
Abstract
The structure and aromatic properties of Rees hydrocarbons 7bH-cyclopent[cd]indene and its benzo-annelated derivative 1a and 2a, respectively, are examined by the B3LYP/6−31+G(d) calculations employing HOMA criterion of Krygowski and coworkers. It is shown that 1a possesses strong π-electron delocalization over the perimeter of the CC bonds, thus forming a quasi-[10]annulene pattern. Its aromatic character is determined to be 83%. In contrast, 2a is less convenient model system for [14]annulene. The reason behind is that the perimeter network of the potentially aromatic 14π-electrons is supplemented by two additional more local aromatic patterns involving 10π and 6π electrons. Consequently, the π-electron delocalization over the molecular rim is incomplete being thus diminished. The aromatic character of the peripheral bonds in both 1a − and 2a − anions formed upon deprotonation of the central C–H bond is decreased, since the role of the smaller rings in forming aromatic subsystems is increasing. Finally, polycyano substitution of 1a and 2a decreases aromaticity due to the price paid for the resonance effect taking place between the carbocyclic π-network and the double bonds of the CN groups. The resonance effect is particularly strong in anions derived by heterolytic cleavage of the C–H bond emanating from the central sp 3 carbon atom.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Aromaticity is one of the most important and at the same time most elusive features of (organic) molecules [1]. It is one of the cornerstones of chemistry just like its counterpart antiaromaticity and yet it cannot be exactly defined. Therefore, strictly speaking, it is a nonobservable. We cannot resist mentioning within context the polarization of the local atomic orbitals called hybridization [2–4], which cannot be rigorously determined either. Nevertheless, it exerts profound influence on a large number of local molecular properties, which are measurable [5, 6]. Since it enables to establish inter-relations between or otherwise unrelated features, we proposed that such concepts are termed quasi-, crypto- or perhaps pseudo-observables [7], because they have some semblance with true observables. The term quasiobservables should be preferred over the other two. Both aromaticity and antiaromaticity belong to this category of notions. Not surprisingly, there are numerous criteria developed to describe aromaticity [1], since none of them is unambiguous. They can be roughly divided into: (a) Thermodynamic, describing extraordinary stability of aromatic compounds [8], (b) Geometric, reflecting some spatial structural characteristics [9, 10], (c) Magnetic, mirroring behavior of the aromatic compounds in external magnetic field [11–13], and (d) Electronic, being related to molecular wave functions or electron densities. The first property (a) is in our view the most fundamental criterion, because it is directly related to chemical reactivity. The second feature (b) is implicitly, but not directly connected to molecular stability, since the equilibrium structures are minima on Born-Oppenheimer potential energy surfaces. There are potential energy functions expressed in terms of structural parameters, but they are rather complex and empirical in nature [10b]. The third group of properties yield response of the ground state electronic structure to perturbation exerted by the applied external magnetic field. On these ground one intuitively assumes that there might be in principle, a relation between the energetic and structural criteria (a) and (b), as elaborated by Krygowski and Cyrański [10b]. On the other hand, the magnetic parameters are interesting on their own right in rationalizing magnetic facets of aromatic compounds, but in our view their correlation with the energy/structure criteria is circumstantial at best [14]. The electronic criteria will be briefly addressed in the last but one section of the article. In this work we discuss expected aromatic character of Rees hydrocarbons [15] 1 and 2 and their anions (Scheme 1), since they might represent model quasi [10]- and [14]annulenes in spe.

This is of some interest because genuine higher annulenes are nonplanar due to angular strain and/or repulsion of inner H atoms. Consequently, they are floppy thus devoiding aromaticity [16–18]. In order to put the present study in a proper perspective, let us mention that a lot of efforts were devoted in the past in preparing quasi-[10]annulenes. Historically, the first quasi-[10]annulene moiety was achieved by introducing central CH2 (or CMe2) bridge yielding 11,11-dimethyl-9,10-methano[10]annulene [19, 20] 3 (Fig. 1). Although the X-ray investigation was interpreted in terms of the bis-norcaradiene structure, subsequent NMR study revealed substantial π-electron delocalization over the carbon skeleton, the bridging C11 atom being excluded for obvious reason. More specifically, the proton chemical shifts indicated the presence of appreciable ring current [21]. The iterative maximum overlap calculations [22] have shown that the bond distances were predominantly determined by the σ-skeleton, while the π-electrons exerted only perturbation corrections [23]. Therefore, it can be safely concluded that the π-delocalization takes part in 3, although there is still some bond alternation left and despite the fact that the bridgehead carbons are pyramidalized. These results were in qualitative agreement with the NMR measurements [21]. Another interesting [10]annulene systems 4 and 5, not yet synthesized, were suggested by Schleyer, Schaefer and coworkers [24] (Fig. 1). Floppy [10]annulene framework was kept rigid and planar by fusion of five three-membered (4) or four-membered (5) rings, respectively. Finally, an example par excellence for quasi-[10]annulene system is given by double fusion of cyclobutene fragments to the benzene moiety yielding benzo[1,2:4,5]dicyclobutadiene 6 (Fig. 2). The DFT [25], MP2 [25], and MR–AQCC [26] calculations provided a conclusive evidence that the π-electrons are completely delocalized over the molecular perimeter in 6 with negligible π-density on the fused bonds. Its heavy substituted derivative 3,6-di-tert-butyl-7,8,9,10-tetraphenyl-benzo[1,2:4,5]dicyclobutadiene is prepared [27] and the X-ray structure [28] is compatible with the aforementioned calculations. It will appear in what follows that Rees hydrocarbon 1a also exhibits high-aromatic [10]annulene character. Unfortunately, the same does not hold for the system 2a, which is not a good model system for quasi-[14] annulene.

Schematic representation of some pseudo-[10]annulenes

Relevant geometrical parameters (in Å) as obtained with B3LYP/6−31+G(d) level of theory
Methodology
We shall employ the structural HOMA criterion [9, 10], which is acronym for the harmonic oscillator model of aromaticity. The HOMA index is defined as:
where the summation is extended over n bonds of the cyclic fragment under scrutiny, α is a free parameter determined by requirement that HOMA = 0 for a reference nonaromatic system like, e.g., the Kekulé structure of benzene. In that case α for C–C bonds is 257.7. Notice that HOMA is dimensionless magnitude assuming one for a perfectly aromatic system without alternation of bond distances, which implies that all CC bond distances are equal to the optimal one d opt. The latter takes a value of 1.388 (in Å), which is close to the true bond distance in the equilibrium D 6h structure of benzene taken as a paradigmatic aromatic molecule. In calculating the structural parameters of Rees compounds 1 and 2 we have employed B3LYP/6−31+G(d) computational scheme in accordance with an earlier study of the acidity of these systems [29]. For this purpose, we have made use of GAUSSIAN03 suite of programs [30].
Results and discussion
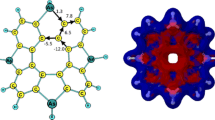
Relevant bond distances of 1a, 1a −, 1b, and 1b − are shown in Fig. 2. The neutral species are not planar. In order to describe nonplanarity, which is caused by the central tetracoordinate carbon atom, we shall estimate its pyramidalization (DP%) [31]:
where (DP%) stands for the degree of pyramidalization and summation goes over three CCC bond angles merging at the pyramidal atom in question. Similarly, a deformation of the ideal tetrahedral arrangement (TD%) of the CC and CH bonds can be defined as [32]:
where α i (given in degrees) denotes one of the six bond angles of the tetrahedral atom. A factor 0.944 is obtained by defining the most distorted tetrahedron, denoted by TDmax, and determined as the one possessing three sharp angles of 90°. The DP(%) values of neutral species 1a and 2a are 28.4% and 27.8%, respectively, thus revealing significant nonplanarity caused by the central four-coordinated carbon atom. This is compatible with its C–C–C angles of roughly 107.3° and 119.9° (in 1a) and 108.0° and 119.6° (in 2a). Cyanation does not affect nonplanarity of the neutral species as evidenced by the DP(%) values given within parentheses: 1b DP (29.2%) and 2b DP (29.5%). The tetrahedral deformations TD(%) of the C(sp 3) center in 1a and 2a are not dramatic being 20.3% and 19.8%, respectively. They are little changed upon CN substitutions assuming values 1b TD (19.5%) and 2b TD (19.4%). However, deprotonation of the central carbon stirres up substantial changes by transforming its sp 3 hybridization into the sp 2 with a consequence that anions are perfectly planar, thus enabling optimal anionic resonance.
Perusal of the bond distances displayed in Fig. 2 reveals that their variation along the molecular perimeter is rather small, since they assume values between 1.40 and 1.42 Å. The same holds for benzene moiety in the anion 1a −. However, the bond distances of the five-membered rings in the latter species exhibit a more pronounced variation in the range of 1.39–1.44 Å. It is noteworthy that CN substitution does incite small changes in the CC bond lengths. In spite of that, the resonance effect is operative being very strong in the anion 1b −. The fusion of the benzene ring in 2a induces a noticeable alternation of the CC bond lengths over the perimeter. Nevertheless, the π-electron delocalization is still effective as pointed out in the Introduction despite differences in bond distances. The rest of the data given in Fig. 2 speak for themselves.
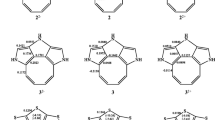
The HOMA indices of 1a and 1a − are given in Fig. 3, where the subscripts refer to the number of CC bonds within the ring, It is interesting that HOMA (0.828)10 of the perimeter CC bonds in 1a is close to one indicating very high aromatic character of 83%. Hence, it is justified to say that Rees hydrocarbon 1a is one more example of quasi-[10]annulene system. The π-electron structure of the planar 1a − anion is more intricate. It can be decomposed into three (aromatic) patterns encompassing: (1) the perimeter π-electron delocalization, (2) the six-membered benzene ring, and (3) two equivalent partially anionic five-membered rings (Fig. 3) with the corresponding HOMA values (0.407)10, (0.859)6 and (0.468)5, respectively. Each of these π-electron substructures is tending to achieve the aromatic (4n + 2)π number of electrons, which implies that the excess electron resides predominantly within two five membered rings. All these rings compete for the aromatic number of π-electrons and none of them is able to ensure the ideal (4n + 2)π pattern due to a presence of the adjacent fused rings. Nevertheless, they represent the leading π-electron spin coupling schemes implying that the rest of the π-electrons are coupled in localized π-bonds (or a lone pair). We shall come back to this point in a little while. It is striking that the HOMA value of the perimeter ring in 1a − is decreased by roughly 50% relative to that in the neutral 1a as a price paid by the increase of aromaticity in the fused central benzene ring to 86%. One should also note that the aromatic character of each of the five-membered ring fragments is 47%, i.e., practically 50% each. This would imply that the excess electron is effectively distributed about 50% over each five-membered ring.

Calculated HOMA indices. Chemical bonds taken into account are given in bold, while their number is given as a subscript next to the HOMA index
The π-electron distribution in Rees hydrocarbon 2a can be partitioned in three aromatic substructures encompassing spin coupling of 14, 10, and 6 π-electrons within the corresponding fragments (Fig. 3). The corresponding HOMA indices are (0.702)14, (0.514)10 and (0.742)6 implying that the perimeter π-electron delocalization seemingly exhibits 70% of the ideal [14]annulene aromaticity. However, the situation is more complex than that, because these π-electrons participate at the same time in the formation of the 10π and 6π electron patterns. The aromatic character of the benzene moiety, for example, is as high as 74%. One can safely conclude that 2a is not a good model system for [14]annulene. The number of aromatic substructures discernible in anion 2a − is increased to six (Fig. 3). The aromatic character of the upper benzene fragment is 87%, whereas the corresponding value in the lower left benzene is 67%. It is interesting to note that the right hand side five-membered ring has the aromatic character of 59%, in contrast with its left counterpart with this character of only 5%.
Inspection of the HOMA values for cyano derivatives reveals that they are invariably lower compared to the corresponding parent neutral and anionic systems, exceptions being the five-membered rings in anions (Fig. 3). For instance, the aromatic characters of 1b and 2b are 62% and 51%, respectively, which is low by 20% when compared with those found in the parent unsubstituted molecules. This is compatible with the resonance effect between the CN fragments and the carbon π-network. It is particularly strong in anions (referred to as the anionic resonance effect), which considerably increases acidity of polycyano substituted Rees molecules [29].
Finally, let us consider the meaning of the predominant spin coupling schemes presented in Fig. 3. If the aromatic substructures are complemented by local electron couples describing localized double bonds or a lone pair, they provide a graphical representation of the valence bond wavefunctions. The linear combination of the latter, optimized by minimizing the total energy would give a very accurate description of the studied systems. This is something similar to Clar VB model treatment of the fully benzenoid structures possessing only aromatic sextets extended over the six-membered rings. This approach was found useful in theoretical treatment of single-walled carbon nanotubes (SWNT) [33–35]. It should be strongly emphasized that Clar's structures serve only as useful book-keeping device, because the extraordinary stability of the benzenoid systems originates in the σ-framework formed by the σ-electrons and the atomic nuclei, as convincingly shown recently [36]. This is in full analogy with the earlier work of Shaik, Hiberty, Jug [37, 38], and others [39], which has conclusively shown that aromaticity occurs due to the σ-type interactions. An empirical way in estimating weights of the VB canonical structure from the bond lengths was developed by Aihara [40]. An authoritative account of the aromatic harmonic oscillator stabilization energy and its relation to the VB canonical structures is given by Krygowski and Cyrański [10b]. It can be safely stated that a lot of information is condensed in the structural parameters, in particular in the (bond) distances.
In concluding this section we would like to comment briefly on some electronic theories of aromaticity that are not mentioned above. A comprehensive review has been published by Poater et al. [41] on a method based on the 3D density picture with particular emphasis on the AIM (Atoms In Molecules) [42] and ELF (Electron Localization Functions) [43] methodologies. They provide orbital-free total electron density description of delocalization. This is an very important feature, because the σ-electrons are crucial in rationalizing aromaticity (vide supra). In contrast, Hückel type calculations [44] must be considered with utmost care—not to mention the electron-free graph theoretical methods [45]—since they might give satisfactory results for wrong reasons.
Concluding remarks
It is shown that Rees hydrocarbon 7bH-cyclopent[cd]indene 1a represents a quasi-[10]annulene aromatic system due to its rigidity introduced by the central sp 3 carbon atom and efficient overlapping of the π-atomic orbitals on the molecular perimeter. Polycyano derivative 1b exhibits a diminished aromatic character due to a resonance effect taking place between the CN double bonds and the carbon π-network. Both 1a and 1b are nonplanar due to the central C(sp 3) atom. Their deprotonation yields planar anions 1a − and 1b − possessing 16π electrons. The annulene character is lost and the number of Pauling’s resonance structures is increased, underlying importance of smaller aromatic π-electron subsystems. The anionic resonance is very strong in the cyano derivative 1b −, which makes the neutral system 1b an extremely powerful organic superacid [29]. Finally, it can be concluded beyond any doubt that Rees hydrocarbon 2a is not a pseudo-[14]annulene, because the aromatic substructures involving a flanked annelated benzene ring and the intramolecular quasi-[10]annulene pattern strongly participate in the stabilization of the system. Hence, π-electron delocalization over the perimeter is decreased.
References
Special issue on aromaticity; Schleyer PvR, (ed) (2001) Chem Rev 101:1115
Pauling L (1928) Proc Natl Acad Sci 14:359; Pauling LJ (1931) Am Chem Soc 53:1367
Pauling L (1960) The nature of the chemical bond and the structure of molecules and crystals, 3rd edn. Cornell Univ Press, Ithaca NY
McWeeny R (1979) Coulson's valence, 3rd edn. Oxford Univ Press, Oxford
Maksić ZB (1983) Pure & Appl Chem 55:307
Maksić ZB (1986) In: Hargittai, (ed) Symmetry—unifying human understanding. Pergamon Press, New York, p 697
Maksić ZB (1990) In: Maksić ZB (ed) Theoretical models of chemical bonding, Part 2. Springer Verlag, Berlin-Heidelberg, p 136
Cyrański MK (2005) Chem Rev 105:3773
Kruszewski J, Krygowski TM, Tetrahedron Lett (1972), 3839; Krygowski TM (1993) Chem Inf Comp Sci 33:70
(a) Krygowski TM; Cyrański M (1996) Tetrahedron 52:1713; (b) Krygowski TM, Cyrański M (2001) Chem Rev 101:1385 and references therein
Chen Z, Wannere CS, Corminbeuf C, Puchta R, Schleyer PvR (2005) Chem Rev 105:3842
Heine T, Corminbeuf C, Seifert G (2005) Chem Rev 105:3889
Lazzeretti P (2004) Phys Chem Chem Phys 6:217
For a controversial discussion consult: Katritzky AR, Karelson M, Sild S, Krygowski TM, Jug KJ (1998) Org Chem 63:5228; Cyrański MK, Krygowski TM, Katritzky AR, Schleyer PvR (2002) J Org Chem 67:1333
Gilchrist TL, Rees CW, Tuddenham D, Williams D (1980) J Chem Commun 691; Gilchrist TL, Tuddenham D, McCague R, Moody CJ, Rees CW (1981) Chem Commun 657; Lidert Z, Rees CW (1982) Chem Commun 499; McCague R, Moody CJ, Rees CW (1982) Chem Commun 497; Gilchrist TL, Rees CW, Tuddenham D (1983) J Chem Soc Perkin Trans. 1:83; McCague R, Moody CJ, Rees CW, Williams D (1984) J Chem Commun 909
King RA, Crawford D, Stanton JF, Schaefer HF III (1999) J Am Chem Soc 121:10788; Price DR, Stanton JF (2002) Org Lett 4:2809
Masamune S, Hojo K, Bigam G, Rabenstein DLJ (1971) Am Chem Soc 93:4966
Masamune S, Dorby N (1972) Acc Chem Res 5:272
Vogel E, Roth HD (1964) Angew Chem 76:145
Bianchi R, Mugnolli A, Simonetta M (1972) Chem Commun 1073
Günther H (1965) Z Naturforsch B 20:948
Kovačević K, Maksić ZB (1974) J Org Chem, 39:539; Maksić ZB, Rubčić A (1977) J Am Chem Soc 99:4233
Maksić ZB, Kovačević K, Vampola M (1981) Z Naturforsch A 36:1196
Schleyer PvR, Jiao H, Sulzbach HM, Schaefer HF III (1996) J Am Chem Soc 118:2093
Despotović I, Eckert-Maksić M, Maksić ZB, Smith DM (2003) J Phys Chem A 107:10396 and references therein
Antol I, Eckert-Maksić M, Lischka H, Maksić ZB (2004) ChemPhysChem 5:975
Toda F, Ohi M (1975) Chem Commun 506
Boese R, Benet-Bucholz J, Stanger A, Tanaka K, Toda F (1999) Chem Commun 319
Vianello R, Maksić ZB (2005) Chem Commun 3412
Frisch MJ et al (2003) Gaussian 03, revision B.03, Gaussian, Inc., Pittsburgh PA
Maksić ZB, Kovačević B (1999) J Chem Soc Perkin Trans 2:2623
Raab V, Gauchenova E, Merkoulov A, Harms K, Sundermeyer J, Kovačević B, Maksić ZB (2005) J Am Chem Soc 1275:15738
Matsuo Y, Takahara K, Nakamura E (2003) Org Lett 5:3181
Ormsby JL, King BT (2004) J Org Chem 69:4287
Lu X, Chen Z (2005) Chem Rev 105:3643 and references therein
Maksić ZB, Barić D, Müller TJ (2006) Phys Chem A 110:10135
Shaik, S, Shurki A, Danovich D, Hiberty PC (2001) Chem Rev 101:1501 and references therein
Jug K, Hiberty PC, Shaik S (2001) Chem Rev 101:1477
Barić D, Kovačević B, Maksić ZB, Müller T (2005) J Phys Chem A 109:10594
Aihara J (1979) Bull Chem Soc Jpn 52:2202
Poater J, Duran M, Solá M, Silvi B (2005) Chem Rev 105:3911
(a) Bader RWF, Stephens ME (1975) J Am Chem Soc, 97:7391; (b) Fradera X, Austen MA, Bader R WF (1999) J Phys Chem A 103:304; (c) Fradera X, Poater J, Simon S, Duran M, Solá M (2002) Theor Chem Acc 108:214
Becke AD, Edgecombe K E (1990) J Chem Phys 92:5397; Savin A, Becke AD, Flad J, Nesper R, Preuss H, Vonschnering HG (1991) Angew Chem Int Ed 30:409; Savin A, Nesper R, Wengert S, Fassler TF (1997) Angew Chem Int Ed 36:1809
Matito E, Feixas F, Solá M (2007) J Mol Struct (Theochem) 811:3
Gutman I, Milun M, Trinajstić NJ (1977) Am Chem Soc 99:1692
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to Professor T. M. Krygowski for his outstanding scientific achievements on the occasion of his 70th birthday.
Rights and permissions
About this article
Cite this article
Vianello, R., Maksić, Z.B. Aromaticity of Rees-type hydrocarbons—a DFT computational study. Struct Chem 18, 821–826 (2007). https://doi.org/10.1007/s11224-007-9233-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-007-9233-3