
Abstract
The crystal and molecular structures of N-(2-carboxyphenyl)-4-dimethylaminebenzylideneimine and its protonated form have been determined. The Schiff base exists as the zwitterion, which is stabilized by the intramolecular ionic hydrogen bond. According to quantum-mechanical calculation results this tautomeric form is energetically unfavorable but in the solid and liquid state the intermolecular interactions support zwitterionic form.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Schiff base compounds possess antifungal, anticancer, anticonvulsant, diuretic, and cytotoxic activities [1–4]. These compounds are often used as ligands in coordination chemistry and some of their first-row transition metal complexes exhibit enhanced biological properties [5–7]. They also have applications in organic synthesis, catalysis, biotechnology, and analytical chemistry [8–11]. Aromatic imines and their derivatives have been used successfully to study resonance-assisted hydrogen bonds [12].
Tautomerism and isomerism phenomena for these compounds are of particular chemical and theoretical interest in the context of their photochromic and thermochromic properties [13]. In general, hydroxy Schiff bases display two possible tautomeric forms, the phenol-imine (or benzenoid) and the keto-amine (or quinoid) forms [14]. Various studies have been carried out with these systems to establish the existence of intramolecular hydrogen bonding, O–H···N or N–H···O, stabilizing one of the tautomeric forms. Another form of Schiff base compounds is their zwitterionic tautomer, which is rarely seen for hydroxy derivatives. The characteristic property of this form is the presence of ionic N+–H···O− hydrogen bond.
Considerable attention has previously been given to amino acid Schiff bases and their complexes since they exhibit a wide range of biological activities [15, 16]. Amino acid Schiff bases are used as a model compounds in the study of enzymatic transformations of amino acids (e.g., decarboxylation, transamination, and racemization), which require pyridoxal-5′-phosphate as a cofactor [17–19]. During this catalytic process, the proton migrates from the hydroxyl group to the imine nitrogen atom and in consequence the O–H···N hydrogen bond becomes the N+–H···O− hydrogen bond. It is known that the tautomerism of this intramolecular OHN hydrogen bond has a crucial influence on the enzymatic processes. The equilibrium between two tautomeric forms (O–H and the N–H) depends on many factors such as temperature, solvent polarity, and the presence of additional hydrogen bonds [20, 21].
In accordance with above-mentioned findings, crystallographic and computational studies have been performed on the model Schiff base compound derived from aromatic β-amino acid—the anthranilic acid (2-aminobenzoic acid) to probe the role of the intramolecular OHN hydrogen bond. This particular amino acid was chosen because the crystals of anthranilic acid can contain both its neutral and zwitterionic form [22]. The anthranilic acid-derived Schiff bases and their transition metal complexes have been studied for their antifungal and antibacterial activities [23, 24]. The structure of N-(2-carboxyphenyl)-4-dimethylaminebenzylideneimine pentahydrate (1) reported herein is an example of a Schiff base compound existing as the zwitterion, in which one of the carboxylic group H atoms is transferred to the imine N atom. Since hydrogen bonding is necessary for ligand activity, the protonated form (2) was obtained to establish the primary site available for molecular interactions.
Experimental
Synthesis
N-(2-carboxyphenyl)-4-dimethylaminebenzylideneimine pentahydrate (1) and N-(2-carboxyphenyl)-4-dimethylaminebenzylideneiminium chloride monohydrate (2) were prepared by one-pot synthesis of aldehyde with anthranilic acid in methanol. A hot solution of anthranilic acid (5 mmol) in methanol (40 mL) was added dropwise to a hot solution of p-dimethylaminobenzaldehyde (5 mmol) in methanol (30 mL). The mixture was heated under reflux for 4 h. The solution was then reduced by evaporation to half-volume and allowed to cool. After 7 days, red crystals of 1 were formed. Yield: 75%. Part of the product was dissolved in methanol–HCl solution (pH 3) and left to stand at 278 K. After 3 days, red crystals of 2 were formed.
X-ray crystallography
The crystals were mounted in turn on a KM-4-CCD automatic diffractometer equipped with CCD detector, and used for data collection. X-ray intensity data were collected with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) at temperature 291.0(3) K, with ω scan mode. A 24-s (1), 21-s (2) exposure time was used and reflections were collected up to 2θ = 72.74° (1) and 50.0° (2) (scan width 0.45°). The unit cell parameters were determined from 2,387 (1), 2,588 (2) strongest reflections. Both the crystals used for data collection did not change their appearance. Lorentz, polarization, and numerical absorption [25] corrections were applied. The structures were solved by direct methods and subsequently completed by difference Fourier recycling. All the non-hydrogen atoms were refined anisotropically using full-matrix, least-squares technique on F 2. The hydrogen atoms were found by difference Fourier methods and treated as “riding” on their parent non-hydrogen atoms and assigned isotropic displacement parameters equal to 1.5 (N imine, carboxylic group, methyl groups, and water molecules) or 1.2 (rest of atoms) times the value of equivalent displacement parameters of the parent atoms. The geometry of hydrogen atoms attached to carbon atoms was idealized after each cycle of least-squares refinement. It must be noted that the combination of solely light atoms existing in structure of 1 and Mo Kα radiation leads to relatively weak diffraction intensities (over 20% of reflections can be considered as effectively unobserved). This affects both the refinement weighting scheme and the goodness-of-fit parameter. Careful inspection of data allows to assume that the lack of fit is also due to an underestimate of the variances of the observations, whose relative values have been correctly assigned. The relatively large values of refinement final R factors of 1 originate mainly from some disagreement between calculated and observed reflections intensities at high angles (for θ larger than 30°). It should be outlined that observed value of goodness-of-fit parameter, as well as final R factors lie in the range required for correctly refined structures (according to prerequisites of International Union of Crystallography). SHELXS97 [26], SHELXL97 [27], and SHELXTL [28] programs were used for all the calculations. Atomic scattering factors were those incorporated in the computer programs. Details concerning crystal data and refinement are summarized in Table 1, selected bond lengths and bond angles are given in Table 2.
Theoretical calculations
Geometry optimization and natural bond orbital (NBO) analysis [29–31] were performed at the B3LYP/6-31++G(d,p) level of theory [32, 33] using the GAUSSIAN03 [34] program package. The geometric parameters were employed from crystal structure data. The optimized geometrical parameters were in agreement with those found from X-ray measurement within three standard deviations. The NBO analysis has been employed to evaluate the stabilization energy of the donor–acceptor interactions. The atomic charges were calculated according to natural population analysis (NPA) [29–31], Merz–Kollman–Singh (MKS) [35, 36], and Breneman [37] schemes.
Spectroscopic and thermogravimetric measurements
The IR spectra (400–4,000 cm−1) were recorded with samples prepared as KBr disks in a Magma 560 spectrophotometer. UV–Vis spectra were recorded on a Jasco 660 spectrophotometer. The protonation constant was established using the DL-50 Graphix Titrator (Mettler-Toledo).
The thermal analysis was carried out in a TG/DTA-SETSYS-16/18 thermoanalyser coupled with ThermoStar (Balzers) mass spectrometer. The sample (4.67 mg for 1) was heated using corundum crucibles up to 1,000 °C, at the heating rate of 10 K min−1 in air atmosphere. The products of decomposition were calculated from TG curves. The temperature ranges were determined by thermoanalyser Data Processing Module [38].
Result and discussion
The description of the structures
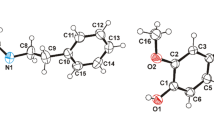
The crystallographic asymmetric unit of 1 consists of one anthranilic acid-derived Schiff base molecule and five water molecules (Fig. 1). The Schiff base exists in the zwitterionic form of the carboxy-imine compound. The proton from the carboxylic group migrates to the azomethine group (–N=CH–) forming a di-polar ion structure similar to that occurring in the pure amino acid. All non-hydrogen atoms of the organic molecule are almost coplanar. The largest deviation from the Schiff base plane is 0.220(2) Å, for atom O2. The dihedral angle between the two benzene rings in the structure is 2.0(1)°.

The molecular structure of 1 showing the atom and ring numbering scheme. The displacement ellipsoids are drawn at 50% probability level and H atoms are shown as spheres of arbitrary radii. Dashed line indicates the intramolecular hydrogen bond
The Schiff base molecule shows the configuration E with respect to the N1=C7 bond, which is stabilized by the intramolecular N1+–H1N···O2− hydrogen bond. The occupancies of the lone pair (n O2), antibonding bond orbital (σ N1–H1N*) and the stabilization energy value (7.94 kcal/mol) indicate medium strength (donor–acceptor) non-covalent interactions between p z lone pairs on the oxygen atom and σ*N–H bond. This is rather low hydrogen-bond energy involving charged donor and acceptor atoms, but deletion of this interaction leads to increase of total molecule energy by 11.6 kcal/mol. Moreover, the N and O atoms of the imine and carboxylate groups, are negatively charged, according to NPA, Breneman, and Merz–Kollman–Singh (MKS) charge values (Table 3). This confirms the existence of an intramolecular hydrogen bond. The geometrical parameters of this interaction (Table 4) are close to those of similar anthranilic acid-derived Schiff bases [14, 39], in which the imine N atom acts as bifurcated donor.
The quantum-mechanical calculations performed for the isolated molecule show that the N-(2-carboxyphenyl)-4-dimethylaminebenzylideneimine is less stable in the zwitterionic form (1) than in the non-ionic form (3) (Fig. 2). The proton transfer from 3 to 1 is an endothermic process with the enthalpy change of 40.9 kcal/mol. It is worth mentioning that during the geometry optimization process, one H atom of the N imine group migrated to the carboxylate anion, forming the non-polar molecule stabilized by the intramolecular O–H···N hydrogen bond. The existence of 1 in the zwitterionic form in the solid state confirms that the presence of solvent molecules and forming of non-covalent interactions influences the equilibrium between tautomeric forms.

The tautomeric forms of 1
In general, the atomic charges do not depend on the method used for calculation (Table 3). The COO− group is negatively charged, whereas the CH3 groups are positively charged. The p-phenylene CH groups have different charges. Those ones lying on the side of the electron-rich group—dimethylamine have negative charge, whereas those ones lying on the side of the imine group have positive charge. This can be explained by the high degree of conjugation, with a strong push–pull effect between the imine—electron withdrawing group and dimethylamine—electron-donating group. According to second-order perturbation theory analysis, the stabilization energy of intramolecular charge transfer interactions between the C8–C13 bonding orbital (which behaves as donors) and N1=C7 antibonding orbital (which acts as acceptors) is equal to 11.51 kcal/mol. The σ NC → σ CC* interactions between the N2–C16 bonding orbital atom and C10–C11 antibonding orbital have the lowest stabilization energy, equal to 2.38 kcal/mol.
The NLMO bond orders [29–31] are equal to 1.53 for the C14–O1 partial double bond, 1.42 for the C14–O2 partial double bond and 1.49 for the N1=C7 bond. The bond order can be also calculated by means of the bond-valence method (BVM), according to which the bond valence (v ij) is defined as a number of electron pairs forming the bond. The BVM can be successfully used for organic compounds on condition that the mean bond length (1.42 Å for the C–O bond and 1.47 Å for C–N bond) is treated as the bond-valence parameter value (R ij) [40]. The bond orders calculated from the Brown–Altermatt equation (ν ij = exp[(R ij − d ij)/0.37] [41]) for bonds of lengths d ij are equal to 1.60 v.u. for the C14–O1 bond, 1.52 v.u. for the C14–O2 bond and 1.58 v.u. for the N1=C7 bond. Although these values are higher than NLMO bond orders, they show the same tendency. The C–O bonds of the carboxylate group are delocalized, whereas the bond order of the NC double bond is lower than expected. A CSD [42] search for the N=C bond length in Ph–HN+=CH–Ph (33 fragments in 30 X-ray structures) and Ph–N=CH–Ph (1,030 fragments in 722 X-ray structures) systems revealed that the azomethine bond (–N=CH–, mean value of 1.277(3) Å) is significantly shorter than the –HN+=CH– bond (1.303(1) Å). Such elongation of the –HN+=CH– bond can be explained by partial transfer of the electron density from the N=C bond to the N+–H bond and thus the bond order of the NC double bond implies weaker character of the N=C bond.
The separation distance between almost parallel (inclined at 2.0(1)º) benzene rings of adjacent Schiff base molecules oriented in opposite directions [symmetry code: (−x, −y + 1, −z + 1)], 3.628(2) Å, indicates a weak aromatic-stacking interaction. The angle between the vector linking the one ring centroid and the normal to the second ring plane is 21.6(2)º. An interesting feature is the presence of a weak C–H···π ring interaction: C16–H16C···ring#1 (#1: C1–C6) with H···ring-centroid distance of 3.270 Å, C···ring-centroid distance of 3.885 Å, C–H···ring-centroid angle of 123.7º. These weak C–H···π ring and π–π stacking interactions form piles of Schiff base molecules along the crystallographic a axis.
The crystal structure of 1 is stabilized by O–H···O hydrogen bonds between the water molecules and carboxylate groups of imine molecules (Table 4). In addition, the water molecules form a hydrogen-bonded network along the crystallographic a axis (Fig. 3), composed of fused rings described by N2 R 12 (8) and N3 R 33 (12) motifs [43].

Part of the packing of molecules in 1. Dashed lines indicate hydrogen bonds
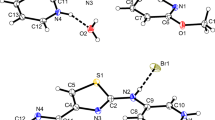
From an acidic environment, the solid-state compound N-(2-carboxyphenyl)-4-dimethylaminebenzylideneiminium chloride monohydrate (2), containing the -COOH group (Fig. 4) can be obtained. The hydrogen atom is bonded to the less negative O1 atom (Table 3), which does not take part in forming the intramolecular hydrogen bond. This hydrogen atom creates the intermolecular O–H···O hydrogen bond with a water molecule (Table 4). As in compound 1, the intramolecular N–H···O hydrogen bond is observed but the C–O bond lengths (Table 2) suggest that it involves the iminium N+–H group and the C=O group of the carboxylic moiety. The molecules of N-(2-carboxyphenyl)-4-dimethylaminebenzylideneiminium cations are linked into dimers through water molecules and chloride ions (Fig. 5). The weak π···π stacking interactions between parallel aromatic rings of adjacent-protonated organic molecules can be observed. The perpendicular distances between the first ring centroid and that of the second ring are 3.976(3) and 3.869(3) Å, and the angles between the vector linking the ring centroids and the normal to the five-membered ring plane are 26.9(2) and 25.7 (2)º, respectively. The protonated iminium cations are oriented in opposite directions [symmetry codes: (−x + 2, −y, −z) and (−x + 1, −y, −z), respectively].

The molecular structure of 2 showing the atom and ring numbering scheme. The displacement ellipsoids are drawn at 50% probability level and H atoms are shown as spheres of arbitrary radii. Dashed line indicates the intramolecular hydrogen bond

Part of the packing of molecules in 2. Dashed lines indicate hydrogen bonds. [Symmetry codes: (i) –x + 2, −y, −z + 1; (ii) x + 1, y − 1, z; (iii) −x + 3, −y − 1, −z + 1]
Protonation constant
The protonation constant of Schiff base (1) was determined potentiometrically in methanol–water mixture of 40% methanol (v/v). Titration was performed at 25 °C and the ionic strength of the medium was maintained at 0.40 mol/dm3 using sodium chloride. The value of logK is equal to 3.81 ± 0.14. The protonation constant is related to the protonation of an oxygen atom of the carboxylate group (as in the solid state, 2). A comparison between the logK values for the Schiff base and the pure amino acid (logK = 2.25 [44]) suggests that the carboxylate group has a lower acidicity in 1 than in the anthranilic acid, because it is stabilized by the hydrogen-bonding interaction with the protonated azomethine group and by the delocalization of the negative charge.
Spectroscopic studies
The vibrational analysis was carried out for the Schiff base compound (1) and its protonated form (2). The IR spectra of 1 and 2 contain characteristic bands of the stretching and bending vibrations of the aromatic CC, NH, OH, and CH groups (Table 5). An important spectral feature that can be used to distinguish protonated and neutral forms of the imine N atom is the C=N stretching vibration that typically occurs between 1,640 and 1,690 cm−1. This frequency displays remarkable bathochromic shift when the N atom of the C=N bond is substituted in such a way that it is able to take on a more polar character [14]. Thus, the band corresponding to the C=N stretching vibration appears at 1,585 cm−1 for 1 and 2. The most significant differences between vibrational frequencies of 1 and 2 were observed for the CO stretching vibrations. The IR spectrum of 2 showed a strong band at 1,680 cm−1 originating from the stretching vibrations of C=O bonds, whereas the medium intensity band at 1,220 cm−1 can be attributed to vibrations of C–O groups. This confirms the presence of carboxylic groups in 2. The observed lowering of C=O frequency is caused by the presence of hydrogen bonds formed by this group in the solid state. In the IR spectrum of 1, the bands corresponding to above-mentioned vibrations are absent. The IR spectrum of 1 showed strong bands at ca. 1,362 and 1,585 cm−1, which can be assigned to the stretching vibrations of carboxylate groups, the νs(COO−) and νas(COO−), respectively. This confirms the presence of these groups in 1.
The UV/Vis spectra of 1 and 2 were recorded in methanol solution. They are almost identical and exhibit three absorption bands. The strong, sharp band at 213 (ε = 25,000 m2/mol) with a shoulder at 244 nm (ε = 9,500 m2/mol) can be attributed to π → π* transitions within the phenyl moieties. The third band located at 354 nm (ε = 19,000 m2/mol) is due to n → π* transitions of the C=N group, influenced by the intramolecular charge transfer effect related to the presence of the intramolecular hydrogen bond. Since the bands corresponding to n → π* transitions are placed at the same wavelength in 1 and 2, thus it can be stated that the anthranilic acid-derived Schiff base exists in solution in zwitterionic form.
Thermal studies
The compound 1 is thermally stable up to 150 °C and its decomposition is a three-stage process. The first endothermic step of thermal decomposition occurs within the temperature range 150–330 °C. The initial mass loss is attributed to the removal of water molecules. The next step of decomposition is characterized by a small peak on the DTG curve at 370 °C. The following step, occurring in the temperature range 450–630 °C, is also exothermic. Above 630 °C, the Schiff base decomposes completely.
Conclusions
The obtained anthranilic acid-derived Schiff base exists as the zwitterion, which is stabilized by the intramolecular N+–H···O− hydrogen bond. According to quantum-mechanical calculation results, this tautomeric form is energetically unfavorable but in the solid and solution states the observed intermolecular interactions support the presence of the zwitterionic form. The protonated azomethine bond is longer than the non-protonated one due to partial transfer of the electron density from the N=C bond to the N+–H bond.
Supplementary data
CCDC-743354 (1) and CCDC-743355 (2) contains the supplementary crystallographic data for this paper. These data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html [or from the Cambridge Crystallographic Data Centre (CCDC), 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44(0)1223-336033; email: deposit@ccdc.cam.ac.uk].
References
Sridhar SK, Pandeya SN, Stables JP, Ramesh A (2002) Eur J Med Chem 16:129
Tarafder MT, Kasbollah A, Saravan N, Crouse KA, Ali AM, Tin OK (2002) J Biochem Mol Biol Biophys 6:85
Kucukguzel I, Kucukguzel SG, Rollas S, Sanis GO, Ozdemir O, Bayrak I, Altug T, Stables J (2004) Il Farmaco 59:839
Vicini P, Geronikaki A, Incerti M, Busonera B, Poni G, Kabras CA, Colla PL (2003) Bioorg Med Chem 11:4785
Mohamed GG, Omar MM, Hindy AMM (2005) Spectrochim Acta Part A Mol Biomol Spectrosc 62:1140
İspir E, Toroğlu S, Kayraldız A (2008) Trans Met Chem 33:953
Adsule S, Barve V, Chen D, Ahmed F, Dou QP, Padhye S, Sarka FH (2006) J Med Chem 49:7242
Tian L, Sun Y, Qian B, Yang G, Yu Y, Shang Z, Zheng X (2005) Appl Organomet Chem 19:1127
Singh HL, Varshney S, Varshney AK (2000) Appl Organomet Chem 14:212
Tantaru G, Dorneanu V, Stan M (2002) J Pharm Biomed Anal 27:827
Fakhari AR, Khorrami AR, Naeimi H (2005) Talanta 66:813
Sobczyk L, Grabowski SJ, Krygowski TM (2005) Chem Rev 105:3513
Hadjoudis E, Mavridis IM (2004) Chem Soc Rev 33:579
Ligtenbarg AGJ, Hage R, Meetsma A, Feringa BL (1999) J Chem Soc Perkin Trans 2:807
Sakıyan I, Loğoğlu E, Arslan S, Sari N, Şakıyan N (2004) Biometals 17:115
Karmakar R, Choudhury CR, Mitra S, Dahlenburg L (2005) Struct Chem 16:611
Sharif S, Powell DR, Schagen D, Steiner T, Toney MD, Fogle E, Limbach H-H (2006) Acta Crystallogr B62:480
Sharif S, Denisov GS, Toney MD, Limbach H-H (2006) J Am Chem Soc 128:3375
Golubev NS, Smirnov SN, Tolstoy PM, Sharif S, Toney MD, Denisov GS, Limbach H-H (2007) J Mol Struct 844–845:319
Tolstoy PM, Smirnov SN, Shenderovich IG, Golubev NS, Denisov GS, Limbach H-H (2004) J Mol Struct 700:19
Sharif S, Schagen D, Toney MD, Limbach H-H (2007) J Am Chem Soc 129:4440
Brown CJ, Ehrenberg M (1985) Acta Crystallogr C41:441
Mohamed GG, Omar MM, Hindy AMM (2005) Spectrochim Acta A62:1140
Abd El-Wahab ZH (2007) Spectrochim Acta A67:25
STOE Cie (1999) X-RED, version 1.18. STOE & Cie GmbH, Darmstadt, Germany
Sheldrick GM (1990) Acta Cryst A46:467
Sheldrick GM (1997) SHELXL97, program for crystal structure refinement. University of Göttingen, Germany
Sheldrick GM (2008) Acta Cryst A64:112
Reed AE, Curtis LA, Weinhold FA (1988) Chem Rev 88:899
Foster JP, Weinhold FA (1980) J Am Chem Soc 102:7211
Reed AE, Weinhold FA (1985) J Chem Phys 83:1736
Becke AD (1993) J Chem Phys 98:5648
Lee C, Yang W, Parr RG (1988) Phys Rev B37:785
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery Jr JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2004) Gaussian 03, Revision E.01. Gaussian Inc., Pittsburgh PA
Singh UC, Kollman PA (1984) J Comput Chem 5:129
Besler BH, Merz KM Jr, Kollman PA (1990) J Comput Chem 11:431
Breneman CM, Wiberg KB (1990) J Comput Chem 11:361
Data processing module, copyright© 1994–1998 SETARAM—FRANCE; version 1.4
Gayathri D, Velmurugan D, Ravikumar K, Devaraj S, Kandaswamy M (2007) Acta Crystallogr E63:o849
Mohri F (2000) Acta Crystallogr B56:626
Brown ID, Altermatt D (1985) Acta Crystallogr B41:244
Allen FH (2002) Acta Crystallogr B58:380
Bernstein J, Davis RE, Shimoni L, Chang N-L (1995) Angew Chem Int Ed Engl 34:1555
Zapała L, Kalembkiewicz J, Sitarz-Palczak E (2009) Biophys Chem 140:91
Acknowledgments
This study was financed by funds allocated by the Ministry of Science and Higher Education to the Institute of General and Ecological Chemistry, Lodz University of Technology, Poland. The GAUSSIAN03 calculations were carried out in the Academic Computer Centre CYFRONET of the AGH University of Science and Technology in Cracow, Poland (Grant No.: MNiSW/SGI3700/PŁódzka/040/2008).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Trzesowska-Kruszynska, A. The zwitterion of N-(2-carboxyphenyl)-4-dimethylaminebenzylideneimine. Struct Chem 21, 131–138 (2010). https://doi.org/10.1007/s11224-009-9547-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-009-9547-4