
Abstract
The recent development and approval of novel oral anticoagulants represents a significant success in the intelligent design of target-specific therapeutics. However, while these agents obviate many of the shortcomings of their predecessor (warfarin), they present novel challenges in pharmacologic management as well. Each was designed to have high oral bioavailability and high affinity for its target molecule, conveying significant anticoagulant effects. Yet, such unique drug–ligand binding, coupled with unfamiliar drug interactions and renal-based clearance, represent challenges to clinical management. The current review describes the development and pharmacokinetic properties of these agents, in the context of their clinical utility and pitfalls.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
For more than half of a century, warfarin has been the only available oral agent to effect systemic anticoagulation. Now it is well-known that several alternatives exist which predominantly extinguish the notorious shortcomings of warfarin (Table 1). In long sought-after successes of pharmaceutical development, dabigatran, rivaroxaban, and apixaban have become approved oral anticoagulants for a variety of indications, including the prevention of stroke or systemic embolism in patients with non-valvular atrial fibrillation (AF) [1–3].
While these drugs do not require the close monitoring characteristic of warfarin therapy, and they have fewer drug and food interactions, there are important distinctions among these new agents. They have unique pharmacokinetic and pharmacodynamic properties that represent a new paradigm in the treatment of patients requiring long-term systemic anticoagulation. Importantly, these biochemical properties can have significant clinical implications. In the current review, we will discuss the development and in vivo pharmacokinetic properties of the novel oral factor Xa inhibitors, as they relate to their clinical utility and effects.
Factor Xa as a target
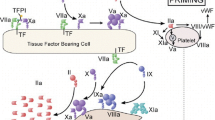
The body’s anticoagulant system exists as a fluid system of biochemical processes balancing bleeding versus coagulation effects. The most acute reaction to bleeding diatheses involves platelets binding to tissue factor (TF) to ultimately form a platelet plug. However, this also serves as one catalyst for the complex coagulation cascade that will ultimately lead to stable fibrin clot formation [4].
The development of fibrin is the result of two parallel and intersecting pathways, the intrinsic and extrinsic anticoagulation cascade. These series of reactions activate various enzymatic factors through proteolysis of their inactive precursors. Along the way, the overall effect is magnified such that a small amount of TF can lead to significant fibrin deposition. This amplification of biochemical effect has been one reason that factors in the cascade have long been targeted for pharmacologic intervention.
Factor Xa is an activated serine protease partially responsible for the conversion of prothrombin to thrombin [5]. The choice of factor Xa for drug development is based on its position at the intersection of the intrinsic and extrinsic pathways. The subcutaneous drug fondaparinux has provided a proof-of-concept example of factor Xa inhibition. Yet, fondaparinux is an antithrombin-dependent agent, and cannot penetrate factor Xa when it is bound within the prothrombinase complex (a complex of prothrombin and factors Xa and Va). This represents a significant shortcoming in its anticoagulant ability, as the prothrombinase complex is responsible for the procoagulant effects of factor Xa. Fondaparinux has historically demonstrated a favorable bleeding profile, and this may be one reason [6].
An additional advantage of targeting factor Xa is that the prothrombinase reaction is a potent effect-multiplier, generating 1,000 thrombin molecules for each Xa molecule [6]. Inhibition of free and bound factor Xa can therefore lead to potent anticoagulant effect. However, the bleeding profile of these agents is thus carefully scrutinized. Studies of patients with genetic factor X deficiencies demonstrate the severe deficiency required for clinical hypocoagulation (<1 % activity) and adverse spontaneous bleeding [5]. Therefore, these agents may have a broader therapeutic window than initially anticipated, particularly in comparison to vitamin K antagonists.
The successful development of agents targeting each aspect of the coagulation cascade represents the culmination of years of work in ‘intelligent’ drug design. Substrate–ligand structures were identified, and inhibitor molecules were designed based on such interactions. Targeted modifications were made to optimize the properties of the resulting inhibitors [5, 7].
Target binding
Proteolytic enzymes throughout the body demonstrate a wide range of target specificities, consistent with their role. For example, serine proteases of the digestive tract demonstrate low target specificity, allowing breakdown of a wide range of ingested nutrients. In contrast, the serine proteases of the coagulation cascade demonstrate significant specificity for a particular target, as would be expected for a crucial equilibrium [8, 9]. Such specificity is often mainly a reflection of active site recognition of a specific substrate, however, factor Xa substrate specificity also depends on interactions with the prothrombinase complex and the cell membrane [9]. Such complex interactions led to the more direct inhibition of prothrombinase via action at the factor Xa active site, more broadly preventing thrombin formation.
Active-site inhibition
This is the major distinction of novel, oral factor Xa inhibitors: their binding within the active site of factor Xa (both free and bound). The design of such compounds was driven by the recognition that the few, naturally-occurring factor Xa inhibitors possessed moieties that acted as mimics of an arginine residue in the natural substrate. This led to development of a variety of candidates, derivatives of oxazolidinones (Fig. 1).

Diagrams of novel, factor Xa inhibitors in development. Reprinted by permission from Macmillan Publishers Ltd [5], Nature Reviews Drug Discovery, © 2011
These inhibitors bind in an L-shaped fashion within the factor Xa active site. The ‘L’ is bracketed by the S1 and S4 binding sites of factor Xa, previously recognized as key constituents of any substrate. At these ends of the ‘L’, natural compounds possess highly polar, charged components, allowing them to bind the target with some specificity. Yet such a charged molecule is poorly-absorbed through the digestive tract. In contrast, synthetic inhibitors instead possess aromatic rings with various moieties attached. These allow for alternative interactions in the S1 and S4 pocket, maintaining binding strength while maximizing bioavailability. Instead of highly-polar ionic interactions, several of the novel agents depend on hydrophobic and hydrogen-bonding interactions with the target. For example, several of the agents approved or in advanced clinical development possess a chlorine-based group which interacts with the Tyrosine of the S1 pocket. Alternatively, apixaban utilizes a methoxyphenyl-based moiety at the S1 binding site [10, 11].
The ‘long’ end of the ‘L’ must conform to a highly-specific tunnel within the target active site. This is accomplished through the common structural backbone of these class of molecules. While there may be little formal bonding between inhibitor and target at this region, the ‘fit’ of such agents contributes to the high specificity for the target.
As noted above, the non-ionic interaction between inhibitor and target maintained oral, bioavailability that could not be achieved with highly-charged molecules. Such agents, which can be very pH sensitive, are not easily-absorbed in the gastrointestinal tract, limiting achievable serum concentrations. However, even within the class, there is variability in this effect, with earlier generations of agents exhibiting relatively more basic chemistry and lower bioavailability, compared with later-developed ones.
Non-active site factor X inhibition
An alternative approach to factor X inhibition is to take advantage of non-active site (‘exosite’) interactions to hinder prothrombinase activity. The requirement of an exosite substrate for prothrombinase activity has been well-described, and is necessary for the catalytic complex to overcome adverse thermodynamic barriers to activity [9]. Furthermore, these data suggest the specificity of the prothrombinase enzyme activity is guided not just by local, active site interactions, but perhaps even more by exosite-inhibits. Krishnaswamy and colleague [12] have previously demonstrated the proof-of-concept that prothrombinase activity can be inhibited by non-active site, surface binding of an experimental monoclonal antibody. This could potentially open a new mechanism and target for factor Xa inhibition—exosite-directed, reversible inhibitors. Theoretically, such agents could have more dynamic pharmacokinetics, as well as perhaps yield novel and more straightforward mechanisms for reversal. RNA or DNA oligonucleotides, also referred to as aptamers effectively inhibit protein–protein interactions through binding to non-active sites (exosites) of coagulation proteins. Their known amino acid sequences, typically 30–50 base pairs, allow for the concomitant manufacturing of complementary oligonucleotides that serve as antidotes, rapidly reversing the molecules anticoagulant properties. To date a formulation that permits oral delivery—specifically absorption and achievement of sufficient plasma concentration for a pharmacodynamics effect—has not been mastered.
Structure–activity relationships
Target-binding specificity was demonstrated to be very closely-tied to specific structures of these molecules (Fig. 2). Various substitutions of S1-pocket binding groups, from chlorine to fluorine, or other carbon-based groups, demonstrated significant variability in inhibitor activity [13]. However, several options maintained target specificity, with a variety of different properties. For example one sister compound to apixaban demonstrated similar, highly-selective factor Xa inhibition, yet had a short half-life [13]. Such considerations open the door for the development of agents tailored to specific applications, based on reversibility, bioavailability, and duration of activity. Several of the compounds furthest along in clinical development will be discussed here (Table 2).

Conceptual diagrams of 4 novel factor Xa inhibitors binding to the active site. Diagrams for rivaroxaban and apixaban are derived from reported structures; [18, 35] betrixaban and edoxaban are extrapolated based on these structures. Not drawn to scale. S1 and S4 represent binding regions of the factor Xa active site; Ox oxazolidinone moiety, Cl chloride, H hydrogen participating in bonding, CH 3 methyl group, H 2 O water, Tyr tyrosine, Gly glycine, Gln glutamine
Rivaroxaban
Rivaroxaban was the first oral, direct factor Xa inhibitor approved in the US. It demonstrates very high selectivity for factor Xa, over other serine proteases in the coagulation cascade (by a factor of >10,000). Notably, rivaroxaban-based inhibition of factor Xa appears independent of thrombomodulin, and therefore does not perturb APC-based negative feedback [5]. This could have significant clinical implications, as enhanced APC activity has been implicated in the ‘rebound’ procoagulant effect of certain direct thrombin inhibitors [14]. Yet, clinical trials have not yet demonstrated such a benefit [2].
Additionally, despite the improved bioavailability over natural compounds [15], pharmacokinetic studies have demonstrated this to be dose-dependent. At the dose approved for prevention of venous thromboembolism, 10 mg, most of the drug is absorbed irrespective of food intake [5]. However, due to lower solubility at higher doses, it is recommended that the dose for stroke prevention in AF, 20 mg, be taken with an evening meal. Clinical studies have dosed rivaroxaban both once- and twice-daily, depending on the dose and indication [2, 16].
Rivaroxaban demonstrates a modest volume of distribution, ~50 L, without affinity for any specific tissue [15]. This probably contributes to its relatively short half-life, and may have important implications for reversal in vivo (discussed later).
Apixaban
Apixaban is the most recently-approved, oral factor Xa inhibitor for the prevention of stroke or systemic embolism in patients with non-valvular AF. Similar to rivaroxaban, it is an oral, reversible, selective inhibitor of factor Xa and has bioavailability and factor Xa selectivity that are both high and comparable to those of rivaroxaban [15, 17]. The binding affinity of apixaban for factor Xa (Ki) is 0.08 nM (compared to an affinity for thrombin of 3,100 nM) [11]. However, binding of apixaban to factor Xa is less dependent on interactions at the S1 and S4 binding pockets of the target [18]. While its structure demonstrates similarities to others in the class, likely accounting for its specificity, there are fewer direct interactions between residues of the S1/S4 pockets and specific subgroups of the drug [18]. This may have important implications for durability of anticoagulant effect, and bleeding rates. Apixaban has been consistently dosed twice daily across indications in studies [3, 19, 20].
Edoxaban
Still in development for the prevention of stroke in patients with AF, edoxaban is the third oxazolidinone-like compound [21]. Like rivaroxaban, it uses a chlorine moiety to modulate the S1 active site interaction. It exhibits a Ki of 0.56 nM, with a 10,000-fold selectivity of factor Xa over IIa, and no cross-reactivity with other coagulation factors [15]. Notably, only 35 % of the circulating drug is excreted in the urine [22]. The current, phase III study of edoxaban for stroke prevention in AF uses a once-daily dosing regimen [21].
Betrixaban
Betrixaban is the third chlorine-based inhibitor, with very high factor Xa specificity (>86,000-fold). It has a Ki 0.12 nM, and is still in early development across indications [23]. However, is it minimally excreted by the kidneys, with mostly bile excretion [15]. This may provide a unique therapeutic option in patients with impaired renal function, and also likely accounts for a higher rate of diarrhea in patients taking betrixaban (compared with warfarin). Thus far, it has been dosed once-daily [24].
Implications for clinical use
Reversal
The lack of reversibility has been noted as one of the most significant drawbacks to the novel factor Xa inhibitors. Currently, there is not an approved mechanism by which to acutely deactivate these drugs, though several recombinant factor combinations have been tested [25]. The competitive binding of factor Xa inhibitors leaves the door open to the development of a novel, small-molecule to more precisely deactivate the effect of one or a class of these agents. One agent in development, PRT064445, was developed as an inert, recombinant, factor Xa mimetic [26]. It has been demonstrated to reduce rivaroxaban-related blood loss in a rabbit liver-laceration model. Notably, administration of PRT064445 not only reduced the measured anti-Xa activity induced by rivaroxaban, but was found to decrease free concentrations of rivaroxaban in plasma. Yet, total concentrations of drug increased with PRT064445, owing to the intravascular migration of rivaroxaban from the extravascular space. This highlights the volume of distribution as one of the key structural properties of factor Xa inhibitors, which could dictate measured ‘reversibility’ of such agents. While betrixaban and apixaban demonstrated similar clinical responses to PRT064445, this effect is likely to be dependent on several properties of the specific inhibitor being reversed.
P-glycoprotein interactions
There are additional clinical implications for the metabolism and drug–ligand properties of novel oral anticoagulants. While they lack many of warfarin’s interactions at the cytochrome P-450 level, clinicians now need to be aware of potential interactions with the P-glycoprotein (P-gp) system. The P-gp system is often responsible for cross-membrane transport in various organs, including the intestinal tract, brain, and kidney. The influence of P-gp transport on drug absorption, metabolism, and excretion is become increasingly important as more novel small molecules become available.
For example, P-gp binding is likely one reason for lower concentrations of factor Xa inhibitors in brain tissue [27]. This could explain one of the major benefits observed with oral factor Xa inhibitors—the significant reduction in intracranial hemorrhage (ICH), compared with warfarin (see below). Yet, as a significant substrate of the P-gp system, P-gp inhibitors (such as ketoconazole and ritonavir) can suppress systemic efflux of factor Xa inhibitors [28]. Importantly, increased brain concentrations of rivaroxaban have been detected in the presence of P-gp inhibitors [29].
Bleeding
The limitations of warfarin are principally related to its narrow therapeutic index, which is subsequently complicated by numerous interactions. The most feared complication of its use remains ICH, which occurs at a rate of approximately 0.7–0.8 % per year in well-controlled cohorts [1–3, 30]. A long-sought objective of novel oral anticoagulants is the reduction of this catastrophic event, while preserving anticoagulant efficacy. Lower rates of ICH have been shown in the well-controlled, randomized studies of rivaroxaban, and apixaban [2, 3]. Yet, this effect is difficult to reconcile with the potent anticoagulant effect of these new agents. One proposed mechanism is the more consistent anticoagulant effect of direct thrombin and factor Xa inhibitors. However, there may be other factors at play. Some have speculated on the necessity of factor VIIa to maintain blood–brain barrier hemostasis [30, 31]. Given that only warfarin affects factor VII activity levels, this could explain such an effect. However, additional possibilities exist, including lower CNS penetration of novel oral anticoagulants.
Monitoring
Closely-tied to the activity of novel anticoagulants is the ability to monitor their effect. Traditional measures of systemic anticoagulation (i.e., the prothrombin time , and activated partial thromboplastin time) are of minimal use in assessing activity of these agents, owing to their mechanism of action. Adoption of these assays for novel agents is heavily dependent on the specific reagents used, and generally are not sensitive [32]. Among widely-available tests, the thrombin time can be used to assess dabigatran activity, however, requires specific calibration. Additional, agent-specific chromogenic assays for rivaroxaban and apixaban have been effectively tested, but are not currently available [33, 34].
Structure, function, and the practicing clinician
While many practicing clinicians are far-removed from the labs where these agents are developed and studied, the clinical implications of their structure–function relationships cannot be underestimated. These micro- and macro-molecular interactions dictate much of the clinical end results. Dynamic equilibrium at the target site can affect potency and side effects (specificity). Availability of an agent for reversibility, either through direct agent binding or competitive target binding, depends heavily on the structure–function relationship. As more agents become available that are broadly labeled as ‘factor Xa inhibitors,’ it will be incumbent upon the clinician to understand the relevant characteristics and distinct properties between each, and select ‘the right drug, at the right dose for the right patient.’
Conclusions
Factor Xa inhibitors represent a marked development in the treatment of patients requiring chronic, systemic anticoagulation. Intelligent, structural design of this new class provides specific benefits in bioavailability and pharmacologic activity, as well as opportunities for reduction in unwanted adverse events, specifically pathological bleeding. Additional data on these interactions, as well as the monitoring and reversal of these agents, is needed.
References
Connolly SJ, Ezekowitz MD, Yusuf S, Eikelboom J, Oldgren J, Parekh A, Pogue J, Reilly PA, Themeles E, Varrone J, Wang S, Alings M, Xavier D, Zhu J, Diaz R, Lewis BS, Darius H, Diener HC, Joyner CD, Wallentin L (2009) Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 361(12):1139–1151. doi:10.1056/NEJMoa0905561
Patel MR, Mahaffey KW, Garg J, Pan G, Singer DE, Hacke W, Breithardt G, Halperin JL, Hankey GJ, Piccini JP, Becker RC, Nessel CC, Paolini JF, Berkowitz SD, Fox KA, Califf RM (2011) Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 365(10):883–891. doi:10.1056/NEJMoa1009638
Granger CB, Alexander JH, McMurray JJ, Lopes RD, Hylek EM, Hanna M, Al-Khalidi HR, Ansell J, Atar D, Avezum A, Bahit MC, Diaz R, Easton JD, Ezekowitz JA, Flaker G, Garcia D, Geraldes M, Gersh BJ, Golitsyn S, Goto S, Hermosillo AG, Hohnloser SH, Horowitz J, Mohan P, Jansky P, Lewis BS, Lopez-Sendon JL, Pais P, Parkhomenko A, Verheugt FW, Zhu J, Wallentin L (2011) Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 365(11):981–992. doi:10.1056/NEJMoa1107039
Davie EW, Fujikawa K, Kisiel W (1991) The coagulation cascade: initiation, maintenance, and regulation. Biochemistry 30(43):10363–10370
Perzborn E, Roehrig S, Straub A, Kubitza D, Misselwitz F (2011) The discovery and development of rivaroxaban, an oral, direct factor Xa inhibitor. Nat Rev Drug Discov 10(1):61–75. doi:10.1038/nrd3185
Weitz JI (2011) Factor Xa and thrombin as targets for new oral anticoagulants. Thromb Res 127(Suppl 2):S5–S12. doi:10.1016/s0049-3848(10)70147-x
Hauel NH, Nar H, Priepke H, Ries U, Stassen JM, Wienen W (2002) Structure-based design of novel potent nonpeptide thrombin inhibitors. J Med Chem 45(9):1757–1766
Rawlings ND, Barrett AJ (1993) Evolutionary families of peptidases. Biochem J 290(Pt 1):205–218
Krishnaswamy S (2005) Exosite-driven substrate specificity and function in coagulation. J Thromb Haemost 3(1):54–67. doi:10.1111/j.1538-7836.2004.01021.x
Perzborn E, Strassburger J, Wilmen A, Pohlmann J, Roehrig S, Schlemmer KH, Straub A (2005) In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939—an oral, direct factor Xa inhibitor. J Thromb Haemost 3(3):514–521. doi:10.1111/j.1538-7836.2005.01166.x
Wong PC, Pinto DJ, Zhang D (2011) Preclinical discovery of apixaban, a direct and orally bioavailable factor Xa inhibitor. J Thromb Thrombolysis 31(4):478–492. doi:10.1007/s11239-011-0551-3
Wilkens M, Krishnaswamy S (2002) The contribution of factor Xa to exosite-dependent substrate recognition by prothrombinase. J Biol Chem 277(11):9366–9374. doi:10.1074/jbc.M110848200
Corte JR, Fang T, Pinto DJ, Han W, Hu Z, Jiang XJ, Li YL, Gauuan JF, Hadden M, Orton D, Rendina AR, Luettgen JM, Wong PC, He K, Morin PE, Chang CH, Cheney DL, Knabb RM, Wexler RR, Lam PY (2008) Structure–activity relationships of anthranilamide-based factor Xa inhibitors containing piperidinone and pyridinone P4 moieties. Bioorg Med Chem Lett 18(9):2845–2849. doi:10.1016/j.bmcl.2008.03.092
Furugohri T, Shiozaki Y, Muramatsu S, Honda Y, Matsumoto C, Isobe K, Sugiyama N (2005) Different antithrombotic properties of factor Xa inhibitor and thrombin inhibitor in rat thrombosis models. Eur J Pharmacol 514(1):35–42. doi:10.1016/j.ejphar.2005.03.009
Eriksson BI, Quinlan DJ, Weitz JI (2009) Comparative pharmacodynamics and pharmacokinetics of oral direct thrombin and factor Xa inhibitors in development. Clin Pharmacokinet 48(1):1–22
Mega JL, Braunwald E, Wiviott SD, Bassand JP, Bhatt DL, Bode C, Burton P, Cohen M, Cook-Bruns N, Fox KA, Goto S, Murphy SA, Plotnikov AN, Schneider D, Sun X, Verheugt FW, Gibson CM (2012) Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med 366(1):9–19. doi:10.1056/NEJMoa1112277
Wong PC, Crain EJ, Xin B, Wexler RR, Lam PY, Pinto DJ, Luettgen JM, Knabb RM (2008) Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies. J Thromb Haemost 6(5):820–829. doi:10.1111/j.1538-7836.2008.02939.x
Pinto DJ, Orwat MJ, Koch S, Rossi KA, Alexander RS, Smallwood A, Wong PC, Rendina AR, Luettgen JM, Knabb RM, He K, Xin B, Wexler RR, Lam PY (2007) Discovery of 1-(4-methoxyphenyl)-7-oxo-6-(4-(2-oxopiperidin-1-yl)phenyl)-4,5,6,7-tetrahydro-1H -pyrazolo[3,4-c]pyridine-3-carboxamide (apixaban, BMS-562247), a highly potent, selective, efficacious, and orally bioavailable inhibitor of blood coagulation factor Xa. J Med Chem 50(22):5339–5356. doi:10.1021/jm070245n
Alexander JH, Lopes RD, James S, Kilaru R, He Y, Mohan P, Bhatt DL, Goodman S, Verheugt FW, Flather M, Huber K, Liaw D, Husted SE, Lopez-Sendon J, De Caterina R, Jansky P, Darius H, Vinereanu D, Cornel JH, Cools F, Atar D, Leiva-Pons JL, Keltai M, Ogawa H, Pais P, Parkhomenko A, Ruzyllo W, Diaz R, White H, Ruda M, Geraldes M, Lawrence J, Harrington RA, Wallentin L (2011) Apixaban with antiplatelet therapy after acute coronary syndrome. N Engl J Med 365(8):699–708. doi:10.1056/NEJMoa1105819
Connolly SJ, Eikelboom J, Joyner C, Diener HC, Hart R, Golitsyn S, Flaker G, Avezum A, Hohnloser SH, Diaz R, Talajic M, Zhu J, Pais P, Budaj A, Parkhomenko A, Jansky P, Commerford P, Tan RS, Sim KH, Lewis BS, Van Mieghem W, Lip GY, Kim JH, Lanas-Zanetti F, Gonzalez-Hermosillo A, Dans AL, Munawar M, O’Donnell M, Lawrence J, Lewis G, Afzal R, Yusuf S (2011) Apixaban in patients with atrial fibrillation. N Engl J Med 364(9):806–817. doi:10.1056/NEJMoa1007432
Ruff CT, Giugliano RP, Antman EM, Crugnale SE, Bocanegra T, Mercuri M, Hanyok J, Patel I, Shi M, Salazar D, McCabe CH, Braunwald E (2010) Evaluation of the novel factor Xa inhibitor edoxaban compared with warfarin in patients with atrial fibrillation: design and rationale for the Effective aNticoaGulation with factor xA next GEneration in Atrial Fibrillation-Thrombolysis In Myocardial Infarction study 48 (ENGAGE AF-TIMI 48). Am Heart J 160(4):635–641. doi:10.1016/j.ahj.2010.06.042
Bathala MS, Masumoto H, Oguma T, He L, Lowrie C, Mendell J (2012) Pharmacokinetics, biotransformation, and mass balance of edoxaban, a selective, direct factor Xa inhibitor, in humans. Drug Metab Dispos 40(12):2250–2255. doi:10.1124/dmd.112.046888
Zhang P, Huang W, Wang L, Bao L, Jia ZJ, Bauer SM, Goldman EA, Probst GD, Song Y, Su T, Fan J, Wu Y, Li W, Woolfrey J, Sinha U, Wong PW, Edwards ST, Arfsten AE, Clizbe LA, Kanter J, Pandey A, Park G, Hutchaleelaha A, Lambing JL, Hollenbach SJ, Scarborough RM, Zhu BY (2009) Discovery of betrixaban (PRT054021), N-(5-chloropyridin-2-yl)-2-(4-(N,N-dimethylcarbamimidoyl)benzamido)-5-methoxybenz amide, a highly potent, selective, and orally efficacious factor Xa inhibitor. Bioorg Med Chem Lett 19(8):2179–2185. doi:10.1016/j.bmcl.2009.02.111
Connolly SJ, Eikelboom J, Dorian P, Hohnloser SH, Gretler DD, Sinha U, Ezekowitz MD (2013) Betrixaban compared with warfarin in patients with atrial fibrillation: results of a phase 2, randomized, dose-ranging study (Explore-Xa). Eur Heart J 34(20):1498–1505. doi:10.1093/eurheartj/eht039
Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M (2011) Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation 124(14):1573–1579. doi:10.1161/circulationaha.111.029017
Lu G, DeGuzman FR, Hollenbach SJ, Karbarz MJ, Abe K, Lee G, Luan P, Hutchaleelaha A, Inagaki M, Conley PB, Phillips DR, Sinha U (2013) A specific antidote for reversal of anticoagulation by direct and indirect inhibitors of coagulation factor Xa. Nat Med 19(4):446–451. doi:10.1038/nm.3102
Wang L, He K, Maxwell B, Grossman SJ, Tremaine LM, Humphreys WG, Zhang D (2011) Tissue distribution and elimination of [14C]apixaban in rats. Drug Metab Dispos 39(2):256–264. doi:10.1124/dmd.110.036442
Gnoth MJ, Buetehorn U, Muenster U, Schwarz T, Sandmann S (2011) In vitro and in vivo P-glycoprotein transport characteristics of rivaroxaban. J Pharmacol Exp Ther 338(1):372–380. doi:10.1124/jpet.111.180240
Weinz C, Buetehorn U, Daehler HP, Kohlsdorfer C, Pleiss U, Sandmann S, Schlemmer KH, Schwarz T, Steinke W (2005) Pharmacokinetics of BAY 59-7939—an oral, direct Factor Xa inhibitor—in rats and dogs. Xenobiotica 35(9):891–910. doi:10.1080/00498250500250493
Hart RG, Diener HC, Yang S, Connolly SJ, Wallentin L, Reilly PA, Ezekowitz MD, Yusuf S (2012) Intracranial hemorrhage in atrial fibrillation patients during anticoagulation with warfarin or dabigatran: the RE-LY trial. Stroke 43(6):1511–1517. doi:10.1161/strokeaha.112.650614
Monroe DM, Hoffman M, Oliver JA, Roberts HR (1997) Platelet activity of high-dose factor VIIa is independent of tissue factor. Br J Haematol 99(3):542–547
Siegal DM, Cuker A (2013) Reversal of novel oral anticoagulants in patients with major bleeding. J Thromb Thrombolysis. doi:10.1007/s11239-013-0885-0
Asmis LM, Alberio L, Angelillo-Scherrer A, Korte W, Mendez A, Reber G, Seifert B, Stricker H, Tsakiris DA, Wuillemin WA (2012) Rivaroxaban: quantification by anti-FXa assay and influence on coagulation tests: a study in 9 Swiss laboratories. Thromb Res 129(4):492–498. doi:10.1016/j.thromres.2011.06.031
Becker RC, Yang H, Barrett Y, Mohan P, Wang J, Wallentin L, Alexander JH (2011) Chromogenic laboratory assays to measure the factor Xa-inhibiting properties of apixaban—an oral, direct and selective factor Xa inhibitor. J Thromb Thrombolysis 32(2):183–187. doi:10.1007/s11239-011-0591-8
Roehrig S, Straub A, Pohlmann J, Lampe T, Pernerstorfer J, Schlemmer KH, Reinemer P, Perzborn E (2005) Discovery of the novel antithrombotic agent 5-chloro-N-({(5S)-2-oxo-3-[4-(3-oxomorpholin-4-yl)phenyl]-1,3-oxazolidin-5-yl}methyl)thiophene-2-carboxamide (BAY 59-7939): an oral, direct factor Xa inhibitor. J Med Chem 48(19):5900–5908. doi:10.1021/jm050101d
Furugohri T, Isobe K, Honda Y, Kamisato-Matsumoto C, Sugiyama N, Nagahara T, Morishima Y, Shibano T (2008) DU-176b, a potent and orally active factor Xa inhibitor: in vitro and in vivo pharmacological profiles. J Thromb Haemost 6(9):1542–1549. doi:10.1111/j.1538-7836.2008.03064.x
Ogata K, Mendell-Harary J, Tachibana M, Masumoto H, Oguma T, Kojima M, Kunitada S (2010) Clinical safety, tolerability, pharmacokinetics, and pharmacodynamics of the novel factor Xa inhibitor edoxaban in healthy volunteers. J Clin Pharmacol 50(7):743–753. doi:10.1177/0091270009351883
Turpie AG (2008) New oral anticoagulants in atrial fibrillation. Eur Heart J 29(2):155–165. doi:10.1093/eurheartj/ehm575
Acknowledgments
Dr. Steinberg was funded by NIH T-32 training Grant #5 T32 HL 7101-37. The authors wish to acknowledge Ms. Seanna Horar for her graphic design input.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Steinberg, B.A., Becker, R.C. Structure–function relationships of factor Xa inhibitors: implications for the practicing clinician. J Thromb Thrombolysis 37, 234–241 (2014). https://doi.org/10.1007/s11239-013-0991-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-013-0991-z