
Abstract
Heparins and VKAs have been the cornerstones of anticoagulation therapy for several decades and these agents have become most important drugs in the primary and secondary prevention of venous and arterial thromboembolic disease. Although effective, their use has been hampered by numerous limitations. In the search for new agents matching the ‘ideal’ anticoagulant profile, a number of different steps in the coagulation cascade have been targeted, including direct thrombin inhibition, and direct inhibition of Factor Xa. There are currently a host of promising new agents at various stages of development and clinical evaluation. With potential benefits including predictable efficacy, rapid onset of action, ability to bind clot-bound coagulation factors and no requirement for therapeutic monitoring, these new agents are set to improve the management of thromboembolic disorders.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Heparins and vitamin K antagonists (VKAs) have been the cornerstones of anticoagulation therapy for several decades and these agents have become important drugs in the primary and secondary prevention of venous and arterial thromboembolic disease. Clinical experience gained with these agents, particularly from large, controlled, randomized trials, has helped the development of evidence-based treatment guidelines such as those from the American College of Chest Physicians [1, 2] and has also helped define the place of these substances in such treatment guidelines [3].
While heparin and VKAs have proven to be effective at inhibiting the coagulation process, they have inherent limitations. This has spurred efforts to develop therapies that will overcome these drawbacks while matching the efficacy of the two classes of anticoagulants. Advances are being made in the development of safer, convenient, more specific treatments, which should provide more predictable anticoagulant responses and substantially improve the prevention and management of thromboembolic disorders. The focus of this paper is on some of the new anticoagulant strategies targeting at Factor Xa or Factor IIa that are in advanced stages of clinical development.
The benefits and limitations of established anticoagulants
Warfarin, the most commonly used vitamin K antagonist (VKA), exerts its anticoagulant effect by interfering with the metabolism of vitamin K, inhibiting the synthesis of several coagulation proteins such as factors II, VII, IX and X (Fig. 1), and proteins C and S. The benefits of warfarin therapy in a wide spectrum of patients with thromboembolic disorders are well established. For example, a meta-analysis of trials involving 2,900 patients demonstrated that dose-adjusted warfarin reduced the relative risk of stroke by 62% compared with placebo in patients with atrial fibrillation [4]. However, warfarin’s use is hampered by numerous limitations (Table 1), such as its narrow therapeutic window, its need for frequent coagulation monitoring and dose adjustments, dietary restrictions, bleeding risk and its delayed on- and off-set of action [5].

Anticoagulant effects of conventional anticoagulants
Unfractionated heparin (UFH) and low molecular weight heparins (LMWHs) are indirect coagulation inhibitors, too. UFH enhances the activity of the plasma cofactor antithrombin that in turn inhibits thrombin and Factor Xa (Fig. 1). While efficacious, UFH, like warfarin, has a number of limitations which restrict its clinical use (Table 1), including its parenteral route of administration, frequent laboratory monitoring and the development of potentially life-threatening heparin-induced thrombocytopenia (HIT) Type II. LMWHs, derived from UFH by enzymatic or chemical depolymerization resulting in shorter heparin chains, have an enhanced affinity for antithrombin-mediated inhibition of Factor Xa relative to thrombin inhibition. LMWHs have overcome several of the limitations of UFH, including a more predictable anticoagulant effect resulting in no requirement for routine coagulation monitoring, but their use is still associated with a risk of HIT—though to a lesser extent than that seen with UFH—while the need for parenteral administration limits their use in the outpatient setting [6].
The ‘ideal’ anticoagulant agent
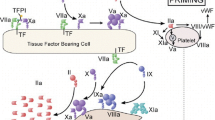
How can anticoagulation therapy be improved? By considering the shortcomings of the current anticoagulation agents, the characteristics required for the ‘ideal anticoagulant’ can be defined and are illustrated in Table 2. In the search for new agents matching the ‘ideal’ anticoagulant profile, a number of different steps in the coagulation cascade have been targeted, including direct thrombin inhibition, and inhibition of Factor Xa, Factor IXa, the Factor VIIa-tissue Factor complex and the Factor Va-Factor VIIIa complex (Table 3). The mode of action of targeted Factor Xa- and Factor IIa-inhibitors is shown in Fig. 2.

Mode of action of targeted Xa- and IIa-inhibitors
Thrombin inhibitors
Due to its central role in the coagulation cascade, thrombin is an attractive target for the development of new anticoagulants. The procoagulant effects can be blocked by several ways: Indirect inhibitors act by catalyzing the physiologic thrombin inhibitors, e.g., heparins require antithrombin as a cofactor. In contrast to the indirect thrombin inhibitors, direct thrombin inhibitors (DTIs) are small molecules that inhibit thrombin by directly binding to the active catalytic site. This allows them to inhibit clot-bound as well as free thrombin without requiring a cofactor—a potential advantage over the indirect thrombin inhibitors [7]. Furthermore, they do not bind to plasma proteins, producing a more consistent anticoagulant response, and do not interact with platelet factor 4, thereby posing no risk of HIT. Hirudin was the first DTI made available for clinical use. Although more effective than heparin, it is associated with an increased risk of bleeding and is parenterally administered. Other DTIs include bivalirudin and argatroban, both of which have a more favorable safety profile than hirudin due to reversible instead of irreversible binding to thrombin [8]. Argatroban has been approved as an alternate anticoagulant in patients with heparin induced thrombocytopenia type II whereas bivalirudin has been licensed for percutaneous coronary intervention. After intravenous administration, bivalirudin shows reversible anticoagulant effects, with coagulation time returning to baseline in approximately 60 min. However, the clinical use of these DTIs is also limited by the need for parenteral administration which makes them not suitable for longterm use. This has led to further advances in the development of oral DTIs such as dabigatran etexilate, which is in phase III of clinical development [8].
Dabigatran etexilate
After oral administration, dabigatran etexilate is rapidly bioconverted to the active form dabigatran which is a novel, direct and specific thrombin inhibitor. Two double-blind, randomized trials were undertaken to investigate the pharmacokinetics, pharmacodynamics and tolerability of orally administered dabigatran etexilate in healthy male subjects. The compound was rapidly absorbed with peak plasma concentrations of dabigatran reached within 2 h of administration. This was followed by a rapid distribution/elimination phase and a terminal phase, with associated estimated half-lives of 8–10 h and 14–17 h with single and multiple dose administrations, respectively. Dabigatran exhibited linear PK characteristics with dose-proportional increases observed in maximum plasma concentration and area under the curve. Steady-state conditions were reached within 3 days with multiple dosing. The time curves for activated partial thromboplastin time (aPTT), prothrombin time (PT), thrombin time (TT) and ecarin clotting time (ECT) paralleled plasma concentration-time curves with values increasing rapidly and in a dose-dependent manner. Of the four assays, TT and ECT exhibited the greatest sensitivity and precision within the anticipated therapeutic dose range. Bleeding events were few and were mild-to-moderate in intensity, occurring only in the higher, multiple dose groups [9].
Wienen et al. [10] assessed and compared the antithrombotic and anticoagulant effects of dabigatran and its oral prodrug dabigatran etexilate, to that of unfractionated heparin, hirudin and melagatran using a rabbit model of venous thrombosis. All compounds demonstrated a dose-dependent reduction in thrombus formation following intravenous administration with complete or almost complete inhibition at the highest doses. Dabigatran (in the dose range 0.03–0.5 mg/kg) had a 50% effective dose of 0.066 mg/kg. By comparison, unfractionated heparin (5–50 U/kg), hirudin (0.01–0.05 mg/kg) and melagatran (0.01–0.3 mg/kg) had a 50% effective dose of 9.8 U/kg, 0.016 mg/kg and 0.058 mg/kg, respectively. Similarly, oral dabigatran etexilate (1–20 mg/kg) inhibited thrombus formation in a dose-dependent manner. Maximum inhibition was achieved within 1 h of administration, suggesting a rapid onset of action. For both routes of administration, inhibition of thrombus formation directly correlated with prolongation of the aPTT. These findings demonstrate a potent anticoagulant and antithrombotic activity of dabigatran as a selective thrombin inhibitor in a rabbit model of venous thrombosis. Notably, dose-dependent and long lasting antithrombotic efficacy was observed after application of its oral form dabigatran etexilate [10].
Dabigatran etexilate is undergoing evaluation for the prevention of venous thromboembolism (VTE) following orthopedic surgery. In a multicenter, parallel-group, double-blind study, 1,973 patients undergoing total hip or knee replacement were randomized to 6–10 days of oral dabigatran etexilate (50, 150 mg twice daily, 300 mg once daily, 225 mg twice daily), starting 1–4 h after surgery, or subcutaneous enoxaparin (40 mg once daily) starting 12 h prior to surgery. The primary efficacy outcome was the incidence of VTE (detected by bilateral venography or symptomatic events) during treatment. Of the 1,949 treated patients, 1,464 (75%) patients were evaluable for the efficacy analysis. VTE occurred in 28.5%, 17.4%, 16.6%, 13.1% and 24% of patients assigned to dabigatran etexilate 50, 150 mg twice daily, 300 mg once daily, 225 mg twice daily and enoxaparin, respectively. A significant dose-dependent decrease in VTE occurred with increasing doses of dabigatran etexilate (P < 0.0001). Compared with enoxaparin, VTE was significantly lower in patients receiving 150 mg twice daily [odds ratio (OR) 0.65, P = 0.04], 300 mg once daily (OR 0.61, P = 0.02) and 225 mg twice daily (OR 0.47, P = 0.0007). Compared with enoxaparin, major bleeding was significantly lower with 50 mg twice daily (0.3% vs. 2.0%, P = 0.047) but elevated with higher doses, nearly reaching statistical significance with the 300 mg once-daily dose (4.7%, P = 0.051). Thus, it can be concluded that oral administration of dabigatran etexilate, commenced early in the postoperative period, was effective and safe across a range of doses. Further optimization of the efficacy/safety balance will be addressed in future studies [11].
In a phase III, multicenter, non-inferiority, double-blind study, patients undergoing total knee replacement were randomized to three treatments. The patients received 8 ± 2 days of oral dabigatran etexilate, 150 or 220 mg once daily starting with a half dose (i.e.,75 or 110 mg) 1–4 h after surgery, or subcutaneous enoxaparin 40 mg once daily starting 12 h prior to surgery. The primary efficacy outcome was the composite of total VTE and all causes of mortality during the treatment period. Efficacy could be evaluated for 1,541 (75%) treated and operated patients. Total VTE and death occurred in 40.5%, 36.4% and 37.7% of patients assigned to dabigatran etexilate 150 or 220 mg once daily or enoxaparin, respectively. Proximal DVT and/or PE occurred in 3.8%, 2.6% and 3.5% of patients receiving dabigatran 150 or 220 mg or enoxaparin, respectively. Safety was evaluated for all 2,076 patients receiving study treatment. The rate of major bleeding was 1.3%, 1.5% and 1.3% of patients receiving dabigatran 150 or 220 mg or enoxaparin. Thus, it can be concluded that oral administration of dabigatran etexilate once daily, given early in the postoperative period, was effective and safe for the prevention of total VTE in patients undergoing total knee replacement surgery [12].
The safety and efficacy of dabigatran etixilate (50, 150 or 300 mg per os b.i.d. for 12 weeks) with or without aspirin (81 or 325 mg/day) or warfarin adjusted to INR 2.0 to 3.0, were examined in a randomized, dose-finding trial conducted in 502 patients with atrial fibrillation on stable warfarin treatment and with at least one additional risk factor for thromboembolic events [13]. Of the patients enrolled, 464 completed the 12 weeks of treatment and 29 and 9 discontinued due to adverse events and other causes, respectively. Aspirin was terminated prematurely in the 300 mg dabigatran etexilate group due to excessive bleeding events (11% of patients). However, coadministration of aspirin in the other dose groups did not significantly increase bleeding events; only 2% and 8% of the patients, respectively, on 50 and 150 mg developed major or relevant bleeding events. The rate of bleeding events in the highest dose group in the absence of aspirin was comparable to in the other groups. Thromboembolic events were seen in two, none and one patient, respectively, in the 50, 150 and 300 mg b.i.d. dose groups and in none patient in the warfarin group. The 150 mg b.i.d. dose of dabigatran etexilate was concluded to have the same anticoagulant activity as the higher dose and warfarin. None of the dabigatran etexilate doses significantly altered liver function tests. From the results of this study, dabigatran etexilate doses of 200–300 mg daily were recommended for phase III testing for the prevention of thromboembolic events in patients with atrial fibrillation.
The promising results from these trials suggest that dabigatran etexilate may represent a significant improvement over existing therapies for the management of various thromboembolic complications. The convenience of oral administration, without the need for individualized dosing or routine coagulation monitoring, mean that the management of patients will be made considerably easier.
Factor Xa inhibitors
Factor Xa also is an attractive target for the design of new anticoagulants as Factor Xa is positioned at the start of the common pathway of coagulation. As the amount of serine protease is amplified at each step of the cascade, it has been hypothesized that the selective inhibition of coagulation factors above thrombin might be a highly effective antithrombotic strategy. Furthermore, by not inhibiting thrombin activity directly, such agents might allow traces of thrombin to escape neutralization, thereby facilitating hemostasis and leading to a favorable safety profile with respect to bleeding. Factor Xa inhibitors include the synthetic pentasaccharides fondaparinux and idraparinux, which act indirectly via activating antithrombin, as well as direct inhibitors such as the synthetic agents DX-9065a, Otamixaban, Rivaroxaban, Apixaban and other compounds [14].
Fondaparinux is the most advanced of the Factor Xa inhibitors (Table 3), having been approved in 2001 for thromboprophylaxis in patients undergoing major orthopedic surgery and in 2004 for treatment of venous thromboembolism, and recent trials have confirmed its efficacy and safety in acute coronary syndrome.
DX-9065a
DX-9065a is a synthetic, non-peptidic, small-molecule inhibitor of Factor Xa that binds reversibly to the active site. It needs to be administered intravenously and prolongs prothrombin time and aPTT as well as inhibiting Factor Xa activity. Overall, the changes in clotting parameters correlate well with the plasma concentrations. In the phase II XaNADU-ACS study, conducted in patients with NSTE ACS, subjects were randomized to low- or high-dose intravenous DX-9065a, or UFH [15]. The composite of death, MI, urgent revascularization and ischaemia on continuous ST-segment monitoring occurred in 34.3%, 31.3% and 34.3% of patients receiving low-dose DX-9065a, high-dose DX-9065a and UFH, respectively. The composite of death, MI and urgent revascularization occurred in 11.9% of the patients receiving high-dose DX-9065a, compared with 19.5% of patients receiving UFH; however, this difference was not significant. Higher plasma DX 9065a concentrations were associated with a significantly lower rate of ischaemic events, but also a tendency towards increased major bleeding [15]. Large-scale, adequately powered studies in patients with cardiovascular disease are warranted to further evaluate this drug.
Otamixaban
Otamixaban is a small-molecule, direct Factor Xa inhibitor that is administered intravenously. Healthy subjects received escalating intravenous otamixaban doses of 1.7–183 μg/kg/h as 6-h infusions, or a bolus of 30 or 120 μg/kg, followed by a 6-h infusion of 60 or 140 μg/kg/h, respectively. Cmax and AUC increased dose dependently, and were slightly more than dose proportional [16]. There was a close correlation between plasma concentrations and pharmacodynamic parameters, and otamixaban was well tolerated without any consistent changes in bleeding time. Anti-Xa activity correlated well with plasma concentrations [17]. PT, aPTT, HepTest and Russell’s viper venom-induced clotting time (RVVT) displayed dose-dependent prolongations at otamixaban doses >27 μg/kg/h. PT, aPTT, and RVVT times returned to baseline within 2–4 h after cessation of the infusion, whereas HepTest remained slightly above baseline for longer. Inhibition of thrombin generation, as measured by endogenous thrombin potential, decreased dose dependently 2–6 h after the otamixaban bolus + 6 h infusion, and returned to baseline 10 h after the start of infusion [17]. Subsequent investigations revealed that the combination of an otamixaban bolus of 25–140 μg/kg followed by a 3-h infusion of 35–200 μg/kg/h would achieve the target effective otamixaban concentrations necessary for a phase II study [18]. A study conducted in healthy male subjects indicated that there was no pharmacokinetic interaction between otamixaban and aspirin; the combination had no additive effects on coagulation or platelet parameters, and no clinically relevant effect on bleeding [19]. These promising pharmacodynamic effects require correlation with outcomes in clinical studies.
Rivaroxaban
Rivaroxaban is a member of a new class of orally available, small-molecule, active-site-directed Factor Xa inhibitors [20]. Rivaroxaban was well tolerated over the whole dose range when administered to healthy male subjects at single oral doses of 1.25–80 mg or multiple doses up to 30 mg twice daily. After multiple dosing, when taken with food, the Cmax and AUC of rivaroxaban were dose proportional. Rivaroxaban was rapidly absorbed, reaching Cmax 2–4 h after oral administration, with a t½ of 5–9 h, and no undue accumulation under steady-state conditions [21]. The relative bioavailability of rivaroxaban was high, reaching approximately 80%. Rivaroxaban has a dual mode of excretion, via the renal (66%) and faecal/biliary (28%) routes, and is mainly excreted as unchanged drug [22].
The onset of inhibition of Factor Xa activity with rivaroxaban is rapid and similar to LMWHs, with the pharmacodynamic effects occurring in parallel to the pharmacokinetics at all doses [21]. The maximum prolongations of PT, aPTT and HepTest due to rivaroxaban are dose dependent and follow the same time profiles as the pharmacokinetic time curves, with some prolongation observed up to 24 h after dosing. The correlation between PT and plasma rivaroxaban concentration is linear, confirming the predictability of the pharmacokinetics and pharmacodynamics of rivaroxaban [21, 22].
Results from phase I studies suggest that dose adjustment of rivaroxaban is not required for gender or in patients with extreme body weight. Co-administration of rivaroxaban with food increased the peak plasma concentrations slightly. Rivaroxaban was shown to have no effect on the QTc interval, and no interaction was observed when rivaroxaban was co-administered with the cardiac glycoside digoxin. No additive effects on platelet aggregation were observed when rivaroxaban was co-administered with either aspirin or the non-steroidal anti-inflammatory drug naproxen [21, 22].
Three phase II double-blind, dose-ranging studies comparing rivaroxaban with enoxaparin for the prevention of VTE after hip or knee replacement surgery have been reported [23–25]. The bid studies examined total daily rivaroxaban doses of 5–60 mg, and the od study tested 5–40 mg. One study was conducted in patients undergoing total hip replacement (THR; N = 722), and one in patients undergoing total knee replacement (TKR; N = 621). In both studies, patients were randomized, doubleblind, to oral, rivaroxaban bid beginning after surgery, or subcutaneous enoxaparin (40 mg od beginning before THR, and 30 mg bid beginning after TKR). Treatment continued until mandatory bilateral venography was performed 5–9 days after surgery. Total VTE including all cause mortality occurred in 16.1–24.4% of per-protocol patients receiving rivaroxaban 5–60 mg, and 27.8% receiving enoxaparin (n = 914). There was a flat dose response relationship between rivaroxaban and total VTE (P = 0.39). Major bleeding (safety population, n = 1,317) increased dose-dependently with rivaroxaban (P < 0.001), occurring in 0.9%, 1.3%, 2.1%, 3.9%, and 7.0% of patients receiving rivaroxaban total daily doses of 5, 10, 20, 40, and 60 mg, respectively, versus 1.7% of patients receiving enoxaparin. No routine coagulation monitoring was performed, and there were no significant differences between dose response relationships with rivaroxaban after THR and TKR [26].
The rivaroxaban once daily study assessed the efficacy and safety of once-daily rivaroxaban relative to enoxaparin for prevention of venous thromboembolism in patients undergoing elective total hip replacement. Patients (n = 873) were randomized to oral rivaroxaban od doses of 5, 10, 20, 30, or 40 mg (initiated 6–8 h after surgery) or a once-daily subcutaneous enoxaparin dose of 40 mg (given the evening before and 6–8 h after surgery). Mandatory bilateral venography was performed between day 5 and 9. The primary end point (composite of any deep vein thrombosis, objectively confirmed pulmonary embolism, and all-cause mortality) was observed in 14.9%, 10.6%, 8.5%, 13.5%, 6.4%, and 25.2% of patients receiving 5, 10, 20, 30, and 40 mg rivaroxaban, and 40 mg enoxaparin, respectively (n = 618, per-protocol population). No significant dose–response relationship was found for efficacy (P = 0.0852). Major postoperative bleeding was observed in 2.3%, 0.7%, 4.3%, 4.9%, 5.1%, and 1.9% of patients receiving 5, 10, 20, 30, and 40 mg rivaroxaban, and 40 mg enoxaparin, respectively (n = 845, safety population), representing a significant dose–response relationship (P = 0.0391). A large phase III program is ongoing and the main focus is on primary prevention and treatment of VTE, secondary VTE prevention, and prevention of stroke in atrial fibrillation.
Apixaban
Apixaban is a small molecule inhibitor with a molecular weight of 460 daltons that targets the active site of Factor Xa. It is a selective and reversible inhibitor of Factor Xa and, like rivaroxaban, it inhibits Factor Xa bound within the prothrombinase complex as well as the free enzyme. The drug is well absorbed from the gastrointestinal tract and peak plasma levels are achieved in about 3 h. With repeated doses, the terminal halflife is between 9 h and 14 h. Therefore, once-daily administration may be possible. Apixaban is oxidized to a phenol metabolite in the liver and CYP3A4 may be involved in this metabolism. However, the potential for drug-drug interactions with apixaban is expected to be low. Like rivaroxaban, apixaban exhibits a dual mechanism of excretion. About 25% is excreted via the kidneys, while the remainder appears in the feces. Apixaban prolongs the INR and the aPTT in a concentration dependent fashion. However, its effect on these tests is minimal at concentrations that are likely to be therapeutic [27]. After an extensive phase I trial program the compound is in clinical development and a large phase II study has recently been completed in patients undergoing total knee replacement.
Patients were allocated randomly to 1 of 6 double-blind doses of apixaban (5, 10, or 20 mg) given as a single or a twice-daily divided dose, or enoxaparin 30 mg bid, or open-label warfarin titrated to an INR of 1.8–3.0. Apixaban and enoxaparin were started 12–24 h after completing surgery; warfarin the evening of the day of surgery. Treatment continued for 10–14 days. Mandatory bilateral venography was performed at the end of treatment. The primary efficacy outcome was a composite of all VTE events comprising asymptomatic and symptomatic deep vein thrombosis (DVT), pulmonary embolism (PE) and all-cause death during the treatment period. Of 1217 subjects treated and included in the safety analysis, 856 were eligible for the efficacy analysis. The composite VTE plus all cause death rate in the apixaban groups combined was significantly lower than the rate in the enoxaparin (P < 0.02) and warfarin (<0.001) groups (8.6%; 15.6%; 26.6%, respectively). A trend for a dose-related decrease (P = 0.09) in the composite VTE plus all cause death rates of 10.6%, 8.6%, and 6.8% was apparent when od and bid apixaban doses were combined as total daily doses of 5, 10, and 20 mg, respectively. When od and bid apixaban doses were assessed separately, dose-dependent decreases in the composite VTE plus all cause death rates were observed for each but did not reach statistical significance. The composite VTE plus all cause death rates for apixaban 2.5 mg bid and 5 mg od were 9.9% (95% CI: 5.1–17.0) and 11.3% (95% CI: 5.8–19.4) respectively, compared with a rate of 15.6% (95% CI: 9.4–23.8) in the enoxaparin group. For the composite outcome of proximal DVT+PE+all-cause death, each apixaban group had a lower event rate (0–2.7%) than the enoxaparin rate (4.6%). The incidence of major bleeding for apixaban ranged from 0.0% (2.5 mg bid) to 3.3% (20 mg od); no major bleeding was observed in either the enoxaparin or warfarin groups. Thus, it can be concluded that apixaban demonstrates a favorable efficacy and an acceptable bleeding safety profile at doses of 5–20 mg per day in the prevention of VTE after knee replacement surgery [28]. Several phase III trials are ongoing in various patient populations including primary prevention of VTE, treatment of DVT, and stroke prevention in atrial fibrillation.
LY517717
LY517717 (Lilly) is an orally active, direct Factor Xa inhibitor with a Ki of 4.4–6.6 nM. The elimination half-life of LY517717 was 25 h in healthy subjects, and the primary route of elimination appeared to be gastrointestinal. The safety and efficacy of LY517717 were evaluated in patients undergoing hip or knee replacement surgery. A total of 507 patients received oral LY517717 doses of 25, 50, 75, 100, 125 or 150 mg od or enoxaparin 40 mg od for 6–10 days. The three lower doses were halted prematurely due to lack of efficacy. Total VTE rates were 24.0%, 19.1%, 17.1% for 100, 125 and 150 mg LY517717, respectively, compared with 22.2% for enoxaparin. Proximal DVT rates were 6.4%, 2.4% and 0% for 100, 125 and 150 mg LY517717, respectively, compared with 4.2% for enoxaparin. Incidences of bleeding events were similar between the higher LY517717 doses and enoxaparin. Dose-related prolongation of PT was observed, reaching a mean prolongation of 4.1 s after 150 mg of LY517717. No other haematological or chemistry abnormalities were associated with LY517717. These data suggested that LY517717 doses of 100–150 mg were as safe and efficacious as a standard enoxaparin regimen for the prophylaxis of VTE. However, as the number of patients included in this study was small, large-scale studies are necessary to further support this conclusion [29].
YM150
YM150 is a once-daily, orally active Factor Xa inhibitor. It was investigated in a phase IIa study at doses of 3, 10, 30 or 60 mg for thromboprophylaxis in 174 patients undergoing hip replacement surgery; the comparator was enoxaparin 40 mg od. Study drugs were administered for 6–10 days. Total VTE occurred in 52%, 39%, 23% and 19% of patients receiving 3, 10, 30 and 60 mg YM150, respectively, compared with 39% of patients receiving enoxaparin. There was a statistically significant dose–response relationship with YM150 for the primary efficacy endpoint. No major bleeding was observed in this study; non-major, clinically relevant bleeding events occurred in 2.9% of patients in the 3 mg group and 5.7% of patients in the 10 mg group. No dose–response relationship was observed for minor bleeding, and the incidence of minor bleeding was similar for the highest YM150 dose and enoxaparin. These promising results warrant further investigations in large-scale studies [30].
Du-176b
Du-176b is an orally available, direct FXa inhibitor that competitively inhibited FXa with a Ki of 0.56 nM. It was ∼10,000-fold more selective for FXa than thrombin, and dose-dependently prolonged PT and aPTT in human plasma, with concentrations of 0.26 and 0.51 μM doubling the clotting times, respectively [31]. A phase I study in healthy human subjects has recently been presented, in which a 60 mg dose of Du-176b showed antithrombotic effects in plasma samples, in a Badimon chamber under venous and arterial conditions. Du-176b had a rapid onset of action with anti-FXa activity peaking 1.5 h after dosing, and returning to baseline 12 h post-dose; PT and aPTT followed a similar pattern [32]. It will be interesting to see how the drug will perform in clinical studies.
Conclusion
Several oral Xa and IIa inhibitors are in phase III of clinical development. These compounds have significant advantages over conventional anticoagulants thereby offering facilitations in prevention and treatment of VTE and improvement of patient compliance. First evidence suggests that these new anticoagulants are set to improve the management of thromboembolic disorders.
References
Geerts WH, Pineo GF, Heit JA et al (2004) Prevention of venous thromboembolism: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 126(3 Suppl):338S–400S
Buller HR, Agnelli G, Hull RD et al (2004) Antithrombotic therapy for venous thromboembolic disease: the Seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 126(3 Suppl):401S–428S
Ansell J, Hirsh J, Dalen J et al (2001) Managing oral anticoagulant therapy. Chest 119(1 Suppl):22S–38S
Hart RG, Benavente O, McBride R et al (1999) Antithrombotic therapy to prevent stroke in patients with atrial fibrillation: a meta-analysis. Ann Intern Med 131(7):492–501
Haas S (2003) Medical indications and considerations for future clinical decision making. Thromb Res 109(Suppl 1):S31–S37
Hirsh J, Raschke R (2004) Heparin and low-molecular-weight heparin: the seventh ACCP conference on antithrombotic and thrombolytic therapy. Chest 126(3 Suppl):188S–203S
Weitz JI, Hudoba M, Massel D et al (1990) Clot-bound thrombin is protected from inhibition by heparin-antithrombin III but is susceptible to inactivation by antithrombin III-independent inhibitors. J Clin Invest 86(2):385–391
Di Nisio M, Middeldorp S, Buller HR (2005) Direct thrombin inhibitors. N Engl J Med 353(10):1028–1040
Stangier J, Rathgen K, Stahle H et al (2007) The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol 64(3):292–303
Wienen W, Stassen JM, Priepke H et al (2007) Antithrombotic and anticoagulant effects of the direct thrombin inhibitor dabigatran, and its oral prodrug, dabigatran etexilate, in a rabbit model of venous thrombosis. J Thromb Haemost 5(6):1237–1242
Eriksson BI, Dahl OE, Buller HR et al (2005) A new oral direct thrombin inhibitor, dabigatran etexilate, compared with enoxaparin for prevention of thromboembolic events following total hip or knee replacement: the BISTRO II randomized trial. J Thromb Haemost 3(1):103–111
Eriksson BI, Dahl OE, van Dijk CN et al (2006) A new oral anticoagulant, dabigatran etexilate, is effective and safe in preventing venous thromboembolism after total knee replacement surgery (The RE-MODEL Trial). Blood 108:Abstr. 573
Wallentin LC, Ezekowith M, Simmers TA et al (2005) Safety and efficacy of a new oral direct thrombin inhibitor dabigatran in atrial fibrillation—a dose finding trial with comparison to warfarin. Eur Soc Cardiol Cong (abstr):P2949
Bauer KA (2006) New anticoagulants. Hematology/the Education Program of the American Society of Hematology. American Society of Hematology, pp 450–456
Alexander JH, Yang H, Becker RC et al (2005) First experience with direct, selective factor Xa inhibition in patients with non-ST-elevation acute coronary syndromes: results of the XaNADU-ACS Trial. J Thromb Haemost 3(3):439–447
Paccaly A, Frick A, Rohatagi S et al (2006) Pharmacokinetics of otamixaban, a direct factor Xa inhibitor, in healthy male subjects: pharmacokinetic model development for phase 2/3 simulation of exposure. J Clin Pharmacol 46(1):37–44
Paccaly A, Ozoux ML, Chu V et al (2005) Pharmacodynamic markers in the early clinical assessment of otamixaban, a direct factor Xa inhibitor. Thromb Haemost 94(6):1156–1163
Hinder M, Paccaly A, Frick A et al (2005) Anticoagulant and anti-platelet effects are maintained following coadministration of otamixaban, a direct factor Xa inhibitor, with tirofiban in healthy volunteers. Thromb Haemost 93(4):794–795
Hinder M, Frick A, Rosenburg R et al (2006) Anticoagulant and anti-platelet effects are maintained following coadministration of otamixaban, a direct factor Xa inhibitor, and acetylsalicylic acid. Thromb Haemost 95(2):224–228
Perzborn E, Strassburger J, Wilmen A et al (2005) In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939—an oral, direct Factor Xa inhibitor. J Thromb Haemost 3(3):514–521
Kubitza D, Becka M, Wensing G et al (2005) Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939—an oral, direct Factor Xa inhibitor—after multiple dosing in healthy male subjects. Eur J Clin Pharmacol 61(12):873–880
Kubitza D, Haas S (2006) Novel factor Xa inhibitors for prevention and treatment of thromboembolic diseases. Expert Opin Investig Drugs 15(8):843–855
Eriksson BI, Borris L, Dahl OE et al (2006) Oral, direct Factor Xa inhibition with BAY 59-7939 for the prevention of venous thromboembolism after total hip replacement. J Thromb Haemost 4(1):121–128
Turpie AG, Fisher WD, Bauer KA et al (2005) BAY 59-7939: an oral, direct factor Xa inhibitor for the prevention of venous thromboembolism in patients after total knee replacement. A phase II dose-ranging study. J Thromb Haemost 3(11):2479–2486
Eriksson BI, Borris LC, Dahl OE et al (2006) A once-daily, oral, direct Factor Xa inhibitor, rivaroxaban (BAY 59-7939), for thromboprophylaxis after total hip replacement. Circulation 114(22):2374–2381
Fisher WD, Eriksson BI, Bauer KA et al (2007) Rivaroxaban for thromboprophylaxis after orthopedic surgery: pooled analysis of two studies. Thromb Haemost 97(6):931–937
Weitz JI (2006) Emerging anticoagulants for the treatment of venous thromboembolism. Thromb Haemost 96(3):274–284
Lassen MR, Davidson BL, Gallus A et al (2006) A phase II randomized, double-blind, eight-arm, parallel-group, dose–response study of Apixaban, a new oral factor Xa inhibitor for the prevention of deep vein thrombosis in knee replacement surgery—on behalf of the Apixaban Investigators. Blood 108:Abst. 574
Agnelli G, Haas S, Ginsberg JS et al (2007) A phase II study of the oral factor Xa inhibitor LY517717 for the prevention of venous thromboembolism after hip or knee replacement. J Thromb Haemost 5(4):746–753
Eriksson BI, Turpie AG, Lassen MR et al (2005) YM150, an oral direct factor Xa inhibitor, as prophylaxis for venous thromboembolism in patients with elective primary hip replacement surgery. A dose escalation study. Blood 106:Abstr. 1865
Morishima Y, Furugohri T, Isobe K et al (2004) In vitro characteristics, anticoagulant effects and in vivo antithrombotic efficacy of a novel, potent and orally active direct factor Xa inhibitor, DU-176b. Blood 104(11):Abstr. 1862
Zafar MU, Gaztanga J, Velez M et al (2006) A phase-I study to assess the antithrombotic properties of Du-176b: an orally active direct factor-Xa inhibitor. J Am Coll Cardiol 47:Abstr. 288A
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Haas, S. New oral Xa and IIa inhibitors: updates on clinical trial results. J Thromb Thrombolysis 25, 52–60 (2008). https://doi.org/10.1007/s11239-007-0108-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11239-007-0108-7