
Abstract
Oats produce a group of secondary metabolites termed avenanthramides (avn). These compounds are biosynthesized through the action of the enzyme hydroxycinnamoyl CoA: hydroxyanthranilate N-hydroxycinnamoyl transferase (HHT) which catalyzes the condensation of one of several cinnamate CoA thioesters with the amine functionality of anthranilic acid, 4-hydroxy- or 5-hydroxy-anthranilic acid. In oat leaf tissue the biosynthesis of avenanthramides appears to result from elicitation by fungal infection. Here we demonstrate the biosynthesis of several avenanthramides in suspension cultures of oat apical meristem callus tissue. This phenomenon appears as a generalized pathogen response, evidenced by the production of PR-1 mRNA, in response to elicitation with chitin (poly-N-acetyl glucosamine). The suspension cultures also produce relatively large quantities of avnA and G in response to chitin elicitation. Under certain culture conditions avnB and C are also produced as well as three additional metabolites tentatively identified as avnH, O and R. These findings portend the utility of oat suspension culture as a tool for more detailed investigation of the mechanisms triggering their biosynthesis as well as the factors dictating the particular types of avenanthramides biosynthesized.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Phenolic antioxidants, termed avenanthramides, are, among the cereals, produced exclusively in oat. Avenanthramides have also been found in butterfly eggs (Blaakmeer et al. 1994a, b) and in carnation (Dianthus caryophyllus) (Ponchet et al. 1988). These metabolites are conjugates of a phenylpropanoid with an anthranilic acid (or hydroxylated anthranilic acid) moiety. The final reaction in their biosynthesis is catalyzed by the enzyme hydroxycinnamoyl-CoA:hydroxyanthranilate N-hydroxycinnamoyltransferase (HHT) (Ishihara et al. 1997, 1999a) (Fig. 1). There are at least twenty-five structural varieties of avenanthramides found in oat (Collins 1989); however, the three forms typically found in greatest abundance in the grain are avenanthramide (avn) A, B, and C (Collins 1989), corresponding to: N-(4′-hydroxy-(E)-cinnamoyl)-5-hydroxyanthranilic, N-(4′-hydroxy-3′-methoxy-(E)-cinnamoyl)-5-hydroxyanthranilic acid, and N-(3′, 4′-dihydroxy-(E)-cinnamoyl)-5-hydroxyanthranilic acid, respectively. Avenanthramides possess potent antioxidant activity in vitro (Bratt et al. 2003; Dimberg et al. 1993; Peterson et al. 2002) and antifungal properties (Mayama et al. 1982). Recently, avnC was demonstrated to be effective in reducing muscle inflammation in exercise stressed rats (Ji et al. 2003). In experiments using in vitro cell culture models for atherosclerosis development, avnC demonstrated potent inhibition of vascular smooth muscle cell proliferation (Liu et al. 2004; Nie et al. 2006b), a hallmark of atherosclerosis. These effects appear to be mediated through inhibition of certain protein phosphorylations necessary for cell cycle progression (Nie et al. 2006a). The anti-irritant properties ascribed in folklore to oatmeal salves have been shown to result primarily from the avenanthramide component (Vollhardt et al. 2000).

Reaction catalyzed by hydroxycinnamoyl-CoA:hydroxyanthranilate N-hydroxycinnamoyltransferase (HHT) the final step in avenanthramide biosynthesis. A Coenzyme A activated phenylpropanoid (p-coumaric, ferulic, or caffeic acid) is conjugated to 5-hydroxy (or 4-hydroxy-) anthranilic acid to form avn A, B, C, G or H or with a Coenzyme A activated avenalumic acid to form avn O and R
Quantitative analysis of avenanthramides in oat grain shows a wide range of concentrations in various genotypes and growing environments. In Swedish cultivars, concentrations in groats ranged between 25–47 mg/kg (avnA), 21–43 mg/kg (avnB), and 28–62 mg/kg (avnC) (Dimberg et al. 1996). The same avenanthramides were found in 80% ethanol extracts from oat grown in Wisconsin, along with additional putative avenanthramides in minor amounts that were not identified (Emmons et al. 1999). Significant differences between cultivars and growing environments were also observed in the Wisconsin grown oats (Emmons and Peterson 2001). Thus, avenanthramides are found constitutively in oat groats (albeit with dynamic metabolic profiles) but only at very low concentrations in healthy leaf tissue (Peterson and Dimberg, unpublished data). Biosynthesis of avenanthramides in leaf tissue occurs in response to fungal pathogens or chemical elicitors (Ishihara et al. 1998, 1999b). Genes encoding HHT have recently been cloned and sequenced and appear to exist as at least four isozymes (Yang et al. 2004).
To further investigate factors affecting the biosynthesis of avenanthramides we have developed a suspension culture system responsive to elicitation by crab shell chitin. These cultured cells produce and secrete avnA (N-(4′-hydroxy-(E)-cinnamoyl)-5-hydroxyanthranilic acid) as well as avnG (N-(4′-hydroxy-(E)-cinnamoyl)-4-hydroxyanthranilic acid), in response to elicitation (Fig. 2). They also demonstrate a generalized pathogen response as evidenced by the production of pathogen related-1 (PR-1) mRNA. Here we describe the time course of avenanthramide biosynthesis as well as the production of HHT mRNA, HHT enzyme activity and the PR-1 mRNA in oat cell cultures, subsequent to elicitation by chitin.

Overlaid HPLC chromatograms monitored by UV adsorption at 340 nm of ethyl acetate extracted media 12 h after elicitation with 0.1 mg/ml chitin (solid) and unelicited callus (dashed). The peaks labeled 1, 2 and 3 had retention times and UV spectra identical to authentic standards of avn C, A and B, respectively. Peak 4 is identified as AvnG (see methods). Peak 5 is unidentified. Both chromatograms are set to the same vertical scale and are slightly offset
Methods
Cell culture
Suspension cultures were initiated from callus obtained from the shoot apical meristem of germinating oat (Avena sativa) cv ‘Belle’ (Forsberg et al. 1999). After sterilization with a 10% solution of chlorox the dehulled groats were imbibed in sterile water for several hours and then allowed to germinate for 2 days. When the emerging coleoptiles reached 4–10 mm in length the shoot apical meristem tissue was cut into approximately 1 mm length sections. The explant meristem was placed on solid (0.2% phytogel) Murashige and Skoog (1962) media (MS), in the dark, for 14 days before transfer to fresh solid media. The callus used in these experiments was subcultured multiple times on solid media. For liquid cultures a minimum of 1.0 g callus was transferred to 25 ml of MS media (MP Biomedicals #2623020) containing: 2 mg/l (4.5 μM) 2,4D (2,4 dichlorophenoxyacetate), 100 mg/l myo-inositol, 2% (w/v) sucrose, and 150 mg/l l-asparagine with 750 μl/l PPM (plant preservative mixture, Plant Cell Technology, (5-chloro-2-methyl-3(2H)-isothiazolone and 2-methyl-3(2H)-isothiazolone active ingredients)), in a 125 ml Erlenmeyer flask and grown an additional 2 weeks at 23°C, shaking at 110 RPM in the dark. Liquid cultures were sub-cultured twice before use. Cultures were induced with sterile chitin at 0.25 mg/ml (unless otherwise stated) by adding an appropriate volume of a sterile suspension of 50 mg/ml chitin in water. Control cultures were treated with an equal volume of sterile H2O.
Effect of time in liquid media on avenanthramide production
In these experiments approximately 2.0 g of tissue was transferred from solid MS media to 25 ml of liquid MS media with PPM. The tissues were conditioned in the fresh media for 24, 48, or 72 h prior to elicitation with 100 μg/ml chitin by adding 50 μl of a sterile 50 mg/ml suspension of chitin. Each treatment was performed in triplicate and media was extracted at 24, 48, and 72 h after elicitation. Control cultures received 50 μl sterile water instead of the chitin solution.
Avenanthramide analysis
Approximately 0.5 ml 1 M HCl was added to the media which was then extracted three times with an equivalent volume of ethyl acetate. The organic phase was pooled, reduced under vacuum (rotoevaporated) and resuspended in MeOH. The methanolic extract was filtered through a 0.22 μm filter, reduced to dryness then resuspended in 0.5 ml MeOH. This fraction was analyzed by HPLC on a 4.6 × 50 mm C-18 column using a diode array spectrophotometer detector (Shimadzu 10A). The mobile phase consisted of buffer A: H20 with 5% acetonitrile and 0.1% formic acid and buffer B: acetonitrile with 0.1% formic acid. A gradient of 13 to 30% B over 20 min at a flow rate of 1.0 ml/min was employed. The peaks corresponding to avnA, B, and C were quantified by comparison of the peak area to standard curves developed using the corresponding authentic, synthesized avenanthramides. The avnG was estimated by comparison of peak areas with the standard curve of avnA. Avenanthramides were extracted from the oat tissue by grinding 0.5–1.0 g (wet weight) with a hand held Polytron for approximately 1 min. The ground tissue was resuspended in 10 ml 80% EtOH and shaken in a water bath at 50°C for 20 min. The tissue was pelleted by low speed centrifugation and the 80% EtOH supernatant decanted. The tissue was extracted twice more in the same manner and the supernatants pooled, reduced to dryness under vacuum (rotovap). The residue was resuspended in 2–3 ml MeOH, filtered through a 0.22 μm filter reduced to dryness, resuspended in 0.5 ml MeOH and stored at −20°C until analyzed by HPLC or LC-MS. LC-MS analysis was performed on an Agilent 1100 liquid chromatography system with a 1946 series ion-trap mass spectrometer. A 2.1 × 30 mm C-18 column (Zorbax SB-C18, Agilent) was employed with the same solvent system as above at a flow of 0.2 ml/min. Detection was made by diode array spectrometry monitoring absorbance at 280 and 330 nm and by ion-trap mass spectrometry. Electrospray ionization parameters were as follows: nebulizer gas (N2) 30 psi, dry gas flow at 8.0 l/min at 350°C with a capillary voltage set at 3,500 V. The ion trap was operated in the positive mode, scanning from m/z 100–1,000 at 13,000 m/z sec−1.
Isolation and NMR analysis of avnG
The media and tissue extracts from the elicitation time course experiments were pooled, concentrated under vacuum and separated on a 10 × 250 mm C-18 column (Beckman ultrasphere) using an isocratic solvent of 30% acetonitrile in water with 0.1% formic acid at 3.0 ml/min. The eluent corresponding to avnG was collected, pooled, extracted into ethyl acetate, dried under vacuum and resuspended in CD3OD for NMR analysis. NMR analysis was performed on a Varian Unity Inova 800 MHz instrument. 1H NMR: δ 6.48 (1H, dd, J = 8.8, 2.4 Hz), 6.54 (1H, d, J = 16.0 Hz), 6.81 (1H, d, J = 8.8 Hz), 7.49 (2H, d, J = 8.8 Hz), 7.56 (1H, d, 16.0 Hz), 7.94 (1H, d, J = 8.8 Hz), 8.15 (1H, d, J = 2.4 Hz). (Note that in the original description of avnG (Miyagawa et al. 1996b) the coupling constants for the signals at δ 6.48 and 6.54 were inadvertently reversed (Miyagawa, personal communication)).
HHT activity
Between 0.5 and 1.0 g of tissue was homogenized with a Brinkman homogenizer in 2 ml of 100 mM BisTris buffer at pH 7.2 with 2 mM DTT. The homogenate was centrifuged at 12,000g for 25 min and the supernatant recovered. Protein concentration was determined by the Bradford method using BSA as a standard. HHT enzyme activity was determined by reacting 10 μl the protein extract with 10 μl of 10 mM 5-hydroxy-anthranilic acid in DMSO and 50 μl of 2.5 mM coumaryl CoA in H2O for 50 min at 30°C in 30 μl BisTris (100 mM, pH 7.2) and 2 mM DTT, total reaction volume 100 μl. The reaction was initiated by adding the protein extract and stopped by adding 20 μl concentrated (17.4 M) acetic acid. The reaction mixture was diluted with 0.38 ml MeOH, filtered through 0.22 μM filter and analyzed by HPLC.
Coumaryl CoA was biosynthesized using a crude buffer extract of corn rind as a source for 4-coumaryl CoA ligase (4-CL). Approximately 50 g of corn rind was ground to a fine power in liquid N2 using a stainless steel Waring blender. The powdered rind was dissolved in 100 ml of MOPS (200 mM with 15% glycerol, 10 mM MgCl2 and 1 mM dithiothreitol), stirred at room temperature for 2 h then filtered through cheesecloth and centrifuged for 15 min at 25,000g. A 50 ml portion of the supernatant was added to a flask containing 200 ml MOPS buffer, pH 7.5 with 15% glycerol, 14 mg p-coumaric acid, 35 mg Coenzyme A and 300 mg ATP (final concentrations of 0.4 mM, 0.2 mM and 2.5 mM, respectively) and incubated at 30°C for approximately 2.5 h (until the UV absorbance at 333 nm no longer increased). The reaction solution was passed through a 12 ml (2 g) C-18 solid phase extraction column pre-equilibrated with 50 mM MOPS, pH 7.5 and washed with 6 volumes of buffer followed by 1 volume de-ionized (MilliQ) H2O. The coumaryl CoA was eluted with 1 volume of MeOH. The MeOH was rotovaped to near dryness and the coumaryl CoA was resuspended in H2O acidified to pH 4.0 with HCl to a final concentration of 2.5 mM (approximately 5 ml) based on the UV absorbance at 333 nm and a molar extinction coefficient of 21,000 l (mol-cm)−1 (Stockgit and Zenk 1975).
mRNA hybridization analysis
Total RNA was isolated from cells elicited with chitin for 18 h, using Triazol (Ambion) according to the vendors instructions. The polyA RNA (mRNA) was extracted from total RNA with a Poly(A) Purist kit (Ambion). cDNA was then generated using Superscript II reverse transcriptase (Invitrogen). A 523 base pair (bp) fragment of HHT corresponding to nucleotides 352 to 874 of AsHHT2 (GeneBank accession #AB076981; (Yang et al. 2004)) was amplified from cDNA isolated from induced tissue using primers HHTF3: 5′-TGGTGCCCTTCTACCCGATG-3′ (forward) and HHTR3: 5′-GCGGGACAGCTTGAAGATGTC-3′ (reverse). This fragment was cloned into Topo TA, sequenced and termed pHHT1F3R3. Plasmid pHHT1F3R3 was digested with EcoRI and the HHT coding insert gel purified on agarose and used as template to generate a 32P labeled probe by random hexamer priming with 32P dCTA using standard methods (Sambrook et al. 1989).
The 454 bp barley PR-1 sequence (GenBank Accession Z21494) was previously cloned by PCR and represents most of the transcript sequence (Federico et al. 2006). Probe synthesis, RNA blot probing and washing were done according to Sambrook et al. (1989). The hybridization and wash temperature for PR-1 was 55°C. Blots were initially probed with HHT, stripped, and then re-probed with the PR-1 probe.
Time course of avenanthramide biosynthesis
Total RNA was isolated from tissue at the indicated times post induction as described above. The RNA from triplicate cultures at each time point was pooled and an aliquot (~5 μg) was electrophoresed on a 1.35% formaldehyde denaturing agarose gel (1.5%) at 15 V overnight. The RNA was transferred to a nylon membrane and probed for HHT mRNA using a 32P labeled probe as described above by standard methods (Sambrook et al. 1989).
Results
Chitin elicitation of avenanthramide biosynthesis
In our preliminary experiments we found that oat callus tissue, suspended in liquid media, biosynthesized substantial quantities of avnA and G in response to treatment with crab shell chitin. Media extracts from chitin elicited cultures yielded maximal quantities of avnA and G 12–24 h after induction. The production of avnA and G was monitored by growing cultures in triplicate for time points (0, 6, 12, 24, 48, and 96 h) subsequent to chitin elicitation. Both tissue and media from these cultures were extracted separately for avenanthramide quantitation. The concentration of extractable avnA in the media increased from 4.6 ± 0.8 μg/g tissue immediately after adding chitin to 62 ± 20 μg/g tissue (wet weight) at 24 h, after which concentrations dropped to 7.9 ± 2.8 μg/g tissue at 48 h and 3.1 ± 2.0 μg/g at 96 h. Similarly avnG increased from 8.6 ± 3.7 μg/g tissue immediately after adding chitin to a maximum of 105 ± 18.6 μg/g at 12 h after elicitation. AvnG levels then dropped to 80.7 ± 13.1 μg/g at 24 h and finally to 3.32 ± 1.7 μg/g tissue at 96 h (Fig. 3a). A similar pattern of avnA and G production was observed in the oat tissue extracts. Starting from an undetectable level immediately after adding chitin, avnA levels reached a maximum concentration of 51.1 ± 7.2 μg/g tissue (wet weight) at 24 h, with the concentration decreasing to 24.3 ± 3.7 μg/g tissue at 48 h then to 8.5 ± 2.1 μg/g tissue at 96 h. AvnG increased from an undetectable level at time 0 to 172 ± 23.2 μg/g tissue at 24 h, then decreased to 15.3 ± 4.0 by 96 h (Fig. 3b). Analysis of the media extracts by LC-MS did not reveal a concurrent increase in either p-coumaric acid or 5-hydroxy-anthranilic acid (data not shown). Thus, this apparent disappearance of avnA does not result from hydrolysis of the pseudo peptide bond linking the phenolic moieties.

Avenanthramide A (●) and G (■) concentration in the media extracts (a) and tissue extracts (b) at the indicated time points after elicitation with chitin. Un-elicited control cultures showed no detectable avenanthramides (▲). Concentrations are represented as μg Avn/g tissue (wet weight). Each point represents the mean of triplicate cultures, bars represent standard error
Time course of HHT induction
Aliquots of the tissue harvested at 0, 6, 12, 24, 48, and 96 h were analyzed by RNA hybridization (Northern) analysis using a 32P-labeled HHT fragment (see Methods). Although a trace of mRNA is discernable in some of the un-elicited control lanes, a dramatic increase in HHT mRNA levels occurs at 6 h after elicitation and reaches a maximum at 24 h (Fig. 4b). The level of HHT mRNA remained elevated for the duration of the experiment. Assay of HHT enzyme activity also shows a pronounced increase from elicitation to 12 h post-induction, followed by a transient decrease at 24 h, then returns to an elevated level where it remains until the end of the experiment at 96 h (Fig. 5).

Northern blot analysis of HHT mRNA. Total RNA isolated from triplicate suspension cultures at the indicated times after elicitation with chitin. I = induced cells, C = control (un-induced) cells. Panel A is the ethidium bromide stained 18S ribosomal band (to assess RNA loading), panel B is the 32P labeled HHT fragment hybridization, panel C is the 32P labeled barley PR-1 cDNA hybridization

HHT activity of crude protein extracts from suspension cultures at indicated times after elicitation with chitin. Each point represents the mean of two assays performed on each of three cultures at each time point. Induced (●), uninduced (▲), bars represent the standard error
Time course of PR-1 induction
Messenger RNA levels of the pathogenesis-related protein gene PR-1 were noticeably elevated by 6 h after elicitor treatment and continued to rise until 48 h, then declined slightly by 96 h (Fig. 4c). PR-1 and HHT mRNA levels increased at similar rates, although HHT continued to increase after 48 h.
PR-1 is a gene associated with pathogen induced defense mechanisms such as systemic acquired resistance (SAR) (van Loon et al. 2006). In our experience PR-1 is a slow-acting gene that produces substantial transcript levels 24–72 h after infection of barley with Fusarium graminearum (Federico et al. 2006). Similar results are also seen in Arabidopsis inoculated with Pseudomonas syringae (Spoel et al. 2003). Elevated salicylic acid (SA) levels commonly follow pathogen infection and these appear to induce increases of pathogenesis related proteins, including PR-1 (Lawton et al. 1995; Malamy et al. 1990). Application of SA through root feeding also induced expression of PR-1 in barley leaves 48 h after treatment (Federico et al. 2005). The up-regulation of a PR-1 like gene suggests a general defense response to a perceived pathogen attack. This, in addition to avenanthramide biosynthesis, demonstrates a capability of this callus tissue for a complex pathogen defense response. We have yet to examine the role for salicylic acid in elicitation of avenanthramide biosynthesis.
Effect of cell time in liquid media on avenanthramide production
In the previous experiment the callus was conditioned in fresh media for seven days prior to elicitation. We found that transfer of callus tissue from solid media to liquid media resulted in secretion of additional avenanthramides (avnB and C), albeit at relatively low levels (1–10 μg/g tissue). We examined the effect of time in fresh media, prior to elicitation, on the quantities and profiles of avenanthramides biosynthesized. Approximately 2.0 g of callus tissue was transferred from solid media to 25 ml of liquid MS media and allowed to condition 24, 48, and 72 h in the media before eliciting with chitin. Due to limitations of culture facilities we only provided un-elicited controls for the 24/24, 48/48, and 72/72 (conditioning time/post-elicitation time) time points (Table 1). Nevertheless, these control cultures illustrate that low, but detectable levels of all four avenanthramides were present in the media without elicitation and that the avenanthramide levels universally diminished under all treatment regimes (in both elicited and un-elicited cultures). Least significant difference (LSD) analysis showed that, at the 95% confidence level, avnA and G were strongly elicited at 24 h after chitin treatment in all three conditioning regimes (Table 1). AvnB concentrations were not significantly elevated relative to the un-elicited control in the cultures conditioned for 24 h in fresh media, however, in the cultures conditioned for 48 and 72 h prior to elicitation there was a statistically significant increase in avnB levels relative to the corresponding un-elicited controls. AvnC concentrations were also significantly elevated relative to the un-elicited controls in all three treatments regimes. However, the levels of avnA and G were dramatically higher than those of the other two avenanthramides. The rate at which the avenanthramides disappeared from the media extracts appears to accelerate with longer conditioning of the callus in the liquid media. Cultures conditioned in fresh liquid media for 24 or 48 h prior to elicitation showed the highest concentrations of avnA and G in the media whereas the cultures conditioned for 72 h had diminished levels of secreted avenanthramide and also showed a more rapid clearing of the avenanthramides from the media. Whether this represents a reduced rate of biosynthesis or an enhanced rate of removal is unclear.
LC-MS analysis of the media extracts from these experiments revealed at least three additional peaks suggestive of avenanthramides. Their UV and mass spectra are consistant with those of avnH (N-(4′-hydroxy-3′-methoxy-(E)-cinnamoyl)-4-hydroxyanthranilic acid), avnO (N[5-(4′-hydroxyphenyl)-(2E,4E)-2,4-pentadienoyl]-5-hydroxy anthranilic acid) and avnR (N[5-(4′-hydroxyphenyl)-(2E,4E)-2,4-pentadienoyl]-4-hydroxy anthranilic acid). From these experiments it is clear that the profile and dynamics of avenanthramide production are affected by the age of the cells and their time in liquid culture.
Growth rate of suspension cultures
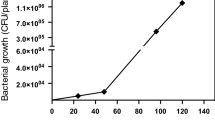
Callus cultured in liquid media was transferred to fresh media at 9–12 day intervals for 45 days and wet weights were determined at each transfer. Two distinct phenotypes were observed; we termed these “friable” and “aggregate”. To determine wet weight the cultures were transferred from their Erlenmeyer flasks to sterile 50 ml Falcon tubes and the large clumps of aggregate tissue allowed to rapidly settle to the bottom of the tube. The friable tissue readily sloughed off the more solid aggregate tissue and remained well dispersed in the culture media. These dispersed cell types were isolated and cultured separately. The aggregate tissue was thoroughly drained of media, weighed, and then transferred to fresh media for continued growth. In six replicate cultures the average wet weights were 1.81 ± 0.31, 1.85 ± 0.14, 2.24 ± 0.17, 2.61 ± 0.44, and 4.47 ± 0.29 (g ± std dev) at 0, 10, 19, 31, and 42 days, respectively. With successive sub-cultures the relative amount of friable tissue declined, as determined by visual observation. Thus the increased rate in tissue weight gain between day 35 and 45 almost exclusively reflects the increase in the aggregate tissue (Fig. 6).

Weight gain of callus tissue in liquid media. Points represent average weight of five replicate 50 ml cultures at approximately 10 day intervals. Error bars represent standard error
In a separate experiment we treated cultures of isolated, finely dispersed (friable) tissue with chitin. Although low levels of avnA and G were produced, no dramatic spike in biosynthesis was observed as with the “aggregate” tissue. The friable tissue cells did not stain with Evans blue, hence appeared to be alive and metabolically active. Thus, the phenotypically different cells are also metabolically distinct. One possibility is that the “aggregate” cells are more highly lignified, which might account for their increased response to chitin elicitation, i.e., they are metabolically equipped to biosynthesize the precursors of avenanthramides. More detailed experiments on the extent of lignification, optimal age and culture conditions for avenanthramide production are being pursued.
Identification of avenanthramides G and putative H, O, and R
Analysis of ethyl acetate extracts from the media of chitin elicited cultures revealed a prominent peak (peak 4), in addition to the avnA peak (peak 2), in the UV chromatogram (Fig. 2). By LC-MS analysis this peak had a retention time of 8.8 min, whereas the peak corresponding to avnA eluted at 7.6 min. Mass spectra of the metabolite showed a pseudo-molecular ion [M + H]+ with m/z 300; this ion fragmented to yield a daughter ion [M2]+ = 147, putatively the acyl carbocation of p-coumarate. These parent and daughter ions are identical to avnA. The NMR data of the HPLC purified compound has a 1H spectra essentially identical to that reported for avnG (Miyagawa et al. 1996b). The retention time relative to avnA, the diode array spectra, the mass spectra, and the NMR spectra provide convincing evidence that this compound corresponds to avnG (N-(4′-hydroxy)-(E)-cinnamoyl)-4-hydroxyanthranilic acid), i.e., the isomer of avnA with the hydroxyl group at the 4 position of the anthranilic acid moiety.
Likewise, in the experiments to determined the effect of time in liquid media some of media extracts from the chitin elicited cultures included a small peak on the total ion chromatogram with a retention time of 9.4 min having a pseudo-molecular ion [M + H]+ with m/z 330 and a daughter ion [M2]+ = 177, putatively the acyl carbocation of ferulate. These ions are identical to avnB, however, the longer HPLC retention time (9.4 min vs. 8.3 min for avnB) suggests that it is the avenanthramide isomer containing 4-hydroxy anthranilic acid, avnH (Collins and Mullin 1988). Two additional peaks eluting at 10.0 and 11.2 min both showed pseudo-molecular ions [M + H]+ with m/z 326 and daughter ion [M2]+ = 173. They also display UV maxima at 340–350 nm. These data are consistent with the structures of avenanthramide O and the 4-hydroxy anthranilic acid homolog avenanthramide R (Collins and Paton 1992). (Note that avnO has also been termed avnL by Miyagawa et al. (1995)). So far we have been unable to isolate sufficient material to confirm their identity by NMR analysis.
Discussion
Chitin, poly-N-acetyl-β-glucosamine, is a component of fungal cell walls. Plants are known to respond to oligomeric chitin by expressing a variety of defense mechanisms including the biosynthesis of phytoalexins (anti-microbial secondary metabolites) (Ren and West 1992; Ryan 1987). In these experiments we demonstrate that the biosynthesis of avnA and G is up-regulated in response to treatment with crab shell chitin. Interestingly, the avenanthramide concentration in both the media and the tissue reaches a maximum at 24 h (avnG reached its maximum in the media at 12 h) but declines precipitously thereafter. We have not, as yet, determined the metabolic fate of this metabolite. Note that the amount of avenanthramide, as a function of tissue wet weight, was comparable in both the media and the tissue, suggesting facile transport of these metabolites across the cell membrane.
Okazaki and co-workers, using whole oat leaves floated on solutions of partially deacylated chitin (chitosan) as elicitor, conducted an incisive investigation of the biosynthesis and metabolism of avenanthramides (Okazaki et al. 2004). They demonstrated that avenanthramides were secreted into the surrounding solution and that, as observed in our suspension cultures, they also disappeared. The dynamics of avenanthramide biosynthesis and disappearance in the suspension cultures were similar to those observed by Okazaki but with some notable differences. Okazaki found avnB to be the most rapidly biosynthesized avenanthramide, its maximum rate of biosynthesis was reached at 24 h subsequent to elicitation after which the rate of biosynthesis slowly declined until the end of the experiment at 96 h. The maximum concentration in the elicitor solution was attained at 72 h after elicitation. Okazaki et al. also found that avnA biosynthesis was up-regulated for the first 24–48 h after elicitation but appeared to cease thereafter. Both metabolites were rapidly metabolized. This is in contrast to previous work by Ishihara et al. (1999b) who found that when whole leaves were elicited with chitin avnA was the major form of avenanthramide to incorporate radio-labeled precursors or to be secreted into the elicitor solution (Ishihara et al. 1999a); avnB was the other major form biosynthesized. Other avenanthramides (D, G and L) were also produced but only at levels about 1–10% of the two major forms (Ishihara et al. 1999a). Moreover, in these experiments, avnA and avnB appeared to decrease about 48 h after elicitation, this decline was more gradual than those observed with the suspension cultures or those described by Okazaki et al. The more complex structure and vasculature of leaf tissue relative to callus might account for the altered dynamics of avenanthramide secretion to the surrounding environment.
The dynamics of avnA and B biosynthesis in the suspension cultures contrasts with those described by Okazaki et al. (2004) where the biosynthesis of avnA completely stops after about 24 h and the biosynthesis of avnB steadily decreases from its maximum at 24 h until 72 h. Ishihara found that HHT activity in chitin elicited leaves reached a maximum at 12 h after elicitation then dropped to about half maximum at 24 h and remained there until the end of the experiment at 60 h post elicitation (Ishihara et al. 1998).
Why we observe the loss of avenanthramide from tissue and media over time is not clear. The avenanthramides are relatively stable at pH 4.0–6.0 (Collins 1989; Dimberg et al. 2001); we found little or no loss of avnA when synthetic avnA was incubated for up to 96 h in MS media alone (data not shown). Okazaki et al. (2004), based on data from feeding radio-labeled avnB to elicited leaves, suggested that some of the radioactivity was bound, through saponifiable bonds, to components of the cell walls. In addition, some of the radio-labeled avnB was converted to dimers and dehydrodimers found in the elicitor solution. We have not observed any ethyl acetate extractable products either from media or tissue suggesting dimerization of the avnA or avnB. We have not attempted saponification of the callus tissue, but the evidence to date indicates that it is exclusively avnB that is dimerized (Okazaki et al. 2007) or incorportated into cell walls (Okazaki et al. 2004). We observe very little avnB in our cultured cells .
Elicitation of leaves from oat cultivars carrying the Pc-2 gene with victorin (a peptide toxin produced by Cochliobolus victoriae), as well as certain heavy metals, results in significant production of avnG (Ishihara et al. 1997; Miyagawa et al. 1996a, b). Although it is unknown if cv Belle, the source of callus tissue used in our experiments, carries this gene, cv Victoria, a source for Pc-2, is in its breeding line (Forsberg et al. 1999). Thus, the high levels of both avnA and G (but not avnB) produced in response to chitin elicitation is novel to this suspension culture system.
Plant suspension cultures may serve two principal purposes. They offer a possibility to produce metabolites of interest, often in a relatively non-contaminated background and they can provide a vehicle with which to investigate the metabolic pathways and basic physiology of the plant. The shoot apical meristem callus tissue employed in these experiments responds to chitin elicitation by up-regulating the biosynthesis of avnA and G. This contrasts with studies using whole leaves in which avnA or B are the principal forms elicited. These cultures also appear to biosynthesize a broad suite of avenanthramides under appropriate culture conditions. The system described here offers an opportunity to more closely investigate the signaling pathways and the role of the various isozymes involved in avenanthramide production and to the general defense response of oat to chitin elicitation. The relatively high expression level of both avnA and G in response to chitin elicitation is unique to these cultures. As avnA and G only differ in the anthranilate hydroxylation position and the likelihood that other 4-hydroxy anthranilate homologs are produced by this system, it might provide a useful tool for investigating the metabolic pathway to these metabolites. We also believe this system offers the potential to selectively biosynthesize the individual avenanthramides with low background contamination.
References
Blaakmeer A, Stork A, van Veldhuizen A, van Beek TA, de Groot A, van Loon JJA, Schoonhoven LM (1994a) Isolation, identification, and synthesis of miriamides, new hostmarkers from eggs of Pieris brassicae. J Nat Prod 57:90–99. doi:10.1021/np50103a013
Blaakmeer A, van der Wal D, Stork A, van Beek TA, de Groot A (1994b) Structure activity relationship of isolated avenanthramide alkaloids and synthesized related compounds as oviposition deterrents for Pieris brassicae. J Nat Prod 57:1145–1151. doi:10.1021/np50110a003
Bratt K, Sunnerheim K, Bryngelsson S, Fagerlund A, Engman L, Andersson RE, Dimberg LH (2003) Avenanthramides in oats (Avena sativa L.) and structure-antioxidant activity relationships. J Agric Food Chem 51:594–600. doi:10.1021/jf020544f
Collins FW (1989) Oat phenolics: avenanthramides, novel substituted N-cinnamoylanthranilate alkaloids from oat groats and hulls. J Agric Food Chem 37:60–66. doi:10.1021/jf00085a015
Collins FW, Mullin WJ (1988) High-performance liquid chromatographic determination of avenanthramides, n-aroylanthranilic acid alkaloids from oats. J Chromatogr A 445:363–370. doi:10.1016/S0021-9673(01)84548-9
Collins FW, Paton D (1992) Methods of producing stable bran and flour products from cereal grains. US patent 5,169,660
Dimberg LH, Theander O, Lingnert H (1993) Avenanthramides—a group of phenolic antioxidants in oats. Cereal Chem 70:637–641
Dimberg LH, Molteberg EL, Solheim R, Frolich W (1996) Variation in oat groats due to variety, storage and heat treatment. I: Phenolic compounds. J Cereal Sci 24:263–272. doi:10.1006/jcrs.1996.0058
Dimberg LH, Sunnerheim K, Sundberg B, Walsh K (2001) Stability of oat avenanthramides. Cereal Chem 78:278–281. doi:10.1094/CCHEM.2001.78.3.278
Emmons CL, Peterson DM (2001) Antioxidant activity and phenolic content of oat as affected by cultivar and location. Crop Sci 41:1676–1681
Emmons CL, Peterson DM, Paul GL (1999) Antioxidant capacity of oat (Avena sativa L.) extracts. 2. In vitro antioxidant activity and contents of phenolic and tocol antioxidants. J Agric Food Chem 47:4894–4898. doi:10.1021/jf990530i
Federico ML, Kaeppler HF, Skadsen R (2005) The complex development expression of a novel stress-responsive barley Ltp gene is determined by a shortened promoter sequence. Plant Mol Biol 57:35–51. doi:10.1007/s11103-004-6769-0
Federico M, Iñiuez-Luy F, Skadsen R, Kaeppler HF (2006) Spatial and temporal divergence of expression in duplicated barley germin-like protein-encoding genes. Genetics 174:179–190. doi:10.1534/genetics.106.058156
Forsberg RA, Kaeppler HF, Duerst RD (1999) Registration of”Belle” oat. Crop Sci 39:878–879
Ishihara A, Matsukawa T, Miyagawa H, Ueno T, Mayama S, Iwamura H (1997) Induction of hydroxycinnamoyl-CoA:hydroxyanthranilate N-hydroxycinnamoyl transferase (HHT) activity in oat leaves by victorin C. Z. Naturforsch. C J Biosci 52c:756–760
Ishihara A, Miyagawa H, Matsukawa T, Ueno T, Mayama S, Iwamura H (1998) Induction of hydroxyanthranilate hydroxycinnamoyl transferase by oligo-N-acetylchitooligosaccharides in oats. Phytochemistry 47:969–974. doi:10.1016/S0031-9422(97)00603-1
Ishihara A, Ohtsu Y, Iwamura H (1999a) Biosynthesis of oat avenanthramide phytoalexins. Phytochemistry 50:237–242. doi:10.1016/S0031-9422(98)00535-4
Ishihara A, Ohtsu Y, Iwamura H (1999b) Induction of biosynthetic enzymes for avenanthramides in elicitor-treated oat leaves. Planta 208:512–518. doi:10.1007/s004250050588
Ji LL, Lay D, Chung E, Fu Y, Peterson DM (2003) Effects of avenanthramides on oxidant generation and antioxidant enzyme activity in exercised rats. Nutr Res 23:1579–1590. doi:10.1016/S0271-5317(03)00165-9
Lawton K, Weymann K, Friedrich L, Vernooij B, Uknes S, Ryals J (1995) Systemic acquired resistance in Arabidopsis requires salicylic acid but not ethylene. Mol Plant Microbe Interact 8:863–870
Liu L, Zubik L, Collins FW, Marko M, Meydani M (2004) The antiatherogenic potential of oat phenolic compounds. Atherosclerosis 175:39–49. doi:10.1016/j.atherosclerosis.2004.01.044
Malamy J, Carr JP, Klessig DF, Raskin I (1990) Salicylic acid: a likely endogenous signal in the resistance response of tobacco to viral infection. Science 250:1002–1004. doi:10.1126/science.250.4983.1002
Mayama S, Matsuura Y, Inda H, Tani T (1982) The role of avenalumin in the resistance of oat to crown rust, Puccinia coronata f. sp avenae. Physiol Plant Pathol 20:189–199. doi:10.1016/0048-4059(82)90084-4
Miyagawa H, Ishihara A, Nishimoto T, Ueda T, Mayama S (1995) Induction of avenanthramides in oat leaves inoculated with crown rust fungus, Puccinia coronata f. sp avenae. Biosci Biotechnol Biochem 12:2305–2306
Miyagawa H, Ishihara A, Kuwahara Y, Ueno T, Mayama S (1996a) Comparative studies of elicitors that induce phytoalexin in oats. J Pestic Sci 21:203–207
Miyagawa H, Ishihara A, Kuwahara Y, Ueno T, Mayama S (1996b) A stress compound in oats induced by Victorin, a host-specific toxin from Helminthosporium victoriae. Phytochemistry 41:1473–1475. doi:10.1016/0031-9422(95)00805-5
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497. doi:10.1111/j.1399-3054.1962.tb08052.x
Nie L, Wise M, Peterson D, Meydani M (2006a) Mechanism by which avenanthramide-c, a polyphenol of oats, blocks cell cycle progression in vascular smooth muscle cells. Free Radic Biol Med 41:702–708. doi:10.1016/j.freeradbiomed.2006.04.020
Nie L, Wise ML, Peterson DM, Meydani M (2006b) Avenanthramide, a polyphenol from oats, inhibits vascular smooth muscle cell proliferation and enhances nitric oxide production. Atherosclerosis 186:260–266. doi:10.1016/j.atherosclerosis.2005.07.027
Okazaki Y, Isobe T, Iwata Y, Matsukawa T, Matsuda F, Miyagawa H, Ishihara A, Nishioka T, Iwamura H (2004) Metabolism of avenanthramide phytoalexins in oats. Plant J 39:560–572. doi:10.1111/j.1365-313X.2004.02163.x
Okazaki Y, Ishizuka A, Ishihara A, Nishioka T, Iwamura H (2007) New dimeric compounds of avenanthramide phytoalexin in oats. J Org Chem 72:3830–3839
Peterson DM, Hahn MJ, Emmons CL (2002) Oat avenanthramides exhibit antioxidant activities in vitro. Food Chem 79:473–478. doi:10.1016/S0308-8146(02)00219-4
Ponchet M, Favre-Bonvin J, Hauteville M, Ricci P (1988) Dianthramides (N-benzoyl and N-paracoumarylanthranilic acid derivatives) from elicited tissues of Dianthus caryophyllus. Phytochemistry 27:725–730. doi:10.1016/0031-9422(88)84083-4
Ren Y-Y, West CA (1992) Elicitation of diterpene biosynthesis in rice (Oryza sativa L.) by chitin. Plant Physiol 99:1169–1178
Ryan CA (1987) Oligosaccharide signalling in plants. Annu Rev Cell Biol 3:295–317. doi:10.1146/annurev.cb.03.110187.001455
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York
Spoel SH, Koornneef A, Claessens SMC, Korzelius JP, Van Pelt JA, Mueller MJ, Buchala AJ, Metraux J-P, Brown R, Kazan K, Van Loon LC, Dong X, Pieterse CMJ (2003) NPR1 modulates cross-talk between salicylate- and jasmonate-dependent defense pathways through a novel function in the cytosol. Plant Cell 15:760–770. doi:10.1105/tpc.009159
Stockgit J, Zenk MH (1975) Chemical synthesis and properties of hydroxycinnamoyl-coenzyme A derivatives. Z Naturforsch [C] 30:352–358
van Loon LC, Rep M, Pieterse CM (2006) Significance of inducible defense-related proteins in infected plants. Annu Rev Phytopathol 44:135–162
Vollhardt J, Fielder DA, Redmond MJ (2000) Identification and cosmetic application of powerful anti-irritant constituents of oat grain. XXIst IFSCC International Congress, Berlin
Yang Q, Trinh HX, Imai S, Ishihara A, Zhang L, Nakayashiki H, Tosa Y, Mayama S (2004) Analysis of the involvement of hydroxyanthranilate hydroxycinnamoyltransferase and caffeoyl-CoA 3-O-methyltransferase in phytoalexin biosynthesis in oat. Mol Plant Microb Interact 17:81–89
Acknowledgments
Thanks are extended to Rachel Angel, Laurie Herrin, John Herbst, and Nick Partridge for expert technical assistance. NMR data was obtained from the National Magnetic Resonance Facility at Madison, which is supported by NIH grants P41RR02301 (BRTP/NCRR) and P41GM66326 (NIGMS). Additional equipment was purchased with funds from the University of Wisconsin, the NIH (RR02781, RR08438), the NSF (DMB-8415048, OIA-9977486, BIR-9214394), and the USDA. This research is supported by the USDA, Agricultural Research Service CRIS #3655-21000-044-00D.
Disclaimer
Names are necessary to report factually on available data; however, the USDA neither guarantees nor warrants the standard of the product, and the use of he name by USDA implies no approval of the product to the exclusion of others that may also be suitable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wise, M.L., Sreenath, H.K., Skadsen, R.W. et al. Biosynthesis of avenanthramides in suspension cultures of oat (Avena sativa). Plant Cell Tiss Organ Cult 97, 81–90 (2009). https://doi.org/10.1007/s11240-009-9501-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11240-009-9501-6