell-based experimental systems. Macromolecules such as DNA, RNA, etc. can be delivered into protoplasts using a variety of methods, including PEG, electroporation, and microinjection. The protoplast system also has some unique advantages for analyzing gene silencing in plants. First, the cells are more homogeneous than are found in planta, which minimizes sample variability. Second, DNA constructs can be directly delivered into protoplasts, making it possible to experiment with different types of DNA vectors. Third, protoplasts isolated from plant tissues retain their differentiated state, allowing detailed time-course studies on the genetics and kinetic features of RNA silencing pathways (Qi et al. 2004; Bart et al. 2006; Chau and Lee 2007).
A number of studies have demonstrated rapid gene silencing measured by transient expression. For example, Bezanilla et al. (2003) developed a rapid identification system for silencing the NLS:GFP:GUS construct of 2 reporter genes and a gene of interest via transiently expressed GUS-RNAi or GFP–RNAi. Yoo et al. (2007) demonstrated transient gene expression in Arabidopsis mesophyll protoplasts. Hanako et al. (2008) found that the degree of RNA silencing and the effectiveness of viral suppressors varied in two protoplast lines derived from different tobacco species. Zhai et al. (2008) showed that transfection of protoplasts with an in vitro-synthesized dsRNA against Arabidopsis thaliana γ-glutamylcysteine synthase (AtECS1) resulted in a 95% depletion of the AtECS1 transcript, a 72% decrease of the AtECS1 polypeptide, and a 60% drop in GSH content.
Although RNA interference is a natural antiviral mechanism, the natural RNAi systems do not provide complete viral. The application of hpRNA as a new antiviral strategy is promising, as its mechanism is different from that of normal transformation. In fact, hpRNA-mediated silencing may strengthen the natural antiviral mechanisms, to create the ideal antiviral transformation against serious crop pathogens. In the present paper, we were interested in studying papaya ring spot virus (PRSV), one of the most common diseases and one of the major causes of severe losses in production worldwide (Fitch et al. 1994; Tripathi et al. 2004). Our interest was to determine the best RNAi vector for increasing viral resistance in papaya breeding stock. A transient expression system using Nicotiana benthamiana protoplasts was use to determine the silencing efficiency of three inverted repeat constructs containing different fragments of a PRSV coat protein gene. Silencing efficiency was determined by testing the fluorescence intensity of the fusion reporter protein GFP:CP, by comparing the relative abundance of transcripts of RNA-targeted CP genes, by detecting the protected RNA fragments by RPA, and by peptide analysis.
Materials and methods
Tobacco protoplast isolation and gene transformation by PEG
Protoplasts were isolated using a modified version of a protocol developed for Arabidopsis (Sheen 1995, 2001). Nicotiana benthamiana seedlings were grown for 20–25 days. The leaves were cut into 0.5 mm wide pieces with sharp razor blades. We then put the leaves into an active enzyme solution (1% Cellulase Onozuka R-10 s, 0.4% Macerozyme R-10 s, 0.2 M mannitol, 20 mM MES, 20 mM KCl, pH 5.7) under vacuum for 10 min. Subsequently, they were incubated in a tube containing 10 ml of filtered-sterilized enzyme solution for 1.5 h with gentle shaking at 40 rpm and 28°C, in the dark. The tissues were then filtered through a 320-mesh stainless steel filter, and the protoplasts were purified in a solution (0.2 M mannitol, 20 mM MES, 20 mM KCl and 21% sucrose) by centrifugation for 5 min at 100 g (800c Shanghai AnTing, China). Protoplasts were allowed to settle at room temperature for 15 min, and then the deep green suspension layer was removed, washed with solution I (0.2 M mannitol, 20 mM MES, 20 mM KCl, 1% BSA) and suspended in solution II (solution I plus 0.2 M CaCl2). Protoplast density was measured with a hemocytometer.
We used a polyethylene glycol (PEG) method for protoplast transformation essentially as described by Huang et al. (2000), He et al. (2006) and Yoo et al. (2007). A 40% (w/v) PEG 4,000 solution was prepared with 0.2 M mannitol and 100 mM CaCl2 (pH 8). Plasmid DNA (10 μg) and carrier calf thymus DNA (20 μg) was added to a 7 ml tube, followed by 500 μl of protoplasts (cell density, 2 × 105) in CaCl2 solution. For co-transfection, equal amounts of the two plasmids were added. Immediately after adding the protoplasts, 530 μl of a 40% PEG 4,000 solution was added and the solution was gently and thoroughly mixed. Protoplasts were incubated at room temperature (23°C) for 25 min, and then the transfection was stopped with the addition of 2 ml W5 solution (154 mM NaCl, 125 mM CaCl2, 5 mM KCl, 4 mM MES, pH 5.7). The solution was then centrifuged at 100 g for 2 min to remove the PEG solution, and the protoplasts were removed gently and added to a 2 ml W5 solution containing 0.2 M mannitol as an osmoticum. Protoplasts were incubated at room temperature (23–25°C) for 36 h, then analyzed for GFP expression. Samples were collected at 36–48 h for molecular analysis. Protoplasts were harvested by centrifuging at 100 g for 5 min. Collected samples were stored at −70°C for several days until RNA extraction.
Vector design and construction
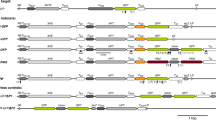
The cloning vector pBS-mGFP(AF078810) was kindly provided by Dr. von Arnim. This vector has a length of 4,780 bp and includes the 35S promoter and the mGFP coding sequence (von Arnim et al. 1998). The entire length of PRSV CP (861 bp) was cloned into the Bgl II and XbaI restriction endonuclease site using primers Cf and Cr (Table 1), yielding the cloning vector pBS–mGFP–CP 861 for the expression of GFP fusion proteins (Fig. 1a). Then, gene fusions were amplified by LA–PCR from the pBS–mGFP–CP 861 based plasmid with primers G1 and C1. The DNA fragment was cleaved using Xba I and BamH I and inserted into the unique Xba I and BamH I sites of the binary vector pGA482 (An 1986), which contains the cauliflower mosaic virus (CaMV) 35S promoter with a duplicated 35S enhancer region, and the 35S terminators (Fig. 1b). The recombinant plasmid was designated pGA. The DsRed gene was amplified with primers Rf and Rr (Table 1); the template was the pDsRed-expression vector (BD Biosciences, Clontech). The DNA fragments were excised and inserted into pGA482 between the restriction endonuclease Xba I and BamHI to form an internal control pGA–DsRed (Fig. 1c).

The cloning vector for pBS–mGFP–CP 861 and the expression cassette for pGA and pGA–DsRed
Construction of inverted repeat expression vectors using GatewayTM technology
We designed attB-flanked PCR primers and PCR-amplified the 861 nt gene for the coat protein (CP) of Papaya ring spot virus. The attB1 PCR primers were
-
CP861 (+): 5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTATGTCCAAGAATGAAGC-3′ (forward) and
-
CP861 (-): 5′-GGGGACCACTTTGTACAAAGCTGGGTTTAGTTGCGCATACCCAG-3′ (reverse).
The pGEM-T easy plasmid containing the cloned CP gene cDNA sequence was used as a template (Jiang et al. 2007). The amplification conditions were as follows: denature at 94°C for 30 s, anneal at 63°C for 30 s, extend 72°C for 1 min and repeat for 35 cycles. We purified the attB PCR product, performed a BP recombination reaction with the attB-PCR products and the donor vector pDONRTM221 to generate an entry clone, and then processed an LR recombination reaction with the entry clone and the destination vector (both pHellsgate12 and pWatergate; provided by CRISO Industry). Positive clones were selected by screening plates with LB medium supplemented with Spec 50 mg/L (Fig. 2a–c). We followed the instructions given in the Gateway® BP Clonase enzyme and LR ClonaseTM II manuals (Invitrogen). Similarly, the predicted RNA structures, containing the 215 nt 3′ end fragment of the target gene sequence, were constructed with the forward primer

The map of the expression vectors pHellsgate12-CP IR (a), pWatergate-CP IR (b) and pHellsgate12-CP 215IR (c)
-
CP215 (+):5′-GGGGACAAGTTTGTACAAAAAAGCAGGCTCTCGCTAGATATGCTTTC-3′ and the reverse primer
-
CP215 (-):5′-GGGGACCACTTTGTACAAAGCTGGGTTTAGTTGCGCATACCCAG-3′.
Fluorescence measurements
Silencing was measured in co-transfection experiments with pGA–DsRed, an internal control, and the following plasmids: (1) pGA, (2) pGA & pHellsgate12-CP IR, (3) pGA & pWatergate-CP IR and (4) pGA & pHellsgate12-CP 215IR, which were transfected into the same batch of protoplasts. The protoplasts (cell density, 2 × 105) in a 500 μl/sample were transfected with plasmids (10 μg each). Three duplicate samples were collected at 36 h post-transfection; three batches of independent transformation experiments were performed. The transient expressions of GFP and DsRed were observed with a fluorescence microscope (Nikon Eclipse E600 Fluorescence Microscope, Japan), using a filter block containing a 450–490 nm excitation filter, a 505 nm dichroic mirror, and a 520 nm barrier filter for GFP fluorescence. DsRed fluorescence was viewed with a filter block containing a 510–560 nm excitation filter, a 575 nm dichroic mirror, and a 590 nm barrier filter.
Samples were extracted with CBS buffer (0.01 M Na2CO3, 0.03 M NaHCO3), and the total fluorescence of 2 × 105 cells in each experiment was measured by a RF-5301PC fluorescence spectrophotometer (Shimadzu, Japan) and associated software (RF-5301PC). Excitation and emission parameters were set as follows: EX 475, EX520 nm for GFP; EX557, EM579 nm for DsRed. The fluorescence values of GFP and DsRed from pGA and pGA–DsRed transfected cells were arbitrarily set to one. Fluorescence measurements for all other treatments are shown as relative values. GFP fusion expression and cellular localization were observed after transfection using a confocal laser-scanning microscope (LSM 510 META, ZEISS, Germany) set to ch2: BP 505, 530 nm, ch3: Lp 560 nm (Pixel Depth: 12-bit; objective: plan-A, Pochromat 63×/1.4 oil DIC) and associated software (Carl Zeiss Image Microscopy Release 4.0). Representative images are presented.
Isolation and purification of total RNA
The transfected cells were first lysed in a denaturing lysis solution, which stabilizes RNA and inactivates RNases. The lysate was then extracted once with Acid–Phenol:Chloroform, which removes most of the other cellular components and leaves a semi-pure RNA sample. This sample was further purified over a glass–fibre filter to yield either total RNA or a size fraction enriched in shorter RNA fragments. We followed the instruction manual for the mirVana™ miRNA Isolation Kit for this procedure.
Detection the abundance of mRNA by RT-PCR
Protoplasts expressing RNAi and the CP gene (2 × 105cells/500 μl) were collected after 36 and 48 h, by centrifugation for 2 min at 100g. Total RNA was extracted with TRIzol®reagent (Invitrogen) following the manufacturer’s protocol. RNA aliquots (2 μg) were subjected to reverse transcription with MMLV reverse transcriptase (TaKaRa). The PCR conditions were as follows: denaturing at 95°C for 5 min, followed by 30 cycles of 95°C for 30 s, 58°C for 30 s, 72°C for 1 min. Primers used for RT-PCR analyses are detailed in Table 1.
Western blot analysis of GFP polypeptide
RNAi and control protoplasts (2 × 105cells/500 μl) were collected by centrifugation at 100 × g for 2 min, then washed by pelleting twice in 0.5 ml PBS, resuspended in 0.3 ml PBS, and disrupted by sonication. Protoplast proteins were separated by SDS–PAGE on 8% (w/v) gels and electro transferred in solution (39 mM Glycine, 48 mM Tris, 0.037% (w/v) SDS, 20% (v/v) methanol) to a nitrocellulose filter. The nitrocellulose blots were probed with the GFP primary rabbit polyclonal antibody (25KD, 1:400 dilution, Clontech) and with secondary AP-conjugated anti-rabbit antibody (1:2,500, GE Healthcare). Immunoreactive bands were visualized with ECL using the Alkphos Labeling and Detection System (Amersham). Duplicated Gel electrophoresis was conducted for the same set of samples with one of the two gels stained by Coomassie Brilliant Blue and used as the protein marker.
RNase protection assays
We prepared samples for the RPA assay from the transfections (1) pGA, (2) pGA & pHellsgate12-CP IR, (3) pGA & pWatergate-CP IR and (4) pGA & pHellsgate12-CP 215IR. We designed DNA Oligonucleotide templates for the transcription of antisense RNA probes for the PRSV CP gene using the manufacturer’s protocol for the mirVanaTM miRNA Probe construction (Table 2). We prepared a master mix for hybridization to yield the following final amounts of each component: 10 μl 2 × Hybridization Buffer, 1–5 × 104 cpm labelled RNA probe, 3 μg sample RNA, 2 μg yeast RNA and nuclease-free water to 20 μl. We denatured the samples at 95°C for 3 min. We then incubated the reactions at 42°C to hybridize the probe to its complement sample RNA for overnight. We prepared a 1:100 working dilution of RNase A/T1 solution in RNase Digestion Buffer, added 150 μl of diluted RNase to each sample RNA tube, and for the negative control we added 150 μl RNase Digestion Buffer without RNase to the remaining no target control tube. These reactions were incubated for 38 min at 37°C; then we added 225 μl RNase Inactivation/PPT Solution followed by 225 μl 100% ethanol. Tubes were placed overnight at −20°C and then centrifuged twice for 25 min at 4°C at 12,000g. All supernatants were removed from the tubes and the pellets were washed with 70% ethanol. The protected fragments were resuspended in 5 μl Gel Loading Buffer II (GLBII) and incubated for 3 min at 95°C. Samples were run on a denaturing 15% polyacrylamide gel at 150 V for 3 h. The gel was exposed to a phosphor screen for 3 h, and signals from radiolabelled protected fragments were detected by a phosphoimager (FUJIFILM IP FLA-5100, JAPAN). Images were captured with the associated software (FLA-5000 Imager). The above protocol was obtained from the mirVana™ miRNA Detection Kit (ambion) instruction manual.
Results and analysis
Isolation and purification of activated protoplasts and protoplast transfection efficiency
The effectiveness of RNAi silencing of genes in protoplasts depends on the viability of the isolated protoplasts and their transfection efficiency. We optimized conditions by employing 20–25 day old Nicotiana benthamiana seedlings for protoplast isolation. The released protoplasts were purified by density gradient centrifugation in a sucrose-enriched buffer and the top green layer of suspension was collected. Using this improved procedure, we obtained pure suspension of protoplasts (Fig. 3a). The cell density of the suspension was measured with a hemocytometer using a Nikon Eclipse E600 fluorescence microscope under an ocular 10×, objective 20× (Fig. 3b) and (oil) 100× (Fig. 3c).

Isolation of protoplasts from Nicotiana benthamiana seedlings. a A tube containing green suspension of protoplasts freshly isolated from Nicotiana benthamiana. b Bright-field microscopy of protoplasts on a hemocytometer under ocular ×10, objective ×20. c enlarged microscopy of protoplasts under ocular ×10, objective (oil) ×100, Bar = 20 μm
An efficient PEG procedure was adopted for the protoplast transfection and the transfection efficiency was examined by a simple visual assay with fluorescence microscopy. Fluorescence microscopy repetitious observation revealed that about 50–60% of the transfected protoplasts expressed the nuclear and cytomembrane-targeted GFP fusion protein at 36 h, suggesting that satisfactory transfection efficiency was achieved (Fig. 4a).

Visualization of GFP fusion protein expression in Nicotiana benthamiana protoplasts and cell localized GFP fusion protein. a Fluorescence image of protoplasts at 36 h under Nikon Eclipse E600 Fluorescence Microscope. Green fluorescence was derived from the pGA vector. Red fluorescence was generated by chloroplasts autofluorescence. Bar = 20 μm. b Confocal micrographs showing transient expression of GFP fusion protein is distributed in the nucleus and cytomembrane in a Nicotiana benthamiana cell. White arrows indicate that GFP was localized in the nucleus primary and cytomembrane in the pGA transfected protoplasts. c Fluorescence micrograph show red fluorescence alone. d Bright field. e Overlays of bright field and fluorescence images on b, c and d
Localization of GFP fusion protein
A GFP fusion reporter was used to examine the cellular distribution of GFP:CP in Nicotiana benthamiana cells. Protoplasts were transformed with PEG using the recombinant construct pGA, incubated for 36 h and then imaged with a confocal laser-scanning microscope. GFP:CP fusion protein was transiently expressed in the nucleus and cytomembrane (Fig. 4b–e).
Comparing the silencing efficiency of three ihpRNAs
We used three constructs for silencing of GFP:CP expression; namely, pHellsgate12-CP IR, pWatergate-CP IR, and pHellsgate12-CP 215IR, which target the CP region. A marked reduction in green fluorescence was observed after 36 h (Fig. 5). The expression of GFP and DsRed proteins in the same cells was visualized by fluorescence microscopy after 36 h. The transformed protoplasts emitted green fluorescence and the addition of hpRNA effectively silenced the expression of the GFP fusion gene (Fig. 5e–h). The red fluorescence protein functioned as an internal control (Fig. 5i–l).

hpRNA silencing of the GFP fusion reporter in Nicotiana benthamiana protoplasts co-transfected with pGA (10 μg) (a, e, i) and together with the plasmids: pHellsgate12-CP IR (10 μg) (b, f, j), pWatergate-CP IR (c, g, k) or pHellsgate12-CP 215IR (d, h, i). pGA–DsRed (10 μg) was used as the internal control and red fluoresce was cytoplasmic and freely diffused into the nucleus. After 36 h of incubation, the protoplasts were observed using fluorescence microscopy; a, b, c and d. Brightfield through Dla-ILL filter; e, f, g and h. B-2A filter; i, j, k and l. G-2A filter. Bar = 20 μm
Three versions of the inverted repeat expression vectors were compared to find the one most competent for gene silencing. Fluorescence intensity of equal amounts of protoplasts co-transfected with each vector was determined and normalized relative to the intensity of the control sample. There was a marked drop in GFP expression compared to the control, while DsRed fluorescence showed almost no change, in protoplasts co-transfected with all three vectors. The silencing effect calculated as 77.7,75.4 and 65.6% for plasmid combinations (1) pGA, (2) pGA & pHellsgate12-CP IR, (3) pGA & pWatergate-CP IR, and (4) pGA & pHellsgate12-CP 215IR. The (2) and (3) presented a silencing efficiency significantly higher than that of the (4) (P < 0.05, LSD0.05 = 8.93, LSD0.01 = 13.52) by least significant difference (LSD) analysis (Fig. 6). The quantitative data agreed with those obtained by fluorescence microscopy.

Quantitative analysis of fluorescence intensity of GFP and DsRed. The intensity in control Nicotiana benthamiana protoplasts was artificially set to a value of 1; those of the treated protoplasts are presented as values relative to the control. 1, Control. 2–4, Plasmids pHellsgate12-CP IR, pWatergate-CP IR and pHellsgate12-CP 215IR
These results confirm that the DNA encoding the inverted repeat RNA structure can trigger transient RNAi-mediated gene silencing. Both pHellsgate12-CP IR and pWatergate-CP IR have the 861 bp of CP and an intron of cat and pdk, but use different promoters. The silencing efficiency of the pHellsgate12-CP IR and pWatergate-CP IR constructs was similar to but higher than that for pHellsgate12-CP 215IR with a shorter fragment of CP gene. The above results indicated the length of the IR fragment might affect the silencing efficiency.
Analysis the relative abundance of mRNA
The effectiveness of RNAi was evaluated by analysis of aliquots from the RT-PCR reaction on 1% gel and comparing the relative abundance of transcripts of RNA-targeted genes, using the Gelscan V 5.0 software (BioScitec GmbH). The housekeeping gene ACTIN was amplified as an internal control to determine the amount of template. Depletion of 90.7, 91.5 and 87.5% of the CP transcripts was observed in protoplasts co-transfected with pGA and with the three ihpRNAs separately (pHellsgate12-CP IR, pWatergate-CP IR and pHellsgate12 -CP 215IR) at 36 h (Fig. 7a, c). Similar trends in expression silencing were detected at 48 h (Fig. 7b, d).

The ihpRNA-specific silencing of the CP gene in transfected Nicotiana benthamiana protoplasts by RT-PCR analysis. a, 36 h after transfection. Lane 1, Protoplasts transfected with pGA. Lane 2–4: Protoplasts co-transfected with pGA and one of three ihpRNAs (pHellsgate12-CP IR. pWatergate-CP IR or pHellsgate12 -CP 215IR.), housekeeping gene ACTIN was amplified as a control for amount of a template. b: 48 h after transfection. Lanes 1–4 correspond to those depicted in Fig. 7 a. Lane 5, WT. C, Analysis of abundance of mRNA level in Fig. 7 a using the Gelscan V 5.0 software, resulted in 90.7, 91.5 and 87.5% depletion of CP transcript from lane 2–4. d Analysis of abundance of mRNA level in Fig. 7 b resulted in 84.5, 84.2 and 81.9% depletion of CP transcript from lane 2–4, 5, WT
Silencing analysis via western blotting
Western blotting was used to analyze whether transient RNAi in protoplasts results in depletion of the GFP:CP fusion polypeptide. RNAi in protoplasts decreased the abundance of the corresponding polypeptide (Fig. 8). A single band of about 40KD revealed a peptide signal that reacted with the rabbit polyclonal GFP antibody in protoplasts transfected with pGA (Fig. 8, lane 1). The co-transfection of pGA and pHellsgate12-CP IR (lane 2), pGA and pWatergate-CP IR (lane 4), and pGA and pHellsgate12 -CP 215IR (lane 5), showed only a weak 40KD band. No signal was detected for the wild type (lane 3).

Gene silencing assay with Western blotting using GFP antibody. 1, Transfected with 10 μg of pGA, the blot was probed with GFP polypeptide antibody, an approximately 40KD band (arrow) corresponding to fusion GFP: CP. 2, 4, 5, Co-transformation with an equal amount of pGA and pHellsgate12-CP IR, pGA and pWatergate-CP IR and pGA and pHellsgate12-CP 215IR, showed weak 40KD bands, the disturbed peptide signal was barely visible. 3, WT, no signals were detected. M, Protein marker cut from a duplicate gel
RNase protection assays of short RNA fragments
To perform highly sensitive RNase Protection Assays, the 37 and 49 nt 32P-labeled CP probes were separately hybridized with a denatured RNA sample. We prepared the antisense RNA probes of the CP gene and evaluated whether siRNA could be efficiently produced in tobacco protoplasts upon co-expression of both the fusion expression vector of pGA and the ihpRNA construct. The short fragment-dependent band signals of small RNA fragments consistent with ~17, 21, and 25 nt markers were observed on a denaturing polyacrylamide gel (Fig. 9a, b). The small RNA fragment specifically complementary to the p6a or p7a antisense probe confirmed the presence of siRNA derived from the CP IR constructs.

Detection of siRNA in Nicotiana benthamiana protoplasts that were co-transfected with a CP gene expression vector and an IR-DNA construct. a RPA analysis using probe P6a. Lane 1, hybridization with the negative control probe labeled with a-32P UTP. Lanes 2 and 3, P6a without digestion by RNaseA/T1. Lane 4, RNA from protoplasts cotransfected with pGA and pHellsgate12-CP IR. Lane 5, RNA from protoplasts cotransfected with pGA and pWatergate-CP IR. Lane 6: γ-32P ATP labeled 21 nt marker. b RPA analysis using probe P7a. Lane 1, co-transformation of pGA and pHellsgate12-CP IR. Lane 2 and Lane 3, co-transformation of pGA and pWatergate-CP IR. Lane 4, co-transformation of pGA and pHellsgate12 -CP 215IR. Lane 5, WT. Lane 6, undigested probe P7a. Lane 7, RnaseA/T1 digested probe. Lane 8, undigested probe P7a. Lane 9, γ-32P ATP labeled 25, 21 and 17 nt marker. Lane 10, 21 nt marker
Discussion
Transient expression systems that use marker genes such as green fluorescent protein to detect the silencing efficiency of RNAi targeted genes of interest have been used for gene function analysis and in plant breeding for virus resistance. Bezanilla et al. (2003) reported the constitutive silencing of fusion protein (NLS:GFP:GUS) with GUS–RNAi or GFP–RNAi and the silencing of NLS-4, leading to loss of function phenotype in a moss. In the present experiment, we studied an RNAi system by co-transfecting both a fusion expression vector of an exogenous GFP:CP gene and the corresponding RNAi expression vectors targeting the CP gene of papaya ring spot virus. The RNAi vector targeting CP clearly silenced the expression of both GFP and CP in protoplasts. The following findings are notable.
First, fluorescence intensity level corresponded to the silencing effect of the gene of interest by ihpRNA vector. Second, the method was a convenient and reliable strategy for fast detection of the efficiency of ihpRNA silencing. Third, silencing efficiency was related to the length of the sense and antisense strands of the RNAi vector. Fourth, the optimum time for detecting RNAi silencing effects was between 36 and 48 h. Fifth, known probes, instead of mixed probes, were used to detect siRNAs by RPA, which allowed a clearer understanding of the sequence of the target siRNAs. Sixth, transfection of hpRNA depleted not only the transcript of a targeted gene, but also the corresponding polypeptide.
Protoplasts isolated from young leaves of 20–25 days-old seedlings of Nicotiana benthamiana were the ideal candidates for transfection. The experimental conditions for protoplast isolation, transfection, and expression were optimized. The entire procedure, from leaf collection to PEG transformation, took only 6–7 h, and the transient expression of green fluorescence could be observed by 36 h with a fluorescence microscope.
SiRNAs are short (usually 21 nt) doubled-stranded segments of RNA (dsRNA) with 2 nt 3′ overhangs on either end. The transfection of a synthetic siRNA can be defective because the gene knockdown effect is transient, particularly in rapidly dividing cells. Considerable research has been conducted to determine the most efficient silencing constructs. Thus far, the most effective dsRNA construct is one that transcribes an hpRNA (Smith et al. 2000). In the present experiment, we used three inverse repeat constructs to induce gene silencing in Nicotiana benthamiana protoplasts. The efficiency of the constructs pHellsgate12-CP IR and pWatergate-CP IR was comparable. Both vectors were slightly more effective than pHellsgate12-CP 215IR. Our data confirmed the idea proposed by Chen et al. (2004) that silencing efficiency was positively correlated to the length of the target gene fragment. The result also indicated there was no significant difference between constructs with the same IR DNA driven by the 35S CaMV or ARbcs promoters.
The ribonuclease protection assay (RPA) is a sensitive method for the detection of specific microRNA or siRNA. The probe set is hybridized in excess to target RNA in solution, and then free probes and other single-stranded RNAs are digested with RNases. The remaining “RNase-protected” probes are purified, resolved on a denaturing polyacrylamide gel, and quantified by autoradiography or phosphorimaging. (Huang, Han and Howell 2000; Agarwal et al. 2007). We used the mirVana™ miRNA Detection Kit (Ambion) to detect siRNA. In this procedure, there is no need to transfer the siRNA to a solid support, and hybridization is performed in solution. This procedure ensures its sensitivity. We designed and selected two antisense probes from four antisense probes (data not shown), small fragment RNA specifically complementary to the p6a or P7a antisense probe was detected and confirmed the presence of short fragment RNA derived from the CP IR construct. Most previous studies have used mixed probes to detect the siRNA (Qi, et al. 2004; Chau and Lee 2007), but we used known probes whose sequence was not homologous with published microRNA MIR162a of papaya (Porter et al. 2008).
Off-target silencing is another challenge, although this problem can be partly addressed with appropriate control experiments. Cordula et al. (2008) found that mRNA was deregulated by a GFP siRNA containing a short sequence that was perfectly matched to the sense or antisense strand of GFP siRNA. Whether ihpRNA introduced into plants causes non-specific silencing is still in question. In the present experiment, we designed an appropriate negative control to avoid the possible errors caused by off-targets.
One drawback with a protoplast transient expression system is that protein behaviour in protoplasts may be different from that in intact cells in the plant. Biological and morphological differences may exist between protoplasts and cells in a living plant. These differences should be taken into consideration and investigations should be carried out regarding this issue.
In summary, we have developed an effective transient expression system that uses a green fluorescent protein marker gene (GFP) fused to inverse repeat constructs targeting a gene of interest, for rapid detection of gene silencing in Nicotiana benthamiana. We have also successfully tested this system in papaya (data not shown), and we believe that this system is readily adaptable for use in other plant species.
Abbreviations
- CP :
-
Coat protein
- dsRNA:
-
Double stranded RNA
- GFP:
-
Green fluorescence protein
- hpRNA:
-
Hairpin RNAs
- IR:
-
Inverse repeat
- NLS:
-
Nuclear localization signal
- PBS:
-
Phosphate buffer solution
- PTGS:
-
Post-transcriptional gene silencing
- RNAi:
-
RNA interference
- RPA:
-
Ribonuclease protection assays
- siRNA:
-
Small interfering RNA
References
Agarwal SK, Gao SH, Smith AV, Jin H (2007) A novel class of bacteria-induced small RNAs in Arabidosis. Genes Dev 21:3123–3134
An G (1986) Development of plant promoter expression vectors and their use for analysis synthase promoter in transformed tobacco cells. Plant Physiol 81:86–91
Bart R, Chern M, Park CJ, Bartley L, Ronalc PC (2006) A novel system for gene silencing using siRNAs in rice leaf and stem-derived protoplasts. Plant Methods 2:13. doi:10.1186/1746-4811-2-13
Baulcombe DC (2004) RNA silencing in plants. Nature 431:356–363
Bernstein E, Caudy AA, Hammond SM, Hannon G (2001) Role for abidentate ribonuclease in the initiation step of RNA interference. Nature 409:363–366
Bezanilla M, Aihong P, Quatrano RalphS (2003) RNA interference in the moss Physcomitrella patens. Plant Physiol 133:470–474
Brodersen P, Voinnet O (2006) The diversity of RNA silencing pathways in plants. Trends Genet 22:268–280
Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O (2008) Widespread translational inhibition by plant miRNAs and siRNAs. Science 320:1185–1190
Chau BL, Lee KA (2007) Function and anatomy of plant siRNA pools derived from hairpin transgenes. Plant Methods 3:13. doi:10.1186/1746-4811-3-13
Chen YK, Lohuis D, Goldbach R, Prins M (2004) High frequence induction of RNA-mediated resistance against Cucumber mosaic virus using inverted repeat constructs. Mol Breeding 14:215–226
Cordula T, Angela S, Armin P, Wiebke W, Axel B, Agnes HW, Leticia SB, Peter L, Daniel M (2008) Off-target effects of siRNA specific for GFP. BMC Mol Biol 9:60. doi:10.1186/1471-2199-9-60
Fitch MM, Pang SZ, Slightom JL, Lius S (1994) Genetic Transformation in Carica papaya L (Papaya). In: Bajaj YPS (ed) Biotechnology in Agriculture and Forestry somatic embryogenesis and synthetic seed, vol 29. Springer, Berlin, pp 237–256
Hanako S, Yuki K, Kazunori G, Chikara M (2008) Degree of RNA silencing and the ability of a viral suppressor vary depending on the cell species in a protoplast system. J Gen Plant Pathol 74:326–330
He P, Shan L, Sheen J (2006) The use of protoplasts to study innate immune responses. In: Ronald PC (ed) From: methods in molecular biology, plant–pathogen interactions: methods and protocols, vol 354. Humana Press Inc., Totowa http://genetics.mgh.harvard.edu/sheenweb/reprints/Protoplast-Innate%20immunity-final.pdf
Huang Z, Han Y, Howell SH (2000) Formation of surface tubules and fluorescent foci in Arabidopsis thaliana protoplasts expressing a fusion between the green fluorescent protein and the cauliflower mosaic virus movement protein. Virology 271:58–64
Jiang L, Zhang FY, Zhang MT (2007) Detection of Papaya ringspot virus (PRSV) by cDNA probes labeled with Digoxgenin labelling and AlkPhos direct labelling. J Agric Biotechnology 15(3):496–502
Lennefors BL, Savenkov EI, Bensefelt J, Elisabeth WW, van Roggen P, Tuvesson S, Valkonen JPT, Gielen J (2006) DsRNA-mediated resistance to beet necrotic yellow vein virus infections in suger beet (Beta vulgaris L. ssp. Vulgaris). Mol Breeding 18(4):313–325
Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH, Senin P, Wang W, Ly BV, Lewis KL, Salzberg SL, Feng L, Jones MR, Skelton RL, Murray JE, Chen C, Qian W, Shen J, Du P, Eustice M, Tong E, Tang H, Lyons E, Paull RE, Michael TP, Wall K, Rice DW, Albert H, Wang ML, Zhu YJ (2008) The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452:991–996
Porter BW, Aizawa KS, Zhu YJ, Christopher DA (2008) differentially expressed and new non-protein-coding genes from a Carica papaya root transcriptome survey. Plant Sci 174(1):38–50
Qi YJ, Zhong XH, Itaya A, Ding B (2004) Dissecting RNA silencing in protoplasts uncovers novel effects of viral suppressors on the silencing pathway at the cellular level. Nucleic Acids Res 32(22):e179
Sheen J (1995) Methods for mesophyll and bundle sheath cell separation. Methods Cell Biol 49:305–314
Sheen J (2001) Signal transduction in maize Arabidopsis mesophyll protoplasts. Plant Physiol 127:1466–1475
Smith NA, Singh SP, Wang MB (2000) Gene expression: total silencing by intron-spliced hairpin. Nature 407:319–320
Tougou M, Furutani N, Yamagishi N, Shizukawa Y, Takahata Y, Hidaka S (2006) Development of resistant transgenic soybeans with inverted repeat-coat protein genes of soybean dwarf virus. Plant Cell Rep 25:1213–1218
Tripathi S, Bau HJ, Chen LF, Yeh SD (2004) The ability of papaya ring spot virus strains overcoming the transgenic resistance of papaya conferred by the coat protein gene is not correlated with higher degrees of sequence divergence from the transgene. Eur J Plant Pathol 110(9):871–882
Von Arnim AG, Deng XW, Stacey MG (1998) Cloning vectors for the expression of green fluorescent protein fusion proteins in transgenic plants. Gene 221:35–43
Yoo SD, Cho YH, Sheen J (2007) Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc 2(7):1565–1572
Zhai ZY, Sooksa-nguan T, Olena K (2008) Vatamaniuk functional analysis of genes in protoplasts by RNAi. Plant Physiol Preview. doi: 10.1104/pp.108.130260 (Published on November 12)
Zrachya A, Kumar PP, Ramakrishnan U, Levy Y, Loyter A, Arazi T, Lapidot M, Gafni Y (2007) Production of siRNA targeted against TYLCV coat protein transcripts leads to silencing of its expression and resistance to the virus. Transgenic Res 16(3):385–398
Acknowledgments
We thank CRIRO industry provided the destination vectors. We are grateful for the State Key Laboratory of Agricultural Microbiology of Huazhong Agriculture University provided the pGA482 vector and the equipments of phosphorimager and isotope label. Thanks for SRF (Student Researching Fund) project of Huazhong Agriculture University supported this work. This work was funded by the national natural science foundation of china projects (No. 30571277 and No. 30771477).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jiang, L., Wang, J., Liu, Z. et al. Silencing induced by inverted repeat constructs in protoplasts of Nicotiana benthamiana . Plant Cell Tiss Organ Cult 100, 139–148 (2010). https://doi.org/10.1007/s11240-009-9629-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-009-9629-4