
Abstract
Stable isotope compositions of biologically cycled elements encode information about the interaction between life and environment. On Earth, geochemical biomarkers have been used to probe the extent, nature, and activity of modern and ancient organisms. However, extracting biological information from stable isotopic compositions requires untangling the interconnected nature of the Earth’s biogeochemical system, and must be viewed through the lens of evolving metabolisms on an evolving planet. In this chapter, we provide an introduction to isotope geobiology and to the geobiological history of Earth. We discuss the isotope biogeochemistry of the biologically essential elements carbon, nitrogen and sulfur, and we summarize their distribution on the modern Earth as an interconnected network of isotopically fractionated reservoirs with contrasting residence times. We show how this framework can be used to explore the evolution of life and environments on the ancient Earth, which is our closest accessible analogue for an extraterrestrial planet.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Living things exist in thermodynamic disequilibrium with their environment. This can be reflected in the stable isotope ratios of materials that are produced by organisms or otherwise impacted by metabolic processes. Isotopic analyses can therefore be used to study life where direct observations are not possible. Isotopic information has been used to understand metabolism on Earth in environments that are otherwise inaccessible in space or time, such as the deep subsurface (e.g., Kotelnikova 2002; Hinrichs et al. 2006) and in the geologic past (e.g., Leavitt et al. 2013). Soon, with the realization of a sample return mission to Mars, isotopic signatures of putative biogenic materials will be an important diagnostic tool in the search for extraterrestrial life (e.g., Franz et al. 2017).
Metabolic processes can broadly be grouped as either anabolic (involving biomass synthesis) or catabolic (purely energy yielding). Both of these are required by all living things on Earth. In life as we know it, all anabolic processes involve carbon. An individual organism can synthesize biomass from existing organic matter (heterotrophy) or from CO2 (autotrophy). Catabolic process meanwhile include all reactions that can be harnessed to provide the energy required to grow, move, and propagate. The energy released—that is not lost as heat—is used to maintain cellular organization and to drive chemical reactions that would be otherwise unfavorable, such as the production of complex organic molecules. Most organisms obtain this energy from their environment by catalyzing systems of net exergonic chemical reactions, which are kinetically inhibited in the absence of life. The important exception to this is photosynthesis, where an additional source of chemical potential energy to the system is generated through the conversion of light energy and which subsequently feeds into a cascade of diverse metabolic activity (see Middelburg 2019, for review). On a planet-wide scale, the ensuing interactions are observable as global biogeochemical cycles. On a sub-cellular scale, enzymes—biology’s catalysts—direct the flow of energy. Information about enzyme structure, localization and regulation within the cell is encoded in an organism’s DNA. Through selective propagation of this information, the efficiency of these catalysts is subject to evolutionary adaptation. Metabolic processes can thus be viewed as sets of stoichiometrically tuned reactions that maximize energy release, biomass synthesis, and propagation over long timescales.
Thermodynamic disequilibrium between the intracellular (/near-cell) and ambient environment is maintained with the energy released from metabolic reactions, and results in spatial heterogeneity. Organisms create microenvironments that are conducive to the production of organic molecules and biominerals (such as calcite, opal, and bone). Organisms also create reactive chemical species that subsequently precipitate as minerals through abiotic processes. Much of the solid material produced at the Earth’s surface at low temperature that makes it into the geologic record is precipitated in these biologically controlled/influenced environments. The isotopic composition of these phases in sediments and sedimentary rocks can thus record the biological processes and environments in which the organisms lived.
In this chapter we discuss the isotopic imprint of life on materials found on Earth. We first describe some general concepts for considering biological isotope fractionations. We then provide summaries of the isotope biogeochemistry of the biologically essential elements carbon, nitrogen and sulfur on the modern Earth and outline the geological evidence for the evolution of the Earth system over the course of its history. Finally, we synthesize the previous sections to discuss how the structures of Earth’s biogeochemical cycles influence their sedimentary isotopic records and speculate on how these cycles and records would differ in the absence of life on Earth.
1.1 Isotope Notation
Carbon, nitrogen and sulfur stable isotope fractionations in natural environments are modest. It is therefore useful to express isotope ratios in terms of deviations from a reference ratio. In this chapter we use \(\delta \) notation, defined as follows:
where
and where \(\mathrm{[^{r}E]}\) = the concentration of the rare isotope of element, E, and of mass r, \(\mathrm{[^{c}E]}\) is the concentration of the common isotope of element, E, and of mass c. In the isotope systems considered here (\({\frac{{}^{13}\text{C}}{{}^{12}\text{C}}}, {\frac{{}^{15}\text{N}}{{}^{14}\text{N}}}\), and \({\frac{{}^{34}\text{S}}{{}^{32}\text{S}}}\)), all rare isotopes are heavier than their common counterparts, so more positive isotopic compositions are more enriched in the heavy isotope. The standard for carbon is the Vienna Pee Dee Belemnite (VPDB), for nitrogen it is the modern atmosphere (AIR), and for sulfur it is the Vienna Canyon Diablo Troilite (VCDT). Here and elsewhere, \(\delta \) values are expressed in permil (\(\permil \); i.e., \(\delta \times 1000\)).
Differences in stable isotopic ratios of substrates (S) and products (P) are given by the fractionation factor (\(\alpha _{\mathrm{P-S}}\)):
The equivalent of \(\alpha _{\mathrm{P-S}}\), in permil fractionation is \(\varepsilon _{\mathrm{P-S}}\), defined as follows:
where \(\Delta \) is an approximation for \(\varepsilon \), valid when \(\alpha \) is close to unity. According to this definition, reactions associated with a positive fractionation factor result in a product that is enriched in the rare isotope compared with the substrate; negative fractionation factors correspond to depleted product. As with \(\delta \) values, \(\varepsilon \) values are expressed in permil (\(\permil \); i.e., \(\varepsilon \times 1000\)).
1.2 Factors Affecting Isotopic Fractionations
Enzymatically catalyzed chemical reactions are often associated with (relatively) large isotope effects, whereby a reaction product is enriched or depleted in the rare isotope of a given element compared with the substrate by tens of permil. Isotopic fractionations are typically categorized as: i) kinetic—where a unidirectional reaction occurs at different rates for compounds containing different isotopes; or ii) equilibrium—where a reversible reaction, occurring at equal rates in each direction, results in the partitioning of isotopes largely as a function of their zero point energy in the reactant vs. product (Criss 1999). Heavy isotopes tend to form stronger chemical bonds than their light counterparts. In chemical reactions, a kinetic isotope effect reflects the difference in energy required to break a bond when it involves a heavy vs. a light isotope of the same element. Kinetic isotope effects are usually negative in both the forward and reverse directions for a given chemical reaction, though there are apparent exceptions (e.g., Casciotti 2009). Equilibrium isotope effects reflect the difference between the forward and reverse kinetic effects, and manifest as an enrichment of the heavy isotope in the phase where the bonding environment is most stable. Owing to the relative strength of the bonding partners involved, for a redox reaction allowed to reach equilibrium, the heavy isotope tends to be enriched in the more oxidized species (Schauble 2004). Kinetic and equilibrium isotopic fractionation factors are idealized, end-member scenarios. Enzymatically catalyzed reactions that occur inside organisms are often characterized by an intermediate state, where the net flux in the forward direction is larger than zero (equilibrium) but smaller than the gross flux (kinetic). While chemical kinetics dictate the gross rate of the forward reaction, the thermodynamic driving force of the reaction determines the ratio of the reverse to forward reaction rates. The net reaction rate, therefore, is a combination of both kinetics and thermodynamics (Britton 1965; Beard and Qian 2007; Noor et al. 2013).
Irreversible reactions cause localized chemical and isotopic disequilibrium that impacts the composition of reaction products. The spatial scale over which these departures from equilibrium occur can range from a physical membrane-bound compartment such as a cell or organelle to, e.g., the entire ocean. In every case, for a reaction flux associated with a given fractionation factor (\(\varepsilon \)), changes in the isotopic composition of the compartmental reservoir result in changes in the net fractionation between the reactant entering the compartment and the product leaving it. Utilization within a compartment is the fraction of substrate entering the compartment that is redirected to the reaction flux (Hayes 2001).
Classically, systems are described as ‘open’ when the removal of substrate by reaction is continually replenished from an external source, or ‘closed’ when the system is isolated and there is no replenishment (Hayes 2001). A one-compartment system can only be at steady state when it is ‘open’. Microbial cells are often treated as open systems and characterized by steady state growth due to the relatively high rates of intracellular substrate turnover (or short residence time—see Sect. 1.3). Higher reaction rates inside the cell (perhaps corresponding to higher growth rates) result in greater utilization, while a higher flux of substrate to the cell (perhaps due to higher concentrations of substrate in the environment) results in lower utilization (Farquhar et al. 1982; Rau et al. 1996). It is therefore common for even simple systems to be characterized by a complex isotopic interplay between reaction kinetics, reversibility, and reservoir effects through substrate and product concentrations. Closed system reservoir effects can result in very large drifts in isotopic composition when not at steady state, due to continued reaction with no replenishment of substrate.
Due to these complexities, the same reaction can express different isotopic fractionations depending on the conditions under which it occurs. Such variations are exacerbated in biological systems, where physiological and environmental factors can influence the reversibility, openness, and utilization of a given reaction. In Table 1, we summarize the isotopic fractionations associated with key biological and abiotic reactions in the cycles of carbon, nitrogen, and sulfur on Earth. These reactions, and their roles in the relevant biogeochemical cycle, are detailed in the following sections. We emphasize that no single number characterizes the isotopic fractionation of any of the reactions listed; rather, reactions express characteristic ranges of isotopic fractionations, and variations within these ranges result from the various factors described above. As expected, variability is greater for biologically catalyzed reactions.
1.3 Sedimentary Isotope Records in the Context of Reservoir Residence Times
An isotope record is a sequence of isotopic measurements made on the same sedimentary phase from samples that have been resolved into a geologic time series. Compilations of sedimentary carbon, nitrogen, and sulfur isotope records are shown in Fig. 1. These records are superpositions of multiple features: secular changes, transient events (excursions), and repeating cycles, each with different amplitudes/magnitudes and different characteristic timescales ranging from shorter than years to billions of years. Such variations are often expressions of the changes in the makeup of the respective elemental cycle at any one time. The carbon, nitrogen and sulfur cycles on Earth comprise pools of varying sizes, and are interconnected by fluxes associated with isotopic fractionations. While the isotopic fractionations and magnitudes of these fluxes set the steady state isotopic composition of each pool, the time it takes for any given pool’s isotopic composition to respond to a change in input or output is approximately the residence time of the element in that pool. Changes in the composition of any given pool are therefore dynamic on approximately the residence time of the pool, which is defined as the ratio of the size of pool (in moles) to the flux through the pool at steady state (in moles per year). Compared with a timescale of interest (e.g., the temporal duration of an isotope excursion), pools with longer residence times change little and can be assumed to be approximately constant, while pools with shorter residence times change rapidly and can be assumed to be approximately in steady state.

The isotopic records of C, N, and S cycling on earth: Compilation of isotope compositions of biomolecules and minerals through time. a) Carbon data from Krissansen-Totton et al. (2015), Zachos et al. (2001) and references therein. b) Nitrogen data from Yang et al. (2019) and references therein. c) Sulfur data from Fike et al. (2015), Crockford et al. (2019), Canfield (2004), and references therein
Most pools do not have geologic records. In the carbon cycle (Sect. 2, below) sedimentary carbonate carbon and organic carbon are the principal archives of past conditions (Fig. 1). Attempts to reconstruct the makeup of a past state of the carbon cycle (i.e., the size and isotopic composition of every pool, plus the size and isotopic fractionation of the fluxes between them) are typically grounded in two observations: the \(\delta ^{13}\)C values of carbonate and organic carbon. Such an exercise is clearly underconstrained. Considering the Earth’s biogeochemical system in this way—as separated into steady state, dynamic, and constant components at a given timescale—provides additional constraints on the system. The relevant timescale determines which pools need be treated dynamically (i.e., where the composition of the pool depends on its previous state) and which can be assumed to be in steady state or constant. Moreover, a clue as to the mechanism behind a specific feature in an isotopic time series lies in the timescale of that feature—e.g., the onset and decay time of an isotope excursion or the period of a repeating cycle. The characteristic timescale of a feature may be imposed by an external ‘forcing’ on the biogeochemical cycle (such as temperature), by an independent process (such as biological evolution) driving a secular change in the factors controlling fluxes or isotopic fractionations between these pools, or by dynamics inherited from a pool with the appropriate residence time. In the following sections, we develop graphical spaces relating isotopic compositions and residence times for the C, N and S pools on the modern Earth (Figs. 2, 3, and 4). We use these spaces as a framework for interpreting the evolving imprint of biology on the isotope chronologies shown above (Fig. 1) and, thus, on Earth’s biogeochemical cycles though time.

a) Quantitative depiction of the exogenic pre-industrial carbon cycle: Carbon reservoirs (circles) are drawn proportional to their size and colored according to their weighted average carbon oxidation state. To highlight differences among surface and near-surface reservoirs, deep mantle and core carbon reservoirs are omitted. Reservoir locations are plotted according to their average \(\delta ^{13}\)C composition versus turnover time (Sec. 1.3). Widths of fluxes (arrows) are proportional to their magnitude. Estimates of flux sizes, reservoir sizes, and isotopic compositions from Falkowski et al. (2000), Ciais et al. (2013), Bar-On et al. (2018). \(\delta ^{13}\)C values shown are estimates of the mean values of these pools today; deviations from these mean \(\delta ^{13}\)C values can be large (especially among organic species; \(\sim 10-20 \permil \)), and are approximated by the ranges in the isotopic fractionations of the relevant reactions shown in Table 1. b) Timescales of isotopic features that may be recorded by various sets of reservoirs in the carbon cycle

A quantitative depiction of the modern (pre-Anthropogenic) nitrogen cycle. Sizes and positions of components are determined in a manner analogous to those in Fig. 2. Nitrogen reservoir sizes, flux sizes, and isotope compositions compiled from Galloway et al. (2004), Seitzinger et al. (2005), Gruber (2008), Sigman et al. (2009), Fowler et al. (2013), Johnson and Goldblatt (2015). \(\delta ^{15}\)N values of pools are estimates of mean modern values; the ranges in the \(\delta ^{15}\)N values of some pools are much larger due to the ranges in the relevant isotopic fractionations expressed, as listed in Table 1. All fluxes (and thus, residence times) have uncertainties in the range of \(\pm 20-50\%\)

A quantitative depiction of the modern (pre-industrial) sulfur cycle: construction of diagram is analogous to Figs. 2, 3. Sulfur reservoir and flux sizes after Canfield (2004), Palme and O’Neill (2013), Brimblecombe (2013), Moran and Durham (2019). For simplicity, less-abundant atmospheric species are omitted, and the marine organic sulfur network—which constitutes its own cycle of metabolites and fluxes (Amrani 2014; Moran and Durham 2019)—is subsumed into a single reservoir. Briefly, sulfur also exists as organic sulfur in sediments, deposited as a constituent of primary organic matter, or fixed via sulfurization reactions between sedimentary organic matter and sulfide in porefluids. This sedimentary organic sulfur pool is of order 0.1–1 Pmol S (for an S:C ratio of around 1–4%), and its isotopic composition is generally close to that of other reduced sulfur species (Raven et al. 2018, 2016; Sinninghe Damsté et al. 1998; Werne et al. 2000). Ordinate of each reservoir is an estimate of its modern mean \(\delta ^{34}\)S value; deviations from these mean \(\delta ^{34}\)S values are common, due to the variability in the underlying isotopic fractionations (Table 1)
2 Carbon
Carbon (C) is central to life on Earth. It forms the chemical backbone of all organic molecules. Transitions between its wide range of oxidation states is ultimately the means by which energy from the sun drives most metabolisms on Earth. Carbon from the mantle enters Earth’s surface environment through volcanic activity mostly in its most oxidized form, CO2 (Ciais et al. 2013). The reduction of CO2 to organic carbon requires a strong reducing agent. On the modern surface Earth, oxygenic photosynthesis dominates primary production (CO2 reduction to organic carbon; also called CO2 fixation). This conversion is powered by sunlight, with water as the terminal electron donor, and results in the production of oxygen. CO2 reduction can also occur via anoxygenic photosynthesis, which is also powered by sunlight but uses sulfide, molecular hydrogen or reduced iron as a terminal electron donor (Blankenship et al. 2004), or via chemoautotrophic processes like methanogenesis, which couples the oxidation of molecular hydrogen to the reduction of CO2 (Demirel and Scherer 2008). These strongly reducing terminal electron donors are often hydrothermal in origin (McCollom and Shock 1997). Molecular hydrogen, in particular, can be also generated through radiolysis of water in sediments (Blair et al. 2007), through serpentenization reactions (Russell et al. 2010), or by organisms from fermentative and hydrolytic metabolisms. Once reduced, organic carbon can be used to generate energy by coupling its oxidation to the reduction of a wide range of oxidized compounds including molecular oxygen, sulfate, nitrate and oxidized metals (e.g., Jørgensen 1982; Thamdrup 2000; Achtnich et al. 1995; Straub et al. 1996; Vargas et al. 1998, and see Sects. 3 and 4 in this Chapter). All known life needs carbon to build biomass, and most life (including all eukaryotes) also uses carbon-based redox chemistry to generate energy.
Two stable isotopes of carbon exist on Earth—12C (six protons, six neutrons) and 13C (six protons, seven neutrons)—in a ratio of ∼90:1. The extra mass of 13C causes it to react more sluggishly and bond more stably. As carbon is cycled between various phases, these differences fractionate carbon isotopes. Fractionations manifest as small differences in the isotopic ratio of different carbon pools, typically on the order of a few to tens of ‰. As the carbon isotope compositions of different reservoirs within the Earth-system depend on their relative sizes, the carbon isotopic compositions of ancient materials are sensitive to the sizes of these reservoirs in the past.
Earth’s 350 million petamol (Pmol = \(10^{15}\) mol) carbon inventory is distributed between the metallic core (\(\sim 90 \%\)), the mantle (\(\sim 8-9 \%\)) and the crust and surface pools (\(\sim 1.5 \%\)) (McDonough and Sun 1995) (Fig. 2). The atmosphere, biosphere and oceans, where we can observe the interactions between life and the carbon cycle, together house only \(0.001 \%\) of Earth’s carbon. The atmospheric carbon pool (\(\sim 50\) Pmol C on the pre-industrial Holocene Earth) exchanges with the other surface reservoirs, in which we include the surface ocean (\(\sim 80\) Pmol C), biosphere (\(\sim 50\) Pmol C), and soils (\(\sim 160\) Pmol C), on a timescale of years to decades (Ciais et al. 2013). The pool of carbon in the deep ocean, which is an order of magnitude larger than the combined surface pools (at around 3100 Pmol C) has a residence time of around 1000 years (Ciais et al. 2013). Carbonate in marine surface sediments in contact with seawater can precipitate or dissolve in response to changes in deep ocean seawater chemistry. Due to these feedbacks the size of this pool is difficult to constrain (nominally \(\sim 140\) Pmol C; Ciais et al. 2013). Given the long residence time of the deep ocean, any change to the ocean–sediment system takes over 10,000 years to reach a new steady state (Sigman and Boyle 2000). Earth’s exosphere comprises the atmosphere, terrestrial biosphere, the ocean, and the reactive surface sediments (note that this usage differs from the other common usage of ‘exosphere’ to describe the outermost layer of a planet’s atmosphere). The combined exospheric pool has a residence time of less than 105 years (Fig. 2), and is thus approximately at steady state on million year timescales, but is dynamic on timescales of < 1 Myr (Sect. 1.3; Walker et al. 1981). The amount of carbon stored as carbonate rocks, terrestrial sediments, and as fossilised organic material in the continental and oceanic crust exceeds that of the deep ocean by 3 orders of magnitude (Ciais et al. 2013). Anthropogenic CO2 emissions through the burning of fossil fuels are unidirectionally short-circuiting the usually slow exchange between the vast pool of buried organic carbon and the atmosphere.
In this section, we describe the phases of carbon—and mechanisms for transitions between them—that comprise Earth’s biogeochemical carbon cycle (see Falkowski et al. 2000; Hayes and Waldbauer 2006; Ciais et al. 2013, for reviews). Our focus is the mechanisms by which biology catalyzes transitions between phases of carbon, the isotopic fractionations associated with these changes, and the timescales over which these changes manifest in the geologic record. While there is a rich literature regarding the systematics and applications of radiocarbon (14C), here we exclusively discuss the stable isotopes of carbon.
2.1 Organic Carbon
The term ‘organic carbon’ describes carbon in compounds containing at least one C–H bond. Carbon atoms exist in a wide variety of redox states (\(+4\) to −4) depending on the electronegativity of available bonding partners. Some of the simplest organic molecules, such as methane (CH4), formaldehyde (CH2O) and methanol (CH3OH), can form spontaneously by bombardment of high energy particles and are found in the interstellar medium and in the tails of comets (Snyder et al. 1969). More complex organic molecules are built upon chains and rings of carbon-carbon bonds. These ‘carbon backbones’ are versatile; the specific configuration of hetero-atoms (typically O, N, S, or P) and functional groups built off of the carbon backbone determine an organic molecule’s physical properties (e.g., polarity, reactivity, solubility) and biologic function. For instance, the arrangement of hydrophobic and hydrophilic functional groups on opposite ends of lipids allows cells to build semi-permeable lipid membranes that selectively exclude the outside world. The rings of CH(OH) units of water-soluble carbohydrates are an efficient, flexible way to store and transport energy that can be polymerized to make larger, stabler compounds like cellulose and starch. The high density of functional groups in amino acids (each containing at least one amine (NH2) and carboxyl (COOH) group) results in reactive molecules that can be linked into the proteins that catalyze reactions, replicate DNA, and perform tasks central to life.
The total carbon biomass of all living organisms on Earth is about 44 Pmol (Bar-On et al. 2018). Of this, about one third comprises metabolically active, high-turnover compounds in plants, animals, and vigorously-growing microorganisms. The remainder is in static tissues, such as the woody tissues of trees and deep biosphere microbes with exceptionally slow (superannual) turnover times (Bar-On et al. 2018; Magnabosco et al. 2018). The components of dead organisms are mostly respired (oxidized to CO2) or assimilated by heterotrophs. Organic carbon compounds that escape consumption, either because they are difficult to degrade (e.g., lipids, lignin) or rapidly buried in inhospitable environments, become sequestered in soils and sediments. About 160 Pmol of organic carbon from recently-dead plants resides in soils (Batjes 2014), with an additional \(\sim 160\) Pmol of older carbon preserved in permafrost and anoxic wetlands (Bridgham et al. 2006; Tamocai et al. 2009). During further sedimentation and lithification, surviving organic compounds are transformed by polymerization and condensation reactions into recalcitrant, insoluble agglomerations of organic matter collectively known as kerogen. Globally, around \(0.1-0.4 \%\) of organic carbon that is cycled by the biosphere escapes remineralization and is buried in sediments (Berner and Canfield 1989; Middelburg 2019). Nonetheless, due to continued burial and sedimentation over billions of years, kerogen disseminated in the crust constitutes the largest organic carbon reservoir on Earth (\(\sim 1.25\times 10^{6}\) Pmol; Tissot and Welte 1984; Falkowski et al. 2000).
2.2 Methane
Methane (CH4), the smallest organic molecule, plays an especially important role in the carbon cycle. As the most reduced form of carbon, it is an energetically-favorable endmember of abiotic and biotic redox reactions. In strongly reducing environments, organic carbon decomposition, microbial fermentation, and Fischer-Troph-type (FTT) reactions produce abundant methane (Reeburgh 2007). In relatively oxidizing environments, methane is oxidized either by aerobic methanotrophs or by microbial consortia performing anaerobic oxidation of methane (AOM) (Hinrichs et al. 2000). Despite its low abundance and short residence time in the modern atmosphere (\(\sim 700\) ppb in the pre-industrial era and less than 10 years respectively; Ciais et al. 2013; Prather et al. 2012), methane is a potent greenhouse gas, so methane that escapes oxidation contributes to the regulation of Earth’s climate.
The sizes of the environmental sources and sinks of atmospheric methane are difficult to constrain, in no small part because they are dwarfed by anthropogenic methane emissions in the Industrial era (Kirschke et al. 2013; Schwietzke et al. 2016; Saunois et al. 2020). Modern abiotic methane fluxes are small compared to those involving biology, but were likely important for fertilizing early Earth with a viable reduced energy source in the pre-biotic world (Russell et al. 2010). Together, marine hydrothermal systems and terrestrial geothermal/volcanic areas contribute ∼0.004 Pmol/yr to the atmospheric methane budget (Etiope et al. 2008). Biotic fluxes are larger; obligately anaerobic archaea produce methane by either completely reducing CO2 with H2 or by adding a fourth hydrogen atom to a methyl-containing organic molecule (usually acetate, but also methanol, methylsulfides, or methylamines) produced by the fermentation of larger organic molecules (Ferry 2010). Worldwide, microbial methanogenesis in wetlands, peatlands, and lakes emits about 0.02 Pmol of methane to the atmosphere each year (Kirschke et al. 2013). These geologic methane fluxes would be larger were it not for microbial degradation; it is estimated that more than 90 % of methane produced in marine sediments is oxidized by cooperative consortia of methanotrophic archaea and sulfate reducing bacteria before it escapes the sediment column (Orphan et al. 2002; Knittel and Boetius 2009; Reeburgh 2007). Aerobic bacteria in soils oxidize and consume about 0.002 Pmol of atmospheric methane per year (Spahni et al. 2011). An additional methane reservoir of uncertain—but potentially significant—size exists in methane hydrates (a solid, high-pressure, cryslalline phase of methane bound to water) in ocean basins (Archer et al. 2009). The methane hydrate reservoir may be as large as 160 Pmol, and is susceptible to release in response to global warming (Archer et al. 2009). Such a feedback has been invoked as a major contributor to prominent disturbances to the ancient carbon cycle (Dickens 2003).
2.3 Carbon Isotopes in Organic Carbon and Methane
A range of factors control the carbon isotopic composition of organic matter. Broadly these factors fall into the following categories: i) processes dictating the isotopic composition of the primary organic matter, including environmental conditions and fixation pathway; ii) metabolic processes downstream of fixation that cause divergences in the isotopic compositions of various classes of organic compounds such as lipids, proteins and carbohydrates; and iii) heterotrophy and metabolic recycling, which lead to further alteration of the chemical and isotopic makeup of organic matter, largely through fermentation and preferential oxidation of certain classes of compounds.
2.3.1 Carbon Fixation Pathways
Almost all organic carbon on Earth is fixed through the Calvin cycle. The fixation reaction step, where an inorganic CO2 molecule is bound, is catalysed by the enzyme ribulose-1,5-biphosphate oxygenase-carboxylase (RuBisCO). This fixation reaction is associated with a large negative fractionation of carbon isotopes (13\(\varepsilon _{f}\)), causing the primary organic matter to be 13C-depleted relative to the source CO2 (Farquhar et al. 1982). Although once thought to carry a universal value of around −25 to \(-28 \permil \) (e.g., Hayes 2001), the absolute magnitude of 13\(\varepsilon _{f}\) has recently been shown to vary significantly between RuBisCOs extracted from different species of cyanobacteria, eukaryotic algae and higher plants (−11.1 to \(\sim -30 \hbox{\fontencoding {U}\fontfamily {wasy}\selectfont \char 104}\); Boller et al. 2011, 2015; Tcherkez et al. 2006). In addition to the enzymatic fractionation factor, a multitude of factors influence the net (i.e., apparent) fractionation between CO2 in the ambient environment and the isotopic composition of biomass produced (13\(\varepsilon _{p}\)).
Reservoir effects tend to dampen the expression of the fractionation by the RuBisCO enzyme at higher utilization, so any process or infrastructure that increases separation of the substrate pool from the site of fixation will result in less-13C-depleted organic products. In photosynthesizing microorganisms, physiological and environmental parameters that determine substrate availability—such as cell size, shape, and growth rate (Rau et al. 1996; Popp et al. 1998)—influence the magnitude of fractionation that is expressed (see: Wilkes and Pearson 2019; McClelland et al. 2017, for reviews). In plants that use the C3 photosynthetic pathway, 13\(\varepsilon _{p}\) is sensitive to the relative rates of photosynthetic carbon fixation and diffusion of CO2 into the leaf (Farquhar et al. 1989). Plants employing the C4-photosynthetic pathway or Crassulacean acid metabolism (CAM) separate the processes of CO2 uptake and carbon fixation in space or time, so the expressed carbon isotope fractionations of these pathways are smaller than the full fractionation of RuBisCO (\(^{13}\varepsilon _{p} \approx 10 \permil \); Farquhar et al. (1989)).
In addition to the Calvin cycle, at least five other carbon fixation pathways exist on Earth (Berg et al. 2010; Ward and Shih 2019). Of these, the reductive citric acid cycle (rTCA) and the reductive acetyl coenzyme A (Wood-Ljungdahl) pathway contribute most significantly to gross primary productivity today, though their importance has probably varied throughout Earth’s history (Berg et al. 2010; Ward and Shih 2019). A handful of bacterial and archaeal clades fix carbon through the rTCA cycle, most notably green sulfur bacteria. The enzymes that catalyze the rTCA cycle are O2-sensitive, limiting the habitat of the reliant organisms to sub-oxic or anoxic environments (Berg et al. 2010). When using H2S, S0, or H2 as an electron donor for anoxygenic photosynthesis, these organisms express a relatively small fractionation against 13C in the range of −2 to \(-12 \permil \) (Hayes 2001). The flux of organic carbon fixed by the rTCA pathway today is estimated at \(\sim 0.08\) Pmol/yr (about two orders of magnitude smaller than the Calvin cycle flux), but was probably more abundant in past periods when atmospheric O2 levels were lower (Brocks et al. 2005; Ragsdale 2018). Due to its moderate carbon isotope fractionation, finding unambiguous carbon isotope evidence for ancient rTCA activity is problematic (Ward et al. 2019).
The Wood-Ljungdahl pathway is a strictly anaerobic carbon assimilation mechanism that enables the existence of autotrophic life in some of the planet’s most extreme environments. Where environmental conditions make it possible (anoxic, H2-replete, with abundant trace metals), the pathway is the least energetically costly of all carbon fixation schemes, and is used by (most notably) acetogens and methanogens to produce these environmentally significant biomolecules (Berg et al. 2010; Ward and Shih 2019). The pathway imparts exceptionally large carbon isotope fractionations; methanogenic archaea typically assimilate carbon with an 13\(\varepsilon \) of −30 to \(-36 \permil \) (Hayes 2001); the 13\(\varepsilon \) of fixation by acetogenic bacteria can be as large as \(-68 \permil \) (Blaser et al. 2013). The methane released by methanogens is also significantly depleted in \(\delta ^{13}\)C. Depending on the substrate (CO2, acetate, or another methyl-bearing compound), and the specific species, fractionations from substrate to methane range from −20 to \(-80 \permil \) (Summons et al. 1998). The modern carbon fixation flux from this pathway is small (\(<0.01\) Pmol/yr), but its distinctive carbon isotope signature allows its presence to be recognized in past environments (Ward and Shih 2019). The widespread occurrence of kerogens with low-\(\delta ^{13}\)C values in late Archean sedimentary rocks, for instance, has been used to argue for the proliferation of reductive acetyl-CoA pathway at the time (Hayes and Waldbauer 2006; Slotznick and Fischer 2016).
2.3.2 Downstream Carbon Isotope Discrimination
Once fixed, primary organic carbon is converted into different classes of biochemicals including lipids, proteins, carbohydrates, and nucleic acids. Biosynthetic networks involve myriad branch-points, recycling processes, and enzymatic reactions with varying degrees of utilization and reversibility. These mechanisms all influence the carbon isotope compositions of metabolites and thus preclude accurate de novo predictions of the carbon isotope composition of any specific metabolic end-product (Hayes 2001). Broad-brush relationships between compound classes, however, are well established: within the same autotrophic organism utilizing the Calvin cycle, the \(\delta ^{13}\)C values of carbohydrates are canonically about \(1 \permil \) higher than proteins and nucleic acids, and about \(6 \permil \) higher than lipids (Blair et al. 1985; Hayes 2001). In general, this ordering is thought of as a function of metabolic path length; the larger number of metabolic steps between primary photosynthate and membrane lipids results in lipids that are relatively depleted in 13C as compared to proteins and carbohydrates, which are metabolically ‘closer’ to primary photosynthate (Hayes 2001). In practice, however, specific metabolic steps—often, branch points—have outsized effects on the \(\delta ^{13}\)C values of metabolic products. For instance, the 13C depletion of acetogenic lipids appears to arise specifically from the kinetic isotope effect associated with decarboxylation of pyruvate by pyruvate dehydrogenase, and the exact \(\delta ^{13}\)C value of such lipids is modulated by changes in the various metabolic demands for pyruvate (DeNiro and Epstein 1977; Monson and Hayes 1982). Environmental conditions and metabolic needs can also overwhelm the isotopic imprint of metabolic networks, such that the same set of biomolecules synthesized by the same organism will retain a different isotopic ordering depending on the conditions under which they are formed (e.g., Zhang et al. 2009; Bird et al. 2019). Some autotrophs, such as green sulfur bacteria, can even exhibit ‘inverse’ carbon isotope ordering (\(\delta ^{13}\)C\(_{\text{lipids}}\) > \(\delta ^{13}\)C\(_{\text{bulk biomass}}\)) due to utilization of the rTCA cycle for lipid synthesis (Van Der Meer et al. 1998). At the scale of the ocean, differences in the preservation potential of compounds from various community factions (e.g., prokaryotes vs. eukaryokes, autotrophs vs. heterotrophs) can overprint native relationships between compound classes to the extent that inverse carbon isotope ordering exists in sedimentary records from throughout the Phanerozoic (Close et al. 2011).
2.3.3 Heterotrophy
Heterotrophy alters the isotopic composition of fixed carbon in two ways: i) CO2 respired by animals tends to have a lower \(\delta ^{13}\)C value than the food they eat (DeNiro and Epstein 1978). By mass balance, the biomass of animals is therefore enriched in 13C compared to the biomass they consume by about \(1 \permil \) (DeNiro and Epstein 1978); heterotrophic microorganisms can also express this behavior (Mahmoudi et al. 2017). On an ecological scale, carbon is isotopically distilled over multiple trophic levels, and has been used as a proxy for position within the food chain. ii) Heterotrophic organisms preferentially consume some biomass components (i.e., proteins, carbohydrates) while leaving tough-to-metabolize compounds behind. In the oceans, the organic carbon that survives biodegradation and is buried in sediments is predominantly in the form of lipids and recalcitrant polymers (Tissot and Welte 1984). On land, the preservation potential of woody tissues exceed that of plant proteins and polysaccharides, leading to compositional bias in the material preserved in anoxic waters to become kerogen and coal (Tissot and Welte 1984). Due to the different \(\delta ^{13}\)C values of these different compound classes, selective degradation can, in principle, fractionate the carbon isotope composition of biomass by a few permil. In practice, variability in the 13C content of biomass and sedimentary organic matter (Table 1) tends to outweigh such compound-based shifts in bulk \(\delta ^{13}\)C value.
2.4 Inorganic Carbon System
Whether in bodies of water, inside cells, or in a thin film of water on the surface of a cell, virtually all biologically mediated reactions occur in the aqueous phase. When CO2 dissolves in water it reacts to form carbonic acid (H2CO3) and subsequently its conjugate bases, bicarbonate (\(\text{HCO}_{3}{}^{-}\)) and carbonate (\(\mbox{CO}_{3}{}^{2-}\)) ions. The two acid dissociation constants (pKas) of carbonic acid straddle the pH of most biologically relevant media such as seawater or the cellular cytosol. As a result, carbonate speciation in these systems is sensitive to pH, and, to a lesser extent, temperature and salinity (via the effect of these environmental parameters on the pKas). In modern seawater the total concentration of dissolved inorganic carbon (DIC) is around 2 mM, and CO2 (CO2 (aq) + carbonic acid), \(\mbox{CO}_{3}{}^{2-}\), and \(\text{HCO}_{3}{}^{-}\) exist in an approximate ratio of 1:10:100. The pH, and thus the speciation of DIC, and other weak acids in solution, is controlled by the ratio of DIC to total concentration of conjugate bases of weak acids in solution (known as alkalinity; reviewed extensively in: Zeebe and Wolf-Gladrow 2001; Dickson and Goyet 1994). Seawater on the modern Earth is buffered dominantly by carbonic acid. There is a complex interplay between the system of reactions internal to the carbonate system, which are constantly relaxing towards a state of equilibrium, and any process involving a specific species (e.g., carbonate precipitation), which push the system out of equilibrium. Indeed, biological processes are often sufficiently fast that disequilibrium and interconversion kinetics need be considered. The interconversion of CO2 and \(\text{HCO}_{3}{}^{-}\) is slow, but in some biological systems is catalyzed by an enzyme called carbonic anhydrase, with important isotopic implications (e.g., Chen et al. 2018). Since CO2 is the only gaseous phase of the carbonate system, equilibration between a body of water and a gas requires all carbon to pass through the form CO2. Therefore, as pH increases, which results in an increase in the fraction of DIC in forms other than CO2, the equilibration timescale between the aqueous and gas phases also increases.
Carbon isotopes are fractionated among the different species of DIC. At equilibrium, \(\text{HCO}_{3}{}^{-}\) is the most 13C-enriched species, followed by \(\mbox{CO}_{3}{}^{2-}\) and then CO2(aq). The sizes of these decrease with increasing temperature. In the range 0–35 ∘C, \(\mbox{CO}_{3}{}^{2-}\) and CO2(aq) are depleted in 13C relative to \(\text{HCO}_{3}{}^{-}\) by \(3-1 \permil \) and \(12-8 \permil \), respectively. CO2(g) is consistently enriched compared to CO2(aq) by about \(1 \permil \). Since the isotopes of carbon are distributed among all DIC species, the isotopic composition of each species depends on its relative abundance (see: Zeebe and Wolf-Gladrow 2001, for further reading). Various biogeochemically important reactions involve specific DIC species, so changes in these fluxes influence both the inorganic carbon speciation and isotope composition of the aqueous medium, on scales from the cellular cytosol to the global ocean. When the DIC system is disturbed by a rapid change, disequilibrium effects come into play (Sade and Halevy 2017; Zeebe 2014; Zeebe et al. 1999b). The rate of exchange between \(\text{HCO}_{3}{}^{-}\) and \(\mbox{CO}_{3}{}^{2-}\) is extremely rapid, and in the majority of cases these species can be assumed to comprise a single equilibrated pool. The comparatively sluggish interconversion of CO2 and \(\text{HCO}_{3}{}^{-}\) can lead to isotopic disequilibrium in a variety of settings, including in the vicinity of phytoplankton (e.g., Rau et al. 1996), zooplankton (e.g., Zeebe et al. 1999a) and speleothems (e.g., Mickler et al. 2006).
2.5 Dynamics and Evolution of Earth’s Carbon Cycle
On timescales of longer than a million years, Earth’s exospheric carbon pools are approximately at steady state (Fig. 2). The principal inputs to the exogenic carbon cycle are CO2 emissions from the crust and upper mantle, mostly from mid-ocean ridge degassing, arc volcanism, and metamorphism (Lee et al. 2019), and the outputs are the permanent burial and sedimentation (and ultimately subduction) of carbonate and organic carbon (Fig. 2). Once it enters the exosphere, CO2 is partitioned with respect to chemical form, oxidation state, and isotopic composition into a number of interconnected reservoirs (Fig. 2). Organic and inorganic carbon move in parallel from more reactive surface reservoirs (e.g., the shallow ocean, the terrestrial biosphere) to progressively less reactive reservoirs with longer residence times (e.g., the deep ocean and sediments). Along the way, various processes interconvert organic and inorganic carbon within the exosphere before their ultimate removal (Fig. 2). The input of volcanic CO2 to the exosphere is approximately constant today, but varies on multi-million year timescales with the cycles of continental rifting and collision. This has been widely invoked as the long-term driver of Earth’s climatic state (Lee et al. 2016; McKenzie et al. 2016), but is an area of active debate (Macdonald et al. 2019; Jagoutz et al. 2016). On sufficiently long timescales, the silicate weathering feedback balances variations in the size of CO2 inputs (Walker et al. 1981), but whether variations in Earth’s climatic states are primarily source-driven or sink-driven remains uncertain (Lee et al. 2019).
Carbon appears in the geologic record in the form of carbonate and organic carbon in sediments and rocks. Most Phanerozoic carbonate sediment is biogenic in origin, and is precipitated in pelagic environments (dominantly by coccolithophores and planktonic foraminifera) and both deep and shallow benthic environments (mostly by benthic foraminifera, corals and molluscs). The carbon isotopic compositions of these carbonates represent that of DIC in the water in which they formed, with small offsets from equilibrium (typically \(2-5 \permil \)) known as “vital effects” (Zeebe et al. 1999a; McClelland et al. 2017; Chen et al. 2018). Unlike carbonate, organic carbon is more reduced than the volcanic influx. Any organic matter that escapes remineralization and is permanently buried therefore renders the exosphere increasingly oxidized. On long timescales (\(>10^{6}\) years), the fluxes of carbon entering and leaving the exosphere from the lithosphere are widely assumed to be maintained at steady state largely through the negative feedback of silicate weathering (Walker et al. 1981), although some models suggest that the surface-sediment carbon cycle may have a response time of as long as 10-100 million years (Rothman et al. 2003). Nonetheless, on sufficiently long timescales the weighted mean isotopic composition of the sequestered carbonates and organic carbon must equal that of the volcanic influx. Because organic matter is depleted in 13C compared to carbonate, an increase in the fraction of carbon buried as organic matter (\(F_{\text{org}}\)) results in an increase in the isotopic enrichment of the buried carbonate.
Under the assumptions of i) steady state in the carbon cycle, ii) the applicability of a simple mass balance, and iii) the invariance of the isotopic composition of the volcanic influx through time (which may not be a good assumption, c.f., Mason et al. 2017), the \(\delta ^{13}\)C values of carbonate (\(\delta ^{13}\)C\(_{\text{carb}}\)) and organic carbon (\(\delta ^{13}\)C\(_{\text{org}}\)) in ancient rocks have been widely used to constrain \(F_{\text{org}}\), and thus the oxidation state of the exosphere and (more tangibly) the concentration of oxygen in the atmosphere (e.g., Hayes and Waldbauer 2006; Krissansen-Totton et al. 2015). As reviewed in the preceding sections, however, the evolving picture is more nuanced: the \(\delta ^{13}\)C value of carbonate depends on variables influencing DIC speciation, such as temperature, pH, and pCO2 (Sect. 2.4), while \(\delta ^{13}\)C\(_{\text{org}}\) depends on the magnitude of the carbon fixation fractionation that is expressed (Sect. 2.3). Both records may be strongly influenced by local effects (Swart 2008), and overprinted by post-depositional effects such as selective organic carbon compound degradation and authigenic carbonate precipitation (Banner and Hanson 1990; Des Marais 2001; Schrag et al. 2013). Use of a simple mass balance to reconstruct changes in organic carbon burial flux is only strictly valid at steady state (Hayes and Waldbauer 2006). Under conditions where the timescale of a perturbation is shorter than the residence time of the combined exospheric carbon pool, the system will evolve dynamically (e.g., Rothman et al. 2003). Given these complexities here we avoid describing quantitative changes in \(F_{\text{org}}\). Instead, we emphasize the biogeochemical advances that best explain the qualitative changes observed.
2.5.1 Precambrian Carbon Cycling
Prior to the evolution of oxygenic photosynthesis, life was limited by the availability of viable terminal electron donors (Des Marais 2001; Ward and Shih 2019). In the Archean eon the most abundant reduced chemical species, which thus limited early primary production, was likely H2 emitted from volcanic vents (Des Marais 2001). Recent estimates indicate a steady state atmospheric pH2 of less than 0.01 bar (Kadoya and Catling 2019; Catling and Zahnle 2020). Even if seafloor spreading were more vigorous in the Archean due to higher radionuclide heat flux, and could have sustained H2 emissions at an order of magnitude larger than today, the dependence on H2 would have placed an upper bound on primary productivity of \(\sim 0.01\) Pmol/yr (<0.1 % of modern GPP) (Des Marais 2001). This remains an active area of debate, with arguments made for stricter (Korenaga 2006) or more relaxed (Höink et al. 2013) limits on H2. Regardless, due to imperfect utilization of volcanic reduced species, and with habitats likely limited to the photic zone (Tice and Lowe 2004), Archean primary productivity was likely orders of magnitude lower than this upper bound (Ward and Shih 2019; Canfield et al. 2006).
The \(\delta ^{13}\)C of early Archean organic matter is similar to that of the Phanerozoic (\(\delta ^{13}\)C\(_{\text{org}}\) \(\approx -28 \permil \)), but with greater variability (\(\approx -50\) to \(-10 \permil \); Fig. 1). Carbonate \(\delta ^{13}\)C values were lower, however, by \(2-4 \permil \). It has been argued that both the average \(\varepsilon _{p}\) of primary autotrophy and \(F_{\text{org}}\) were lower than today (e.g., Krissansen-Totton et al. 2015). The small \(\varepsilon _{p}\) of the rTCA cycle—canonically an ancient fixation pathway (e.g., Ragsdale 2018)—is compatible with isotopic evidence for a muted primary 13C fractionation in the early Archean, but not uniquely so, given the overlap of this \(\varepsilon _{p}\) with those of other fixation pathways (Ward and Shih 2019). Mean organic carbon \(\delta ^{13}\)C values in the late Archean are lower than the early Archean by about \(10 \permil \); with some values as low as \(-60 \permil \) (Strauss et al. 1992; Hayes and Waldbauer 2006) (Fig. 1). High variability—and possible bimodality—in \(\delta ^{13}\)C\(_{\text{org}}\) records is observed at various spatial scales at this time (Williford et al. 2016). This depletion in \(\delta ^{13}\)C\(_{\text{org}}\) has been canonically attributed to an ‘age of methanotrophy’ (Hayes 1994), but carbonate \(\delta ^{13}\)C records from the same sections are more consistent with the dominance of organisms employing a metabolism that does not produce such \(\delta ^{13}\)C-depleted DIC (and thus \(\delta ^{13}\)C-depleted authigenic carbonate) upon remineralization (Slotznick and Fischer 2016). Instead, the late Archean may have been an era when multiple primary carbon fixation metabolisms proliferated: a metabolism with a small \(\varepsilon _{p}\) (such as rTCA-utilizing anoxogenic photoautotrophs) and a metabolism with a large \(\varepsilon _{p}\), such as the Wood-Ljungdahl pathway (by early acetogens and/or methanogens) (Des Marais 1997; Slotznick and Fischer 2016; Ward et al. 2019).
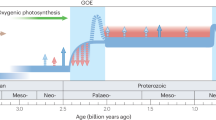
Multiple lines of evidence document a substantial rise in atmospheric O2 levels about 2.4 Ga, in the so-called Great Oxidation Event (GOE) (Holland 2006; Gumsley et al. 2017). Although low levels of oxygen may have existed up to 50 million years prior (Anbar et al. 2007), the disappearance of sulfur mass-independent fractionations (S-MIF) in sedimentary pyrites and sulfates at this time requires that O2 concentrations were higher than 10\(^{-5} \times \) present atmospheric levels (Farquhar et al. 2000). The appearance of Oxygen-MIF measured in contemporaneous sulfate evaporites further requires an oxygen concentration of greater than 10\(^{-3} \times \) present levels (Crockford et al. 2019), though it has been suggested to have risen higher still (potentially even greater than present levels; Bekker and Holland 2012). The introduction of O2 is attributed to the proliferation—but necessarily the evolution (Anbar et al. 2007)—of oxygenic photosynthesis (Falkowski 2011; Fischer et al. 2016). Unlike their volcanism-dependent predecessors, oxygenic phototrophs had a comparably-limitless supply of terminal electron donors in the form of water. As a result, primary productivity may have reached \(1 \%\) of modern levels (Crockford et al. 2019).
Despite the apparent increase in primary production across the GOE, a secular change in the carbon isotopic composition of organic matter is conspicuously absent (Fig. 1; Catling and Claire 2005). One explanation may be that an increase in primary production was compensated for by an increase in heterotrophy, and therefore there was no net change in the organic carbon exported from the exosphere (Catling and Claire 2005). Importantly, although localized ‘oases’ of the surface ocean may have been oxygenated by this time (Eigenbrode and Freeman 2006), these putative heterotrophs would have likely been anaerobes: aerobic heterotrophy appears to have postdated the evolution of oxygenic photosynthesis (Soo et al. 2019), and the deep ocean seems to have remained effectively anoxic for the remainder of the Proterozoic (Stolper and Keller 2018; Planavsky et al. 2011). An alternative explanation suggested that the appearance of atmospheric O2 was due to a change in the sinks of O2 instead of the sources (Catling and Claire 2005; Kump and Barley 2007). By decoupling a rise in O2 from a rise in \(F_{\text{org}}\), the GOE may have been a threshold response to a gradual (i.e., secular) change in the carbon cycle. Some primary pO2 proxy evidence suggests that the GOE was characterized by a gradual rise (Murakami et al. 2011; Kump et al. 2011), while other evidence contends that it was a geologically stepwise event (Luo et al. 2016).
The oxygenation of Earth’s atmosphere led to the removal of methane via photochemical oxidation reactions, scrubbing this potent greenhouse gas and cooling the exosphere (Pavlov et al. 2000; Kopp et al. 2005). The early Proterozoic saw at least two, and perhaps as many as four (Rasmussen et al. 2013), massive, global glaciations (so-called ‘snowball Earth’ events, Kirschvink and Kopp 2008; Hoffman 2013). Between and in the aftermath of these glacial intervals, major, globally-expressed perturbations to the carbon cycle are observed (Karhu and Holland 1996, Fig. 1). During one of these, the \(\sim 2.2-2.0\) Ga Lomagundi-Jatuli Excursion, \(\delta ^{13}\)C\(_{\text{carb}}\) values exceed \(+16 \permil \) (Karhu and Holland 1996; Martin et al. 2013). This excursion has been interpreted as a period of enhanced organic carbon burial (Karhu and Holland 1996; Des Marais 2001), but both the high scatter among \(\delta ^{13}\)C\(_{\text{carb}}\) values and the lack of a matching positive excursion in \(\delta ^{13}\)C\(_{\text{org}}\) values precludes a simple rise in \(F_{\text{org}}\) (Karhu and Holland 1996). Barring a diagenetic origin for these elevated \(\delta ^{13}\)C\(_{\text{carb}}\) values (Planavsky et al. 2012), some degree of basin-specific effects or non-steady state behavior may be required to account for this period. The Lomagundi-Jatuli Excursion is followed by a globally-synchronous negative carbon isotope excursion mirrored in both carbonate carbon and organic carbon at ca. 2.0 Ga (Kump et al. 2011). This event has been interpreted as a widespread oxidation of sedimentary organic carbon culminating in a reduction of global primary productivity of at least 80 % (Kump et al. 2011; Hodgskiss et al. 2019). Such a productivity crash may have ushered in an era in which nutrient-limited conditions prevented further substantial changes to the global carbon cycle (as observed by \(\delta ^{13}\)C\(_{\text{carb}}\) and \(\delta ^{13}\)C\(_{\text{org}}\) records) for much of the subsequent billion years (Buick et al. 1995; Hodgskiss et al. 2019; Laakso and Schrag 2019).
The apparent relative stability of the global carbon cycle for the duration of the mid-Proterozoic (\(\sim 1.8-0.8\) Ga) is terminated by a series of climatic and biogeochemical changes reminiscent of those seen in the Paleoproterozoic. The carbon isotope record from 0.8 to 0.54 Ga is interrupted by two ‘snowball Earth’ glaciations and one large regional glaciation (Kirschvink 1992; Hoffman et al. 1998). These glaciations are characterized by a gap in the sedimentary record (Partin and Sadler 2016). Large positive and negative excursions are seen throughout this interval, which may represent dramatic swings in the marine DIC reservoir, and may or may not have been productivity related. The timing and regional coherency of these excursions remains poorly constrained and the relationships to glaciations uncertain. Still, it is likely that the glaciations shut off marine autotrophy and the silicate weathering feedback until volcanogenic CO2 inputs contributed sufficient atmospheric insulation to thaw the icehouse (Kaufman and Knoll 1995; Kirschvink et al. 2000). The terminations of these global glaciations are demarcated by ‘cap carbonate’ intervals. In the recovery from the Sturtian glaciation, eukaryotic algae may have replaced cyanobacteria as the dominant primary producers in the ocean (Brocks et al. 2017). This putative rise of eukaryotes was echoed by the proliferation of animals—and the restructuring of the carbon cycle that accompanied the evolution of predation and grazing—in the Ediacaran following the smaller Marinoan glaciation (Butterfield 2007).
2.5.2 Phanerozoic Carbon Cycling
The appearance of biomineralization in the late Neoproterozoic and its rapid convergent evolution throughout the tree of life in the Cambrian led to a step change in the way geochemical information was preserved in sediments. In sediments where carbonate is preserved in the form of fossils, taxonomic information is available—this can be used to infer habitat, and therefore the position in the water column where the carbonates formed. Through a comparative biology approach involving extant organisms, aspects of physiology can also be inferred, enabling ‘vital effect’ corrections to be applied. The start of the Phanerozoic saw a dramatic decrease in the amplitude of variation in the carbonate carbon isotope record compared to the Neoproterozoic (Hayes et al. 1999; Krissansen-Totton et al. 2015). This change suggests an increase in the relative stability of the Earth system (Bachan et al. 2017), possibly due to the emergence of multicellularity and complex, and relatively robust, ecosystems (Schobben and van de Schootbrugge 2019).
The marine sedimentary record prior to \(\sim 180\) Ma is biased towards continental shelf sediments, and is thus dominated by fossils produced by coastal, and shallow water species (Veizer et al. 1999; Müller et al. 2008). Acknowledging this caveat, the broad features of the marine Phanerozoic carbon isotope records begin with a secular trend in \(\delta ^{13}\)C\(_{\text{carb}}\) towards heavier values throughout the entire Paleozoic, followed by a relatively abrupt decline and stabilization of the long term mean at the present-day value for the duration of the Mesozoic and Cenozoic (Veizer et al. 1999). \(\delta ^{13}\)C\(_{\text{org}}\) approximately tracks \(\delta ^{13}\)C\(_{\text{carb}}\) throughout the Paleozoic and Early Mesozoic, probably representing an increase and subsequent decrease in the fraction of carbon buried as organic matter, and a negligible change in the fractionation of carbon into phytoplankton organic matter (Berner and Kothavala 2001; Krissansen-Totton et al. 2015). \(\delta ^{13}\)C\(_{\text{org}}\) undergoes a secular drift towards heavy values in the Cenozoic that is independent of \(\delta ^{13}\)C\(_{\text{carb}}\), which has been widely interpreted to reflect a global decrease in atmospheric CO2 (Pagani et al. 1999; Witkowski et al. 2018).
Some of the largest perturbations to the global carbon cycle throughout the Phanerozoic are ocean anoxic events (OAEs). These ∼ million year intervals are characterized by high (relative) rates of organic carbon burial in thick shale deposits, and associated with widespread ocean anoxia, high temperatures and mass extinction of marine organisms (Schlanger and Jenkyns 1976; Jenkyns 2010, see Sect. 3.5 for further discussion on OAEs). Most of these organic matter burial events are associated with positive carbon isotope excursions and are thus consistent with the classic mechanism of global ocean 13C enrichment via an increase in \(F_{\text{org}}\). However, a few are preceded by large negative carbon isotope excursions, which suggest more nuanced mechanisms, such as recycling of isotopically light carbon from depth into the surface ocean (Schouten et al. 2000), or global methane release and oxidation (Hesselbo et al. 2000). The Phanerozoic also saw two massive glaciations, the Ordovician (∼ 440 Ma) and Gondwanan (\(\sim 250-320\) Ma) glaciations, which were probably less intense than the Neoproterozoic glaciations, and not associated with the same decreases in apparent \(\varepsilon _{p}\) (Hayes et al. 1999).
The Cenozoic carbonate carbon isotope record is more complete than older intervals, owing largely to the ubiquitous presence of the calcitic shells of foraminifera, which are microscopic predators that have diversified to inhabit the surface (planktonic) ocean and benthic environments. The deep ocean carbonate carbon isotope record, generated by targeting benthic species of foraminifera, is rich in environmental information and low in noise due to the relative homogeneity and low energy of the environment (Zachos et al. 2001). The planktonic record is comparatively sparse and noisy. A group of planktonic calcifying algae called coccolithophores contribute similar amounts of calcite to the deep marine sedimentary record, but their carbonate exhibits large and variable vital effects, limiting their utility as compared to the foraminifera whose well-characterized vital effects are small and relatively constant. The Cenozoic benthic \(\delta ^{13}\)C\(_{\text{carb}}\) record contains a \(\sim 2.5 \permil \), 10 million year oscillation in the aftermath of the K-Pg mass extinction, followed by 30 million years of relative quiescence, terminated by an increase towards the heaviest values of the last 50 million years at the ‘Middle Miocene Climatic Optimum’ (16–14 Ma) (Zachos et al. 2001). Following this peak in \(\delta ^{13}\)C\(_{\text{carb}}\), values decrease towards the Plio-Pleistocene, likely driven in part by the expansion of C4 grasses (Cerling et al. 1997), which have a smaller isotopic fractionation than the previously dominant C3 metabolism (Farquhar et al. 1989), and a global decrease in CO2, which modulates the net fractionation by plants and phytoplankton. Punctuating this curve are a number of hyperthermals, most notably the Paleocene/Eocene thermal maximum (PETM) (Zachos et al. 2001; Pagani et al. 2006). This event, thought to be the most rapid natural (i.e., not anthropogenic) CO2 increase in the observable geologic record, is characterized by a sharp spike towards isotopically depleted values that represents a massive release of isotopically depleted organic matter, potentially supplemented by the mobilization of extremely isotopically depleted methane (Zeebe et al. 2009).
For at least the last 4 million years, the Earth system has oscillated between glacial periods, characterized by cold temperatures and high ice volume, and interglacial periods characterized by warmer temperatures and low ice volume (Broecker and van Donk 1970; Shackleton 1987). These cycles have a characteristic saw-tooth shaped oscillation: a gradual descent towards glacial maximum conditions and rapid emergence (Zachos et al. 2001; Lisiecki and Raymo 2005). The mixing ratio of CO2 in the atmosphere, as measured in bubbles trapped in antarctic ice, has varied in concert with temperatures and ice volume over at least the last 800 thousand years (Lüthi et al. 2008; Neftel et al. 1982) and most likely beyond (Hönisch et al. 2009). During peak glacial periods, pCO2 was around 180 ppm; the rapid end of glacial periods is associated with similarly rapid increases in CO2, known as ‘terminations’, elevating pCO2 to around 280 ppm (Lüthi et al. 2008). These glacial cycles appear to be paced by periodic variation in insolation (Hays et al. 1976), but the resonant frequency of the Earth system has shifted at least once, from a 41 kyr period prior to 1 Ma to 100 kyr period after 800 ka with no apparent change in insolation forcing (Lisiecki and Raymo 2005). During glacial periods, a third of the CO2 that resides in the atmosphere at peak interglacial times is stored in the deep ocean (Sigman and Boyle 2000). These changes are reflected in the carbon isotopic composition of gaseous CO2, possibly due to a smaller glacial terrestrial biosphere and a polar ocean with depressed productivity (Marino et al. 1992). Atmospheric pCO2 can be estimated from the carbon isotope compositions of alkenones, a set of lipids that are specific to a single known family of coccolithophorid algae, in ocean sediments (Popp et al. 1998; Pagani et al. 2005, 2011). Although bulk \(\delta ^{13}\)C\(_{\text{org}}\) appears to respond to glacial cycles (Jasper and Hayes 1990), the carbon isotopic composition of alkenones do not vary predictably across glacial cycles (Badger et al. 2019), perhaps due to a trade-off between CO2 concentration and growth rate of this phytoplankton group through glacial cycles.
The last glacial period reached a maximum around 20 ka, and the most recent interglacial period has persisted since 10 ka (Lisiecki and Raymo 2005). The recent period of increasing anthropogenic activity, which may become formally recognized as the Anthropocene epoch (Lewis and Maslin 2015), has been characterized by a rapid rise in atmospheric pCO2 from less than 280 ppm before 1750 A.D. to exceeding 410 ppm by May, 2020 (Hartmann et al. 2013). The full impacts of this rise on Earth’s biogeochemical carbon cycle are still being developed, but include a clear drop in the \(\delta ^{13}\)C of atmospheric CO2 as a result of burning 13C-depleted organic matter that constitutes fossil fuels (Keeling 1979), and a possible rise in terrestrial photosynthesis rates due to CO2-fertilization (Keeling et al. 1996; Ciais et al. 2013).
3 Nitrogen
Nitrogen (N) is another biologically essential element that exists across a wide spectrum of redox states and speciations, reflecting its major involvement in both abiotic and biotic redox-driven transformations. In its most reduced form (–3), N exists as either ammonium (\(\text{NH}_{4}{}^{+}\)) or organic nitrogen, while in its most oxidized form (+5), N is found predominantly as nitrate (\(\text{NO}_{3}{}^{-}\)). In its zero-valent form nitrogen exists as the triple-bonded di-nitrogen molecule (N2), which comprises the majority of Earth’s atmosphere and constitutes the dominant form of N found in the exosphere (Gruber 2008; Johnson and Goldblatt 2015; Bebout et al. 2013). In general, N is most commonly found in one of these three oxidation states. Other more transient oxidation states are represented in several N species including aqueous species such as nitrite (\(\text{NO}_{2}{}^{-}\)), hydroxylamine (NH2OH), and hydrazine (N2H4) as well as gaseous and aerosol phases including ammonia (NH3), nitrous oxide (N2O), nitric acid (HNO3), nitrous acid (HNO2), peroxynitrite (\({\mbox{ONOO}^{-}}\)), and nitrogen oxides such as (NO2, N2O3, N2O4, and N2O5). Many of these intermediate oxidation states arise as metabolic products or intermediates. For example, during the microbial oxidation of \(\text{NH}_{4}{}^{+}\), NH2OH is the first intermediate produced, followed by oxidation to NO and \(\text{NO}_{2}{}^{-}\) (Caranto and Lancaster 2017; Lehnert et al. 2018; Martens-Habbena et al. 2015)
Among these intermediate species, \(\text{NO}_{2}{}^{-}\) is a common redox ‘crossroads species’ with much evidence pointing to tight biological coupling of its oxidative and reductive production and consumption at sharp redox boundaries (e.g., sediment–water interfaces), with a pronounced relationship with ambient carbon cycling. Nitrite can be produced by both heterotrophic nitrate reduction or autotrophic ammonia oxidation, as well as be consumed by heterotrophic denitrification or autotrophic nitrite oxidation—highlighting the complexity of one of many key interfaces between the redox cycling of carbon and nitrogen.
On Earth’s surface, after the atmospheric reservoir of N2 (0.7809 mol fraction) and its dissolved counterpart in the global ocean, the most abundant form of N is organic N buried in ocean sediments and sedimentary rocks (Bebout et al. 2013). N2 fixation into bio-available (reduced) forms is an energetically expensive process that can only be performed by a specialized group of prokaryotes (see below). As a result, more labile forms of dissolved inorganic nitrogen, such as \(\text{NH}_{4}{}^{+}\) and \(\text{NO}_{3}{}^{-}\), play the primary role in regulating levels of biological productivity in N-limited surface waters on Earth. Given the oxidizing nature of the modern Earth, however, it is perhaps not surprising that the primary pool of inorganic nitrogen in the ocean is \(\text{NO}_{3}{}^{-}\). On land, most N is bound in soil organic matter or living biomass.
When taking into account the entirety of the Earth system, the geologic reservoirs of N dwarf those of the actively cycled surface Earth biosphere, similar to carbon. These include N in crustal rocks, the mantle and the Earth’s core, with estimates of reservoir sizes varying over orders of magnitude (e.g., Johnson and Goldblatt 2015), and residence times of tens of millions to billions of years (Fig. 3).
3.1 Abiotic Processes in the N Cycle
Nitrogen transformations can occur in the absence of biological systems and would have played an important role in N cycling on a pre-biotic Earth. In particular, a wide range of photochemical and thermochemical processes are known to occur under Earth surface conditions that largely mimic those of the better-known biological nitrogen cycle (see review by: Doane 2017). For example, much attention has been given to the photochemical degradation of organic compounds producing free ammonium. Beyond this process, however, photochemical fixation of N2 into the more bioavailable NH3 has been demonstrated to occur in connection with surface reactions on transition metal oxides (especially those containing Fe, Ti, Mo, Ni and Zn; Medford and Hatzell 2017; Sclafani et al. 1993; Tennakone et al. 1991). At the same time, studies have also documented photochemical oxidation of N2 into \(\text{NO}_{x}\), \(\text{NO}_{2}{}^{-}\) and \(\text{NO}_{3}{}^{-}\), highlighting their potential importance in desert-like, light-intensive settings (Bickley and Vishwanathan 1979; Medford and Hatzell 2017). In addition to surface-catalyzed fixation of N2, transformation of N2 into \(\text{NO}_{x}\) by lightning through Earth’s atmosphere has also been shown, and argued to play a role in the transformation of N on prebiotic Earth (Kumar et al. 1995; Ridley et al. 1996; Rakov and Uman 2003). Meteorite impacts, volcanic lightning, and (potentially) cosmic rays may have been additional sources of fixed N in this prebiotic world (Laneuville et al. 2018; Navarro-González et al. 1998; Tabataba-Vakili et al. 2016).
Non-photochemical reactions with transition metals also play a key role in catalyzing abiotic transformations of N compounds with notable experimental and observational attention having been given to reactions involving Fe and Mn (e.g., Buchwald et al. 2016; Hansen et al. 1996; Heil et al. 2016; Luther et al. 1997; Ottley et al. 1997; Samarkin et al. 2010; Zhu-Barker et al. 2015). While approaches to teasing apart the relative environmental contribution of such transformations from those of enzyme-driven processes have proven challenging, many laboratory studies have established their feasibility under surface conditions. A number of factors influence reactivity, including: i) coordination environment of metal cations (e.g., surface bound, mineral form), ii) presence, abundance, and nature of organic molecules, iii) solution pH, iv) reactant concentrations, and v) presence of catalytic metals (e.g., Cu, Ni) (Buresh and Moraghan 1976; Dhakal et al. 2013; Grabb et al. 2017; Ottley et al. 1997; Postma 1990; Rakshit et al. 2008; Sørensen and Thorling 1991). Notably, many reactions involve the production and release of reactive intermediate and radiatively absorptive N species, such as NO and N2O (e.g., Cavazos et al. 2018; Coby and Picardal 2005; Kampschreur et al. 2011; Klueglein and Kappler 2013; Nelson and Bremner 1970), prompting some speculation on the potential role of chemodenitrification in atmospheric N2O budgets of Earth in the past (Robertson et al. 2011; Stanton et al. 2018).
3.2 Enzymatically Mediated Processes in the N Cycle
Similar to carbon, enzymatically catalyzed redox processes drive the short-term global nitrogen cycle. Biological nitrogen fixation comprises the enzymatic reduction of N2 to \(\text{NH}_{4}{}^{+}\) for nutritive N2 assimilation and synthesis of biomass (Hoffman et al. 2014). Ecologically, this pathway of nutritive N assimilation is relatively restricted to specialized organisms known as diazotrophs, that are conventionally thought to be favored for lifestyles in environments otherwise devoid of bioavailable forms of N (e.g., the center of nutrient-poor—oligotrophic—ocean gyres) since catalysis by the metalloenzyme nitrogenase is energy intensive. More recent work, however, suggests that other more N-replete environments may also host nitrogen fixation (Bertics et al. 2010; Dekas et al. 2009; Knapp 2012; Mehta and Baross 2006). The most widespread form of nitrogenase contains an iron-molybdenum (Fe-Mo) reaction center, while other so-called alternative nitrogenases contain Fe-V and Fe-Fe reaction centers. The relative distribution and activity of these disparate enzyme forms may have important implications for isotope budgets under some conditions and remains an active area of research (McRose et al. 2017; Zhang et al. 2014).
Nitrification, the biological oxidation of \(\text{NH}_{4}{}^{+}\) (the most reduced form of inorganic N) to the more oxidized forms \(\text{NO}_{2}{}^{-}\) and \(\text{NO}_{3}{}^{-}\) (also nitritation and nitratation), comprises the single most important oxidative flux of N on the Earth surface. This process is carried out by a group of specialized chemoautotrophic archaea and bacteria that use ammonia monooxygenase to harness the oxidizing power of molecular O2 for transforming NH3 into an intermediate hydroxylamine (NH2OH). The conversion of NH2OH to \(\text{NO}_{2}{}^{-}\) proceeds via the enzyme hydroxylamine oxidoreductase (HAO), with recent work suggesting an NO intermediate (Caranto et al. 2016; Caranto and Lancaster 2017; Lehnert et al. 2018). A separate group of chemoautotrophic nitrite-oxidizing bacteria (NOB) have been conventionally considered responsible for the ultimate oxidation of \(\text{NO}_{2}{}^{-}\) to \(\text{NO}_{3}{}^{-}\), also through the use of molecular O2. Recent cultures of a novel bacterial lineage have also demonstrated that conversion of \(\text{NH}_{4}{}^{+}\) to \(\text{NO}_{3}{}^{-}\) by nitrification can also occur within a single species (Daims et al. 2015; Van Kessel et al. 2015).
From the large oxidized pool of \(\text{NO}_{3}{}^{-}\) in the modern global ocean and terrestrial biosphere, the largest return flux of N to the atmosphere occurs through reductive microbial processes known as denitrification and anammox (anaerobic ammonia oxidation) (Gruber 2008; Galloway et al. 2004; Alhaique et al. 1975). Denitrification comprises the stepwise reduction of nitrate and its loss to the atmosphere as gas phase N2, proceeding through the intermediate species nitrite, nitric oxide and nitrous oxide. As a microbial respiration process, denitrification is frequently coupled to the oxidation of organic carbon (heterotrophy), and proceeds only in the absence of more thermodynamically viable oxidants (e.g., O2, \(\text{Mn}^{4+}\), \({\mbox{Fe}}^{3+}\) at low pH). Although denitrification generally occurs only in the absence of O2, the presence of O2 in the biosphere is a necessary precursor for \(\text{NO}_{2}{}^{-}\) formation and hence for the advent of denitrification as a respiratory metabolic pathway. Notably, biological denitrification can also be coupled with oxidation of inorganic substrates (autotrophic denitrification) including reduced sulfur and iron species, and molecular hydrogen (Batchelor and Lawrence 1978; Frey et al. 2014; Melton et al. 2014; Sievert and Vetriani 2012).
Anammox, the production of N2 through microbially coupled oxidation of \(\text{NH}_{4}{}^{+}\) with nitrite reduction, is conducted by specialized chemoautotrophic bacteria. In contrast to denitrification, in which respiratory nitrate reduction is often facultatively engaged after O2 levels are drawn down, anammox is very sensitive to the presence of O2. Anammox has been detected in a wide range of anoxic environments including ocean oxygen minimum zones, soils, wastewater treatment reactors, and marine sediments. Stoichiometric examination of ocean carbon and nitrogen budgets suggests that the relative contribution of anammox likely does not exceed \(30 \%\) of N loss in most cases (Babbin et al. 2014; Dalsgaard et al. 2012).
In addition to reductive transformation and return of inorganic forms of N to the atmosphere, dissimilatory nitrate reduction to ammonium (DNRA) represents an additional microbially catalyzed pathway in which N is retained in bioavailable form. Ecologically, DNRA therefore represents an important N retention pathway for maintenance of N in biological reservoirs. DNRA appears to generally be favored over denitrification when organic carbon is abundant relative to nitrate (Hardison et al. 2015; Koop-Jakobsen and Giblin 2010; Kraft et al. 2014; van den Berg et al. 2017). This trend could be tied to growth yields, where denitrification yields more energy per electron, but DNRA yields more energy per nitrate molecule (Strohm et al. 2007).
All known living organisms require nitrogen for biosynthesis and growth. Thus, biochemical pathways of N assimilation are, at least in part, central to the ecological success of all organisms. The biological assimilation of N for synthesis of organic biomolecules (amino acids, proteins, nucleic acids, chlorophyll, etc.) represents an important link between actively cycling N pools and those that may ultimately be stored in the geological record of sedimentary rocks. Heterotrophic organisms that consume complex organic molecules generally possess a host of metabolic pathways for funneling organic N moieties into cellular assimilatory machinery. In general, both single-celled prokaryotes as well as a wide array of eukaryotic organisms have evolved means of assimilating ambient forms of inorganic N, including \(\text{NO}_{2}{}^{-}\), \(\text{NO}_{3}{}^{-}\) and \(\text{NH}_{4}{}^{+}\). As biomolecules generally comprise N in its most reduced form, assimilative pathways typically involve the necessary reduction of oxidized forms (\(\text{NO}_{2}{}^{-}\) and \(\text{NO}_{3}{}^{-}\)) prior to shuttling of \(\text{NH}_{4}{}^{+}\) into biosynthesis pathways.
3.3 Nitrogen Isotopes in the N Cycle
The majority of the biological processes driving the biogeochemical nitrogen cycle produce measurable fractionations between the 15N/14N ratios of products and substrates. For example, biological nitrogen fixation results in organic matter with \(\delta ^{15}\)N values of \(\sim -4\) to \(+2 \permil \) (relative to atmospheric N2, which is also the \(\delta ^{15}\)N standard) (Bauersachs et al. 2009; Carpenter et al. 1997; Minagawa and Wada 1986; Delwiche and Steyn 1970). While it appears that the Mo form of nitrogenase dominates N-fixation budgets under the more oxidizing conditions of modern surface Earth, the so-called alternative nitrogenases (V and Fe forms) impart a stronger fractionation, yielding biomass that is up to \(7-8 \permil \) lower in \(\delta ^{15}\)N than N2 (Zhang et al. 2014). Denitrification and anammox both return 15N-depleted N2 to the atmosphere, leaving the residual nitrate enriched in 15N by \(10-25 \permil \) (Delwiche and Steyn 1970; Granger et al. 2008; Brunner et al. 2013, and references therein). Loss of low-\(\delta ^{15}\)N N2 to the atmosphere via denitrification and anammox, therefore, exerts the dominant control on \(\delta ^{15}\)N values of nutrients and biomass in the modern oceans, resulting in sedimentary organic matter with average \(\delta ^{15}\)N values of +6 to \(+7 \permil \) (Peters et al. 1978).
As with carbon, the extent to which nitrogen isotope effects associated with biological processes are expressed in the environment depends on the relative fraction of the N pool that is transformed or consumed. For example, uptake of dissolved inorganic nitrogen as nitrate or ammonium can also produce large fractionations in \(\delta ^{15}\)N (Hoch et al. 1992), but these isotope effects are not expressed in N-limited ecosystems where assimilation proceeds to completion. Similarly, in environments with low nitrate and high organic carbon flux, denitrification can consume the entirety of the nitrate pool, erasing any isotopic trace of its existence. This consideration becomes important when interpreting isotopic evidence for N cycling processes from ancient sediments on Earth, and would be similarly important when considering isotopic evidence for biological N cycling on other planets.
Nitrogen is preserved in the sedimentary rock record primarily as organic N or as ammonium in phyllosilicates, and generally records the \(\delta ^{15}\)N of the input biomass. Thus the sedimentary record of \(\delta ^{15}\)N has been used to infer the evolutionary history of N cycling metabolisms on Earth. Care must be taken, however, to consider that sedimentary \(\delta ^{15}\)N values can be modified by post-depositional processes, including diagenesis, metamorphism, and hydrothermal or hydrocarbon fluid migration. In particular, degradation of organic matter during early diagenesis, high-temperature metamorphism above greenschist facies and/or thermal maturation during hydrothermal fluid migration can all increase the absolute \(\delta ^{15}\)N values of the residue (Freudenthal et al. 2001; Bebout and Fogel 1992; Boyd and Philippot 1998). Low-temperature metamorphism can even cause mineral and organic \(\delta ^{15}\)N values to diverge within the same sample (Stüeken et al. 2017). The possible impacts of post-depositional alteration must therefore be considered before interpreting sedimentary \(\delta ^{15}\)N data as reflecting primary signals, particularly when applying these to Precambrian sediments. Additionally, different combinations of N cycling processes can produce identical or non-unique \(\delta ^{15}\)N patterns, meaning that additional depositional or redox context is often required for a convincing interpretation of ancient N cycling. We expand on these ideas in our following discussion of the evolution of Earth’s early N cycle.
3.4 Evolution of the N Cycle on the Precambrian Earth
Abiotic sources of N to the early biosphere (e.g., from lightning or hydrothermal sources, as described above) would have been limited (Navarro-González et al. 2001), requiring biological N2 fixation to have evolved very early in Earth history in order to keep pace with the N requirements of a rapidly evolving biosphere. Indeed, phylogenetic reconstructions suggest that the last universal common ancestor of extant life had proteins for N2 fixation (Weiss et al. 2016), and \(\delta ^{15}\)N records have been interpreted as evidence of diazotrophy as far back as \(\sim 3.2\) Ga (Stüeken et al. 2015a). Once biological N2 fixation evolved, this enzymatic innovation would have provided a virtually unlimited source of nutrient N to an increasingly efficient biosphere, assuming no other nutrient (phosphorus or bioactive trace elements) became limiting.
Prior to ∼2.7 Ga, the early to mid-Archean \(\delta ^{15}\)N record exhibits a wide range of values, spanning from −6 to \(+13 \permil \), and centered around \(0 \permil \) (Fig. 1). The most negative end of these \(\delta ^{15}\)N values are generally interpreted as reflecting a nitrogen cycle with inputs dominated by N2 fixation and the efficient recycling of ammonium deriving from breakdown of diazotrophic biomass, with the higher \(\delta ^{15}\)N values attributed to the influence of metamorphism and other post-depositional effects (Beaumont and Robert 1999). However it has also been suggested that this range of \(\delta ^{15}\)N values in the pre-2.7 Ga world—which are too high to be buffered by primitive mantle N (\(\delta ^{15}\)N\(_{\text{mantle}}\)\(<-40 \permil \))—could reflect isotopic enrichment due to atmospheric escape (Cartigny and Marty 2013; Lammer et al. 2018). Regardless, rocks from around 2.7 Ga have a comparably large range of \(\delta ^{15}\)N values, spanning −11 to \(+50 \permil \). The extremely high \(\delta ^{15}\)N values have been alternatively interpreted to reflect incomplete ammonium oxidation coupled to methanotrophy under low O2 conditions (Thomazo et al. 2011) or ammonia volatilization from alkaline lakes (Stüeken et al. 2015b). The \(\delta ^{15}\)N values lower than \(0 \permil \) were recently interpreted as evidence for non-quantitative uptake of a pool of bioavailable ammonium available in excess sourced from enhanced primary production and export of organic matter following the proliferation of oxygenic photosynthesis (Yang et al. 2019). This ammonium would have provided an ample source of N to fuel the Late Archean biosphere and associated oxygen production, with its incomplete consumption implying that some other critical nutrient (e.g., phosphorus) must have been important in limiting primary productivity.
The timing of the evolution of the aerobic N cycle, driven by the oxidation of ammonium (nitrification), is highly debated. Nitrification would have been a prerequisite for the development of the major N loss pathways, since these require a supply of oxidized N in the form of nitrate (for denitrification) or nitrite (for anammox). Several studies have interpreted small increases in \(\delta ^{15}\)N in single rock sections (Garvin et al. 2009; Godfrey and Falkowski 2009) or statistical trends in the temporal \(\delta ^{15}\)N dataset (Stüeken et al. 2016) as evidence for episodic or localized aerobic N cycling as far back as 2.5 Ga. Interpretations of these data are complicated by the fact that these processes produce a non-unique signal in \(\delta ^{15}\)N. For example, similar small and short-lived positive shifts in \(\delta ^{15}\)N could be produced by the incorporation of 15N-enriched ammonium that has undergone partial nitrification or partial assimilation (Yang et al. 2019), or by nitrogen redox cycling independent of surface oxygenation, such as with Fe-oxides (Busigny et al. 2013).
Consideration of \(\delta ^{15}\)N data within depositional and redox context suggests that a clear isotopic fingerprint of nitrification/denitrification is not evident in the rock record until ∼2.3 Ga, immediately following the Great Oxidation Event (Zerkle et al. 2017). However, as alluded to above, the isotopic signal of denitrification/anammox is lost if the reaction runs to completion in the environment. Therefore, the preservation of this signal in the rock record does not directly pinpoint the timing of the evolution of these metabolisms, but rather indicates when nitrate/nitrite availability became widespread enough for partial N loss processes to impart a long-lived isotopic imprint on the sedimentary \(\delta ^{15}\)N record.
Following the GOE, a hypothesized oxygen overshoot associated with the Lomagundi-Jatuli carbon isotope excursions could have increased the availability of nitrate, leading to enhanced denitrification rates and widespread loss of fixed N (Kump et al. 2011). The resulting nitrogen limitation has been implicated in hindering biological evolution, as well as the continued oxygenation of Earth surface environments, throughout much of the Proterozoic (Fennel et al. 2005). It is possible that N2 fixation could have kept pace with N-loss (Luo et al. 2019), although biospheric N limitation could have been exacerbated by the drawdown of molybdenum (Mo) under more widespread sulfidic conditions, since Mo is a key cofactor of nitrogenase and is required for marine N2 fixation (Anbar and Knoll 2002).
The remainder of the Proterozoic is characterized by apparent stasis in O2, with concentrations held between the anoxic and “overshoot” conditions of the Archean, and less than near-modern levels realized in the Phanerozoic. Though data are limited, the \(\delta ^{15}\)N record is also muted, resting between \(+2\) to \(+8 \permil \), consistent with a mildly oxidized surface ocean (Ader et al. 2014), and perhaps reflecting prevailing N limitation (Koehler et al. 2017). The mechanisms for reduced N availability during the Mesoproterozoic may be linked to trace metals (Anbar and Knoll 2002) or redox related loss of N (Fennel et al. 2005) as O2 increased. What remains unclear is whether higher O2 in the later Neoproterozoic had little effect on the \(\delta ^{15}\)N record (Ader et al. 2014) and suggests that planetary oxygenation was not the sole control on the N cycle. One hypothesis states that O2 concentrations may have been sufficiently high to inhibit nitrogenase expression, reducing N2 fixation rates and limiting productivity until evolution of physiological changes in cyanobacteria allowed for N2 fixation at higher O2 concentrations in the Phanerozoic (Allen et al. 2019).
3.5 Phanerozoic N Cycling
Nitrogen cycling in the Phanerozoic stands in contrast with the Precambrian in that variability in the N-cycle and \(\delta ^{15}\)N record reflects changes in the redox state within the ocean as opposed to direct forcing by the oxygen content of the atmosphere. However, the primary issues ultimately still concern the balance between N2 fixation, denitrification and anammox (Galbraith et al. 2013), and how these processes relate to changes in the physical and chemical structure of the ocean and nutrient delivery from land (phosphorus). An additional aspect that distinguishes the Phanerozoic from the Precambrian eras is the preservation of a broad range of primary organic phases within rocks and sediments, such as N-containing biomarkers (Chicarelli et al. 1993) and organic nitrogen preserved in the carbonate skeletons of invertebrates (e.g., Ren et al. 2009; Wang et al. 2015; Tornabene et al. 2017) that open a whole new range of potential areas of study.
Despite the relatively oxygenated atmosphere that characterized the Phanerozoic, its geologic record is punctuated by intervals of global-scale oceanic oxygen deprivation known as Oceanic Anoxic Events (OAEs) that are largely responses of the Earth system to global climate perturbations (Jenkyns 2010). Classically, OAEs are considered to be a Mesozoic phenomenon but recognition of similar depositional patterns and carbon cycle signatures have expanded to include intervals in the mid-Cambrian, Late Devonian, Early Triassic and during the Paleocene Eocene Thermal Maximum (Meyer and Kump 2008; Jenkyns 2010; Junium et al. 2018).
OAEs have been common targets for study into the role of nutrient cycling. Organic matter contents in black shales deposited during OAEs can be in excess of 30 wt.% and were, to some degree, the byproduct of increasing nutrient availability coupled with enhanced preservation of organic matter under anoxic conditions. Anomalous P geochemistry in black shales largely supports the hypothesis that OAEs were accompanied by perturbations to nutrient cycling (e.g., Van Cappellen and Ingall 1996; Rau et al. 1987). Reduced sedimentary retention of P in mineral-oxides and authigenic P minerals is indicated by high C:P values (Ingall and Jahnke 1994) and would have resulted in the accumulation of P in anoxic deep waters. If water-column oxygen deprivation stimulated nitrate reduction or anammox, N deficits may have resulted in overall N:P imbalance.
Nitrogen isotope records from OAEs are consistent with profound changes in water-column nitrogen cycling, with \(\delta ^{15}\)N values typically less than \(0 \permil \) and often as low as \(-5 \permil \). The low \(\delta ^{15}\)N has been attributed to expanded N2 fixation in response to fixed N-loss and P-excess (Rau et al. 1987; Kuypers et al. 2004). However, with \(\delta ^{15}\)N values below the average isotopic composition of diazotroph biomass (\(\sim -1 \permil \)), a role for incomplete utilization of ammonium has become a favored model (Higgins et al. 2012; Junium and Arthur 2007). Alternatively, 15N-depletion may reflect high Fe availability for diazotrophs (Zerkle et al. 2008) or utilization of alternative nitrogenases (Zhang et al. 2014). Regardless of the exact mechanism, compound-specific \(\delta ^{15}\)N of chlorophyll a-derived porphyrins confirm that 15N-depletion was a shallow water signal, and not a byproduct of diagenesis or primary production by phototrophic sulfide oxidizers within the chemocline (Junium et al. 2015).
4 Sulfur
Sulfur (S) is a third versatile, bio-essential element that exists in solid (inorganic and organic), liquid and gaseous forms at Earth’s surface, and spans redox states from −2 to \(+6\). Atmospheric sulfur sourced from volcanoes in the form of SO2 (± H2S) constitutes a relatively minor component of the modern atmosphere, but can produce sulfate aerosols that have important climatic impacts. Sulfate (\({\mbox{SO}_{4}{}^{2-}}\)) is the dominant form of sulfur in well-oxygenated aqueous environments, and forms a major anion in seawater, at concentrations of 28 mM (Canfield and Farquhar 2009). Aqueous sulfide (H2S or \(\text{HS}^{-}\)) is less abundant but central to active biogeochemical cycling of S, and can build up in anoxic sediments and oxygen minimum zones. A variety of sulfur compounds with intermediate redox states, such as elemental sulfur (S0), polysulfides (\(\text{S}_{n}{}^{2-}\)), thiosulfate (\(\mbox{S}_{2}\mbox{O}_{3}{}^{2-}\)), and sulfite (\({\mbox{SO}_{3}}{}^{2-}\)) are additionally produced during abiotic and metabolic processes, but are generally recycled very quickly in most environments (Brimblecombe 2013). Sulfate can be incorporated into sediments and the rock record in the form of barite (BaSO4), gypsum (CaSO4⋅2H2O), anhydrite (CaSO4), or bound in the lattice of carbonate minerals (carbonate associated sulfate, or CAS). Sulfide, elemental sulfur, and polysulfides can be preserved as Fe-monosulfides (e.g., pyrrhotite) and/or pyrite (FeS2) (Fike et al. 2015).
Every living thing known to us requires sulfur. Sulfur is incorporated into organic compounds through both biological and abiological processes (Amrani 2014; Fike et al. 2015). Key organic sulfur compounds in living cells include the proteinogenic amino acids cysteine and methionine. The majority of important biological organosulfur compounds (e.g., coenzyme A, glutathione, S-adenosylmethionine) are biosynthesized from cysteine or methionine. However, some organosulfur compounds, such as sulfoquinovose (a sulfur-containg lipid important in photosynthesizing organisms) are produced from oxidized inorganic sulfur compounds like sulfite. Inorganic iron-sulfur clusters are required constituents of many electron-transfer proteins.
The abiotic reaction of organic compounds with reduced sulfur species is responsible for the formation of a wide range of organic sulfur compounds that can also build up in the environment (Amrani 2014). Hydrogen sulfide can react with hydrocarbons to produce thiols (mercaptans), acyclic and cyclic sulfides, polysulfides, and thiophenes (Sinninghe Damste and De Leeuw 1990). These reactions are widespread in sediments. The cross-linking of small organic compounds through sulfur bridges (natural vulcanization) is hypothesized to be one of the important processes in the formation of macromolecular carbon compounds with decreased lability, such as asphaltenes and kerogen (Vandenbroucke and Largeau 2007). Such macromolecular carbon compounds comprise the bulk of organic carbon in the exosphere.
4.1 Abiotic Redox Reactions in the S Cycle
As noted above, volcanic SO2 forms an important flux of sulfur from the mantle to Earth’s surface (Fig. 4). Volcanic SO2 has a short residence time in the modern atmosphere, on the order of years, as it oxidizes to sulfate aerosols and deposits on the surface (Robock 2000). The dominant input of sulfate to the modern oceans is a riverine flux sourced from dissolution of sulfates and oxidative weathering of pyrite (Kump and Garrels 1986; Canfield 2004; Brimblecombe 2013; Fig. 4). Hydrothermal vents are thought to provide another important source of sulfur to the oceans, but this flux is largely unconstrained. Hydrothermal vent fluids can contain up to 12 mM sulfide, up to \(89 \%\) of which could be derived by leaching of sulfur from seafloor basalt (Ono et al. 2007). The remaining sulfide, which may dominate in some hydrothermal vent fluids, is thought to come from thermochemical reduction of seawater sulfate, as seawater percolates into the oceanic crust and heats up. Given the ubiquitous nature of hydrothermal systems along mid-ocean ridges, these could therefore provide a significant source of sulfur to the deep oceans, and were potentially even more important earlier in Earth history prior to widespread oxidative weathering.
The majority of processes that act to transform sulfur in marine and terrestrial systems are biological. However, several abiotic processes can also contribute to sulfur cycling in the environment. Sulfate can be reduced to sulfide abiotically at high temperatures by reaction with organic compounds, during thermochemical sulfate reduction (TSR). In modern marine sediments, most of this sulfide is subsequently oxidized mainly by interaction with molecular oxygen, and Fe- and Mn-oxides (Goldstein and Aizenshtat 1994).
4.2 Enzymatically Mediated Reactions in the S Cycle
A diverse set of enzymatically-catalyzed metabolic processes interconvert sulfur compounds among redox states (e.g., see review in Jørgensen et al. 2019). Most of these metabolic processes are dissimilatory in nature, i.e., they do not result in the direct incorporation of sulfur molecules into biological materials. However, all living cells require sulfur, and assimilatory processes incorporate abiotic sulfur into organic material, as mentioned previously.
Dissimilatory sulfate reduction (DSR) couples the reduction of sulfate to the oxidation of an electron donor, which may be an organic compound (typically the conjugate base of a small organic acid, e.g., lactate) or an inorganic compound (e.g., H2). This process can be carried out by a diverse set of bacteria or archaea, all of which use the same set of enzymatic steps to catalyze the process (Pereira et al. 2011; Fike et al. 2015; Rabus et al. 2013; Oliveira et al. 2008). After sulfate is imported into the cell, it is converted to an organosulfate compound adenosine-5’-phosphosulfate. The sulfate moiety of this compound is subsequently reduced to sulfite, and then reduced again through a complex enzymatic cascade to hydrogen sulfide (Venceslau et al. 2014; Santos et al. 2015). Byproducts of this metabolic process can include thiosulfate and possibly trithionate (Drake and Akagi 1977; Bradley et al. 2011), though it is possible that these byproducts result from the reaction of excess sulfite with sulfide (Chambers and Trudinger 1975), and may not be relevant under most plausible environmental conditions. Of all the biological transformations that occur in the sulfur cycle, dissimilatory sulfate reduction exhibits the strongest discrimination against the heavier isotopes (Eldridge and Farquhar 2018). Observed fractionations during DSR can range from nil (when electron donors are abundant and DSR is fast or quantitative) to the equilibrium maximum of approximately \(72 \permil \) (Leavitt et al. 2013; Canfield et al. 2010; Sim et al. 2011, 2012; Zaarur et al. 2017).
The most reduced sulfur compound, H2S, has the IUPAC-preselected name sulfane (Favre and Powell 2014). This reflects an updated systematic naming for parent hydrides, so that sulfane (H2S) is consistent with other hydrides such as methane (CH4), silane (SiH4), phosphane (PH3), etc. This term is easily confused with the traditional usage of ‘sulfanes’ to refer to (hydro)polysulfides and polysulfanes (Favre and Powell 2014). For consistency with the geochemical literature, we use the terms ‘sulfide’ and ‘hydrogen sulfide’ here. This compound can be oxidized by an electron acceptor with a suitably high redox potential, most commonly molecular oxygen, nitrate or Fe-oxides. Such oxidation of sulfide is common in marine sediments, where it is suggested that the majority (∼90 %) of sulfide produced by microbial sulfate reduction is reoxidized to sulfate, often via compounds with intermediate oxidation states of sulfur (Jørgensen 1982, 1990; Thamdrup et al. 1994). The product of this reaction can include sulfur compounds at a variety of redox states, including various allotropes of elemental sulfur, thiosulfate, sulfite, and sulfate (Canfield 2001; Fike et al. 2015). Under permissive thermodynamic conditions, these intermediate compounds can also be oxidized by sulfide-oxidizing microbes (Jørgensen and Des Marais 1986). Reduced sulfur compounds such as S0 and H2S can also be oxidized by some photosynthetic organisms (Frigaard and Dahl 2008). Just as cyanobacteria split water to generate electrons for carbon fixation, releasing O2 as a byproduct, anoxygenic phototrophs including green- and purple-sulfur bacteria use H2S as an electron source and release more oxidized sulfur compounds such as S0 or sulfate. Processes that oxidize reduced sulfur compounds are observed in laboratory cultures to involve only small discriminations against 34S (Canfield 2001, and references therein), although this has been challenged recently, as discussed below.
Disproportionation of sulfur compounds involves the simultaneous oxidation and reduction of the same reactant, yielding two products (Bak and Pfennig 1987; Thamdrup et al. 1993; Finster 2008). For example, thiosulfate or S0 can be disproportionated into sulfate and H2S. The intermediate-valence compounds that are the substrates of disproportionation reactions are generally the products of partial oxidation of H2S. Under many conditions, disproportionation is not thermodynamically viable, but it can be if H2S concentrations are kept sufficiently low, e.g., by removal through environmental reactions producing iron sulfide.
4.3 Isotopic Records of the Sulfur Cycle
4.3.1 Sulfur Isotopes
The largest enzymatic fractionations of sulfur isotopes in the sulfur cycle occur during DSR (Canfield 2001). In principle, this process can generate fractionations of up to \(72 \permil \) in \(\delta ^{34}\)S, the equilibrium sulfate-sulfide fractionation (e.g., Eldridge and Farquhar 2018). In practice, fractionations as large as \(70 \permil \) are rarely directly observed in laboratory settings (Canfield et al. 2010; Sim et al. 2011), but have been inferred in sedimentary pyrite records and in sulfate-limited environments (Crowe et al. 2014; Bryant et al. 2019). Observed fractionations during DSR depend on a range of biological and environmental factors. In general, large fractionations occur at higher ambient sulfate concentrations (Habicht et al. 2002), and when the sulfate-reduction rate by individual cells is low (Canfield 2001; Sim et al. 2011; Leavitt et al. 2013; Zaarur et al. 2017). These patterns are related to the energetics of the enzymatically-catalyzed reactions and the sizes of intracellular metabolite pools (Wing and Halevy 2014; Wenk et al. 2018) and to bulk cellular-level processes related to importing sulfate into cells (Bradley et al. 2011, 2016). Differences exist between different strains of sulfate-reducing microorganisms, so that under identical conditions different organisms will produce different fractionations (Sim et al. 2011; Leavitt et al. 2013; Zaarur et al. 2017). The origins of these strain-specific differences is not well understood.
In laboratory cultures, sulfide oxidation generally produces fractionations of only a few permil in \(\delta ^{34}\)S (see review in Zerkle et al. 2009). Both chemotrophic and phototrophic sulfide oxidation have usually been associated with fractionations of the order of \(1-2 \permil \), although fractionations as large as \(12 \permil \) during chemotrophic sulfide oxidation have recently been observed in the laboratory (Pellerin et al. 2019) and in a natural environment (Zerkle et al. 2016).
Disproportionation of S0 has been reported to produce H2S that is depleted in 34S by \(4-9 \permil \) relative to the reactant (Habicht et al. 1998; Johnston et al. 2005). However, since oxidation of H2S is not associated with a large fractionation, repeated cycles of the production of H2S via disproportionation, its subsequent oxidation to 34S-depleted sulfate, and ultimate reduction of this sulfate to even more 34S-depleted H2S have been invoked to explain large sulfate-sulfide sulfur isotope fractionations that have sometimes been observed in nature (Canfield and Teske 1996). In recent years, these large fractionations have often been attributed to DSR occuring at slow rates (Canfield et al. 2010; Zerkle et al. 2010; Wing and Halevy 2014).
In addition to producing large changes in \(\delta ^{34}\)S\(_{\text{,}}\) biological processes can produce changes in the minor isotopes of sulfur, 33S (\(0.75 \%\)) and 36S (\(0.02 \%\)) (Farquhar et al. 2003; Johnston et al. 2005; Zerkle et al. 2009, 2016). Isotope effects in the minor sulfur isotopes are expressed as deviations from \(\delta\)33S or \(\delta\)36S values predicted by a reference value, often with a theoretical basis, e.g., \(\Delta ^{33}{\mathrm{S}} \equiv {\delta ^{33}\mathrm{S}_{\mathrm{measured}}} - {\delta ^{33}\mathrm{S}_{\mathrm{predicted}}}\). These differences, termed mass conservation effects, are generated during complex cycling of sulfur compounds between different S pools in the environment or within individual cellular metabolisms (e.g., Farquhar et al. 2007). Mass conservation effects are generally orders of magnitude smaller than mass-independent fractionations of 33S and 36S formed in the atmosphere (Sect. 4.4), but can be very useful in unraveling sulfur cycling processes in natural environments and at the cellular level (e.g., Johnston 2011).
4.3.2 Oxygen Isotopes in Sulfate
Exchange of oxygen between sulfate and water is extremely slow unless biologically mediated or associated with oxidation of compounds with sulfur of a lower oxidation state (Lloyd 1968; Kusakabe and Robinson 1977). Thus, the triple oxygen isotopic composition (18O/16O and 17O/16O, expressed in \({\delta}\)-notation and permil units, as \(\delta ^{18}\text{O}\) and \(\delta ^{17}\text{O}\)) of atmospheric, dissolved and mineral-bound sulfate has been used to study both biotic and abiotic sulfur redox transformations. In sulfate aerosols, where redox transformations are strictly abiotic, the combined sulfur and the oxygen isotopic compositions of the sulfate have been extensively used to identify its source. The different oxygen isotopic compositions of the possible oxidants of atmospheric SO2, some of which also display “mass-independent” oxygen isotope compositions, allow distinction among sources of atmospheric and riverine sulfate, both in the modern atmosphere (e.g., Holt et al. 1982; Lee and Thiemens 2001; Alexander et al. 2005, 2012) and hydrosphere (e.g., Waldeck et al. 2019), and in geologic repositories such as ice cores (e.g., Alexander et al. 2002; Sofen et al. 2011, 2014), soils, crusts and rock varnishes (e.g., Bao et al. 2001a,b; Bao and Marchant 2006), volcanic ash beds (e.g., Bindeman et al. 2007; Martin and Bindeman 2009; Bao et al. 2010) and massive sulfate deposits (Bao et al. 2000). The oxygen isotopic composition of sulfate has also been used to constrain the oxidants and the (biotic and abiotic) oxidation mechanisms of pyrite in laboratory experiments and natural environments (Balci et al. 2007). Using these insights, the triple oxygen isotopic compositions of seawater sulfate have been used to constrain the sources of sulfate to the ocean (Waldeck et al. 2019), whereas the isotopic composition of sulfate-bearing minerals in sedimentary rocks have been used to constrain the atmospheric abundance of CO2 during the Neoproterozoic “snowball Earth” events (Bao et al. 2008, 2009; Bao 2015), the paleoproductivity of the biosphere over the Proterozoic Eon (Crockford et al. 2018), and the atmospheric chemistry on early Mars (Farquhar and Thiemens 2000).
The oxygen isotopic compositions of aqueous sulfate have also been used to study microbial redox cycling in laboratory and natural settings, where they are thought to record information complementary to that revealed by sulfur isotopes. The sulfate–water oxygen isotope fractionations associated with a subset of sulfur metabolisms, including DSR (e.g., Böttcher et al. 1998; Brunner and Bernasconi 2005; Turchyn et al. 2006; Wortmann et al. 2007), elemental sulfur disproportionation (e.g., Böttcher et al. 2001, 2005), and pyrite oxidation (Balci et al. 2007) have been studied in laboratory cultures and natural environments. On the basis of these studies, it is understood that the \(\delta ^{18}\text{O}\) values of sulfate in aqueous environments with microbial redox cycling reflect a combination of disequilibrium isotope effects associated with enzymatic reactions and equilibration of oxygen isotopes between sulfur species of intermediate valence state (e.g., sulfite, \(\mbox{SO}_{3}^{2-}\)) and water, both intracellularly and extracellularly (e.g., Böttcher et al. 1998; Brunner and Bernasconi 2005; Turchyn et al. 2006, 2010; Wortmann et al. 2007; Wankel et al. 2014; Bertran et al. 2020).
The relationship between sulfate \(\delta ^{18}\text{O}\) and \(\delta ^{34}\text{S}\) values has been shown to reflect the intensity of microbial sulfur redox cycling, whether intracellularly during the multi-step reduction of sulfate to sulfide, or in the environment by concurrent activity of DSR and oxidative sulfur metabolisms. The consensus from laboratory cultures, environmental observations and models is that the slope of an array of sulfate \(\delta ^{18}\text{O}\) vs. \(\delta ^{34}\text{S}\) values is sensitive to the rate of sulfate reduction vs. that of reoxidation of intermediate sulfur species. Specifically, under conditions where sulfate reduction is slow relative to the reoxidation of intermediates, the isotopic (Rayleigh) distillation of sulfur isotopes in the sulfate (i.e., 34S enrichment of the sulfate) is outpaced by the exchange of oxygen isotopes between the sulfur intermediates and water, resulting in a steeper slope in sulfate \(\delta ^{18}\text{O}\) vs. \(\delta ^{34}\text{S}\) (e.g., Böttcher et al. 1998; Aharon and Fu 2000; Böttcher et al. 2001; Brunner and Bernasconi 2005; Wortmann et al. 2007; Antler et al. 2013; Wankel et al. 2014; Johnston et al. 2014; Mills et al. 2016; Antler et al. 2017). Thus, oxidative systems, such as sediments with a low content of organic matter and/or a high content of iron oxides, in which a large fraction of the sulfate reduced during DSR is reoxidized display a steep \(\delta ^{18}\text{O}\)–\(\delta ^{34}\text{S}\) slope. In contrast, more reducing systems, such as gas seeps and estuaries, in which only a negligible fraction of sulfate is reoxidized, are characterized by shallower slopes of sulfate \(\delta ^{18}\text{O}\) vs. \(\delta ^{34}\text{S}\) (Aharon and Fu 2000, 2003; Antler et al. 2014; Antler and Pellerin 2018).
4.3.3 Preservation of S Isotope Records
In geological materials, sulfate is usually preserved in sulfate minerals (barite, gypsum, anhydrite), and carbonate-associated sulfate (CAS). Barite is a mineral of low solubility, and is generally considered to be a high-fidelity recorder of sulfur isotopes, particularly through the Cenozoic (Paytan et al. 1998; Griffith and Paytan 2012; Masterson et al. 2016). Gypsum and anhydrite are often deposited as massive beds that precipitate from seawater in evaporative basins. CAS is a more widespread record (Kampschulte et al. 2001; Kampschulte and Strauss 2004; Gill et al. 2007; Wotte et al. 2012; Wu et al. 2014), but is subject to recrystallization and variable concentration among various carbonate components (Swart 2015; Present et al. 2015; Fichtner et al. 2017; Rose et al. 2019). Recent work has shown that recrystallization of carbonate cements expels CAS from rocks, decreasing its bulk-rock concentrations (Fichtner et al. 2017; Rose et al. 2019). This has not been demonstrated to have an effect on the isotopic composition (\(\delta ^{34}\)S) of CAS, but neither can this possibility be excluded (Gill et al. 2008; Rennie and Turchyn 2014; Fichtner et al. 2017), especially if recrystallization is accompanied by DSR and a change in the isotopic composition of porewater sulfate.
Pyrite is the most commonly interrogated phase of reduced sulfur in sedimentary rocks. The ratio of sulfur isotopes in pyrite is the outcome of a complex suite of early diagenetic processes, including DSR, various other sulfur metabolisms that may cycle sedimentary sulfur among its oxidation states, and interaction with sedimentary iron-bearing phases (Claypool 2004; Rickard and Luther 2007; Leavitt et al. 2013; Aller 2014; Fike et al. 2015; Shawar et al. 2018; Jørgensen et al. 2019). The early diagenetic ratio of sulfur isotopes in pyrite can be disturbed by late-stage processes, including pyrite overgrowth and, more generally, precipitation of iron sulfides when sulfide-bearing fluids flow through iron-bearing sedimentary rocks. A number of studies have also pointed out a relationship between the variability in \(\delta ^{34}\)S of pyrite and depositional environment (Fike et al. 2015, and references therein), where sediments that have undergone remobilization and/or bioturbation have more variable \(\delta ^{34}\)S values and pyrite that is on average more enriched in 34S (Aller 2014; Fike et al. 2015; Pasquier et al. 2017). The mechanisms that result in this enrichment have not been elucidated, but suggest that the determination of \(\delta ^{34}\)S value of primary pyrite in sedimentary rocks is rarely simple.
4.4 Evolution of the Precambrian Sulfur Cycle
The record of sulfur isotopes preserved in pyrite and sulfate minerals over geologic time can provide a window into past sulfur cycling processes. This record shows a generally small spread in pyrite \(\delta ^{34}\)S values (\(\sim 20 \permil \)) in the Archean, increasing to a \(\sim 40 \permil \) spread in the early Proterozoic (Canfield and Raiswell 1999; Halevy et al. 2010; Fike et al. 2015; Fig. 1). Even the small spread of Archean pyrite \(\delta ^{34}\)S values has been interpreted to represent active sulfur cycling and DSR at \(> 3.0\) Ga (Shen et al. 2001, 2009; Zhelezinskaia et al. 2014). The increased spread in pyrite \(\delta ^{34}\)S values in the early Paleoproterozoic is roughly coincident with pervasive oxygenation of Earth’s surface oceans and atmosphere during the Great Oxidation Event, \(\sim 2.4\) Ga. This change has therefore been interpreted to reflect enhanced sulfate delivery via oxidative weathering of pyrites on land, allowing for preservation of larger fractionation effects during microbial sulfate reduction, and the spread of sulfide-rich conditions (euxinia) on continental shelves (Canfield 1998; Habicht et al. 2002). An additional increase in the spread of \(\delta ^{34}\)S values near the end of the Proterozoic, to values spanning more than \(55 \permil \), has been interpreted to reflect larger biological fractionation effects associated with an expansion in oxidative sulfur cycling and widespread oxygenation of the deep oceans in the Neoproterozoic (Canfield and Teske 1996; Canfield et al. 2010; Poulton et al. 2010). Trends in minor sulfur isotopes have pushed the environmental expression of oxidative sulfur cycling and disproportionation back even farther in Earth history (Johnston et al. 2005). However, these data have been challenged by recent evidence from experiments and natural environments suggesting that DSR alone can produce very large fractionations in \(\delta ^{34}\)S without the need for oxidative S cycling (Canfield et al. 2010; Sim et al. 2011). Furthermore, experiments, observations from natural environments and models all suggest that isotopic fractionation during DSR may be large even when sulfate concentrations are low (Canfield et al. 2010; Crowe et al. 2014; Wing and Halevy 2014), casting doubt on the inference of sulfate-limited microbial fractionation of sulfur isotopes prior to the oxygenation of the atmosphere.
A unique feature of the Archean sulfur isotope record is the existence of mass-independent sulfur isotope compositions, preserved mostly in sedimentary pyrite (Farquhar et al. 2000, 2003; Thiemens 2006; Johnston 2011; Ono 2017). Most physical and chemical processes fractionate the heavy isotopes of sulfur proportionally to their mass, such that an enrichment or depletion in 33S is approximately half of the enrichment or depletion in 34S. Sulfur isotope mass-independent fractionations (S-MIF) in the Archean deviate significantly from these canonical relationships (Thiemens 2006; Johnston 2011). Although S-MIF is observed in present-day atmospheric sulfate aerosols and preserved in glacial ice following stratospheric volcanic eruptions (Romero and Thiemens 2003; Savarino et al. 2003; Burke et al. 2019), it is absent from sedimentary sulfate minerals and pyrite younger than \(\sim 2.4\) Ga (Farquhar et al. 2000, 2003; Bekker et al. 2004; Luo et al. 2016; Warke et al. 2020). The abrupt disappearance of S-MIF from the sedimentary pyrite record \(\sim 2.4\) Ga coincides with abundant geological and geochemical evidence for the oxygenation of the atmosphere around this time (Holland 1984, 2006; Lyons et al. 2014). In contrast to other proxi es for the oxidation state of the Earth’s surface that are temporally discontinuous, sedimentary pyrite is a relatively continuous record, offering (potentially) more refined insight into the redox evolution of the surface environment, provided the mechanism(s) governing the existence of S-MIF are understood. Although it is generally agreed that the disappearance of S-MIF is related to the increasing oxidation state of Earth’s oceans and atmosphere, the exact mechanism(s) are a topic of ongoing debate. While laboratory photolysis experiments can broadly reproduce the direction of sulfur isotope fractionations observed in Archean sediments (Farquhar et al. 2001; Masterson et al. 2011; Whitehill and Ono 2012; Whitehill et al. 2013, 2015; Endo et al. 2016), and models can constrain the magnitude of atmospheric O2 level at which the S-MIF signal would be suppressed (Pavlov and Kasting 2002; Zahnle et al. 2006; Claire et al. 2014), fundamental aspects of the Archean S-MIF record remain highly uncertain (e.g., see reviews by Ono 2017; Stüeken et al. 2020). For instance, there is ongoing debate as to the specific atmospheric sulfur species and reaction(s) responsible for the generation of S-MIF signals and the delivery to Archean sediments (e.g., Farquhar et al. 2001; Lyons 2007; Danielache et al. 2008, 2012, 2014; Ono et al. 2013; Whitehill et al. 2013; Endo et al. 2015; Babikov 2017; Harman et al. 2018), plus the role of non-sulfur-bearing gases (e.g., CH4, O2) and biological activity in regulating (or erasing) these effects (Zahnle et al. 2006; Domagal-Goldman et al. 2008; Zerkle et al. 2012; Halevy 2013; Izon et al. 2015, 2017; Liu et al. 2019). For a review of the latest understanding of the Archean S-MIF record, we direct the reader to Stüeken et al. (2020) in this volume.
4.5 Late Neoproterozoic and Phanerozoic Sulfur Cycles
Large differences between \(\delta ^{34}\)S of sedimentary sulfate and sulfide minerals persisted from the late Neoproterozoic into the Phanerozoic Eon (Fike et al. 2015). The 34S-enrichments of sedimentary sulfate minerals reached a maximum in the late Neoproterozoic, with \(\delta ^{34}\)S values approaching \(+40 \permil \) (Fig. 1). This has been interpreted to reflect the values of seawater sulfate during this time period. The origins of these 34S-enrichments are not well understood. Generally, they are assumed to reflect steady-state conditions requiring either addition to the oceans of 34S-enriched sources (e.g., sulfate evaporites) or the preferential removal of 34S-depleted sinks (e.g., sedimentary sulfides) (Fike et al. 2015). However, an alternative to a radical change in the global sulfur cycle is that the extremely 34S-enriched sulfate phases represent the action of sulfate reducing bacteria in partially-closed (likely, sedimentary) environments, which would leave sediment porewater (but not global seawater) sulfate concentrations low and sulfate \(\delta ^{34}\)S values enriched through isotopic (Rayleigh) distillation (Fike et al. 2015).
Sedimentary rocks deposited during the early Phanerozoic show a long-term secular decline in the \(\delta ^{34}\)S of marine sulfate from the Neoproterozoic maximum to a value of around \(+10 \permil \) at 300 Ma (Canfield and Farquhar 2009; Fike et al. 2015; Fig. 1). This decline is followed by a secular increase through the mid to late Phanerozoic to modern values of \(+21 \permil \). Sedimentary sulfide minerals also reach maximum values of \(\delta ^{34}\)S in deposits of the late Neoproterozoic, up to \(\delta ^{34}\)S of \(>+40 \permil \). The Phanerozoic records a long-term secular decline in these values, with values of \(-40 \permil \) or lower common through most of the Phanerozoic (Canfield and Farquhar 2009; Wu et al. 2010; Halevy et al. 2012). These changes are thought to reflect the increasing reservoir of marine sulfate over the course of the Phanerozoic (Canfield and Farquhar 2009; Algeo et al. 2015), and variations in the proportions of sulfate removal from the ocean as sulfate minerals or as pyrite (the product of microbial sulfate reduction) (Garrels and Lerman 1984; Berner 1987; Canfield 2004; Kampschulte and Strauss 2004; Halevy et al. 2012; Fike et al. 2015). Sulfur isotopic fractionations expressed during these biological processes may have changed in response to large-scale environmental shifts that were imposed during the onset of the Phanerozoic (Wu et al. 2010; Leavitt et al. 2013; Rennie et al. 2018). Relevant changes include secular changes, such as the long-term increase in marine sulfate concentrations, and evolutionary innovations, such as the advent of bioturbation or of organic carbon derived from terrestrial plants. The long-term changes in the isotopic records since the end of the Neoproterozoic may also reflect changes in the sulfur isotopic composition of the sulfate inputs to the ocean.
5 Synthesis: Carbon, Nitrogen, and Sulfur Cycles in the Absence of Life
One way to consider how isotopic evidence may be used to detect life on other planets is to compare how the (bio)geochemical C, N, and S cycles on Earth would look without it. Here we synthesize the above material to speculate on how these cycles could be structured.
At temperatures and pressures close to that of Earth’s surface, the most thermodynamically stable heteronuclear phases of carbon, oxygen, and hydrogen are water, carbon dioxide, and/or methane, depending on the oxidation state of the system (Saleh and Oganov 2016; Krissansen-Totton et al. 2018). The equilibrium assemblages of N- and S-bearing species are similarly sparse (Lewis and Randall 1923). Where life is present, it acts to catalyse exergonic reactions that may otherwise be kinetically sluggish. These catabolic processes convert chemical potential energy to heat, accelerating the descent of the entire system towards the equilibrium state, despite often producing metastable intermediates in the process. On the other hand, life’s anabolic reactions store chemical potential energy, and may contribute to prolonging a state of chemical disequilibrium in the system as a whole. For example, cell walls compartmentalize the chemicals of the Earth’s surface against the flow of entropy, autotrophic metabolisms bolster the lifetimes of calorie-dense organic molecules while the organism is alive, and leave a recalcitrant form of reduced material after death. A continued input of chemical potential energy from an external energy source, such as hydrothermal vent H2, which may have been short-lived in the absence of life, may therefore not only sustain, but also be sustained by, life itself. In other words, through kinetic inhibition, anabolic reactions can act to slow the inexorable conversion of chemical potential energy (or light energy, when phototrophy exists) to heat. Whether the net effect of life is to maintain disequilibrium in a system or accelerate a system in its entirety towards equilibrium is therefore a balance between the anabolic and catabolic reactions in a system. On Earth, by sequestering reduced carbon, nitrogen, and sulfur in recalcitrant kerogens and minerals, life separates electrons from their previous owners, which (today) allows O2 to persist in the atmosphere.
The simultaneous presence of certain chemical combinations in a planetary atmosphere that represent chemical disequilibrium has been suggested to be a biosignature in extraterrestrial settings. For example the coexistence of atmospheric methane and CO2 is thermodynamically unstable at Earth surface-like conditions, and would require large, possibly biogenic, fluxes of the unstable component (likely methane) in order to be sustained (Krissansen-Totton et al. 2018). Isotopic compositions of these gases could provide additional constraints on the degree of disequilibrium in the system.
An exploration of isotopic compositions of solid phases in the surface environment would provide an additional layer of information. If sediment cores were obtained and dated, and isotopic time series reconstructed, characteristic timescales may be diagnostic of the past existence of differentiated pools with contrasting residence times (i.e., a (bio)geochemical cycle). Comparing the structures of the C, N and S cycles in the modern Earth (Figs. 2, 3 and 4), it is likely that they would respond differently to a drastic reduction in biologic activity. The modern pre-industrial carbon cycle features connected oxidized and reduced reservoirs (Ccarb and Corg) that occupy the same physical spaces (e.g., deep ocean, sediments, and crust) and have similar residence times (Fig. 2), so they often respond in complementary ways to the same perturbations. It is not uncommon for Phanerozoic carbon isotope excursions to be mirrored in \(\delta ^{13}\)C\(_{\text{carb}}\) and \(\delta ^{13}\)C\(_{\text{org}}\), and situations that induce non-steady-state behavior often do so in both records (Rothman et al. 2003). In the absence of a biosphere, the input fluxes of new organic carbon to these reservoirs would be reduced (at least to ca. Archean levels). Predicting the resulting changes to the size and residence time of the marine organic carbon reservoir is less straightforward because these would depend on the exact response of the output fluxes. Nonetheless, barring stark differences in the way organic carbon is buried in a biosphere-free world, we might expect the size of the marine organic carbon pool to be reduced as well. If the biosphere-free organic carbon pool were orders of magnitude smaller than the corresponding carbonate carbon pool, it would likely make it easier to decouple the two; perturbations to the former, such as a burst of hydrothermal abiotic organic synthesis, may not register if oxidized and added to the latter.
One key difference between the structures of the biogeochemical C and N cycles is that the latter is buffered by a well-mixed inorganic pool with a >Myr residence time (atmospheric N2; residence time ca. \(10^{8}\) yr), while the former is not (Figs. 2, 3). In the existing world, the atmospheric N2 pool buffers the exogenic N cycle against some types of continuous, coupled isotope excursions like those observed in the C cycle. These contrasting behaviors are reflected in the differences in the dynamics of the Phanerozoic C and N isotope records (Fig. 1). In the absence of life, with no N2 fixers, the fixation flux would be smaller and more reliant on lightning and other minor abiotic fixation fluxes (reviewed in Sect. 3.1). However, in the absence of denitrifiers, there also would have been no mechanism to return N back to the atmosphere. Without the strong negative isotopic fractionation associated with the modern denitrification flux, the nitrate pool in the ocean may not be as isotopically enriched as it is today (note, however, that atmospheric escape of low-\(\delta ^{15}\)N N2 could still have altered the nitrate record; Sect. 3.4).
Like the carbon cycle, the sulfur cycle is principally recorded in two sedimentary reservoirs that are distinguished in oxidation state and isotopic composition by (largely) a single, biologically mediated reaction (i.e., primary CO2 fixation vs. dissimilatory sulfate reduction; Figs. 2, 4). Like the nitrogen cycle, the sulfur cycle is buffered against sub-Myr changes by a large reservoir with a long residence time (i.e., the atmospheric N2 and marine sulfate pools). Unlike both C and N cycles, however, in the modern (oxidized) world, the principal metabolic reaction of the S cycle that introduces reduced sulfur to the sedimentary record—dissimilatory sulfate reduction—largely occurs in depositional environments that are in inconsistent communication with the global substrate pool. This variability in the degree of openness at the site of sulfur cycling is reflected in the high variability in the \(\delta ^{34}\)S composition of the Phanerozoic sedimentary pyrite record at any one time (Fig. 1). On an abiotic earth, in addition to a lack of microbial sulfate reduction, thermal sulfate reduction—which requires reaction with an electron donor (often, hydrocarbons)—would also have been reduced. The oxidation of volcanic sulfide would have been reduced, so the isotopic composition of any pyrite in the geologic record would more likely reflect its mantle source, rather than carry a metabolic signature of sulfate reduction. Gas-phase chemistry in an anoxic atmosphere is expected to produce S-MIF, which may be observable both in the atmospheric and the sedimentary components of the surface sulfur reservoir. Under an anoxic atmosphere, which is expected to produce S-MIF, the absence of this isotopic anomaly in sedimentary sulfur repositories (e.g., sulfate evaporites, sedimentary pyrite) may be indicative of intense microbial sulfur cycling.
Within all three of these cycles, the absence of phototrophy may all but remove the dependence of nutrient consumption on light penetration in the ocean. The lack of a biological pump would probably result in a more homogeneous ocean column due to a weaker decoupling between the surface and deep ocean. Furthermore, without nitrate utilization by organisms and burial in organic form, and without sulfate reduction in sediments, the concentration of nitrate and sulfate may have accumulated in the oceans over geologic time. In response to such accumulations, burial/subduction in the form of evaporitic salts may become an increasingly important means of export from the surface to the mantle.
Synthesizing the above discussion, in the absence of life, across the C, N and S cycles, we might expect co-localized pools to be dominantly either reduced or oxidized because their simultaneous existence is unstable. On the other hand, in the absence of enzymes to catalyze catabolic reactions, sluggish kinetics may nonetheless allow disequilibrated species to coexist. Where such oxidised and reduced pools do coexist, we might expect this disequilibrium to be reflected in non-equilibrium isotope fractionations. On a global scale, without similarly sized pools with significant partitioning of isotopes between phases, we might also expect isotopic excursions to be more modest in size. However, without the buffering capacity of differently sized organic reservoirs, we expect isotopic perturbations to be rapid with slow recoveries, and without a parallel expression in paired pools (such as \(\delta ^{13}\)C\(_{\text{org}}\) and \(\delta ^{13}\)C\(_{\text{carb}}\)). Though limited by temporal resolution, there is some evidence for this behavior in the Archean (Fig. 1).
Our framework for interpreting geochemical observations as biosignatures on another planet is informed by—and limited by—our understanding of the imprint of biology on the geochemical records of our own. In this chapter, we have described present understanding of how life on Earth influences and is influenced by the exospheric cycles of carbon, nitrogen and sulfur. We have reviewed evidence for past changes in Earth’s biosphere as told through the sedimentary isotopic record of these three elements. Clearly, many aspects of these records remain unresolved. In order to resolve these various uncertainties, we expect that research into the C, N, and S isotopic signatures in past terrestrial environments will continue, and continue to serve as guides for these elemental cycles beyond this planet.
References
C. Achtnich, F. Bak, R. Conrad, Competition for electron donors among nitrate reducers, ferric iron reducers, sulfate reducers, and methanogens in anoxic paddy soil. Biol. Fertil. Soils 19(1), 65–72 (1995)
M. Ader, P. Sansjofre, G.P. Halverson, V. Busigny, R.I. Trindade, M. Kunzmann, A.C. Nogueira, Ocean redox structure across the late neoproterozoic oxygenation event: a nitrogen isotope perspective. Earth Planet. Sci. Lett. 396, 1–13 (2014)
P. Aharon, B. Fu, Microbial sulfate reduction rates and sulfur and oxygen isotope fractionations at oil and gas seeps in deepwater Gulf of Mexico. Geochim. Cosmochim. Acta 64(2), 233–246 (2000)
P. Aharon, B. Fu, Sulfur and oxygen isotopes of coeval sulfate–sulfide in pore fluids of cold seep sediments with sharp redox gradients. Chem. Geol. 195(1), 201–218 (2003)
B. Alexander, J. Savarino, N.I. Barkov, R.J. Delmas, M.H. Thiemens, Climate driven changes in the oxidation pathways of atmospheric sulfur. Geophys. Res. Lett. 29(14), 30-1–30-4 (2002)
B. Alexander, R.J. Park, D.J. Jacob, Q.B. Li, R.M. Yantosca, J. Savarino, C.C.W. Lee, M.H. Thiemens, Sulfate formation in sea-salt aerosols: constraints from oxygen isotopes. J. Geophys. Res., Atmos. 110(D10), D10307 (2005)
B. Alexander, D.J. Allman, H.M. Amos, T.D. Fairlie, J. Dachs, D.A. Hegg, R.S. Sletten, Isotopic constraints on the formation pathways of sulfate aerosol in the marine boundary layer of the subtropical northeast Atlantic Ocean. J. Geophys. Res., Atmos. 117(D6), D06304 (2012)
T. Algeo, G. Luo, H. Song, T. Lyons, D. Canfield, Reconstruction of secular variation in seawater sulfate concentrations. Biogeosciences 12(7), 2131–2151 (2015)
F. Alhaique, D. Giacchetti, M. Marchetti, F.M. Riccieri, Effect of surfactant monomers on chloramphenicol association to an albuminlecithin complex: a model for modified drug absorption. J. Pharm. Pharmacol. 27(11), 811–817 (1975)
J.F. Allen, B. Thake, W.F. Martin, Nitrogenase inhibition limited oxygenation of Earth’s proterozoic atmosphere. Trends Plant Sci. 24(11), 1022–1031 (2019)
R. Aller, Sedimentary diagenesis, depositional environments, and benthic fluxes, in Treatise on Geochemistry (Elsevier, Amsterdam, 2014), pp. 293–334
A. Amrani, Organosulfur compounds: molecular and isotopic evolution from biota to oil and gas. Annu. Rev. Earth Planet. Sci. 42(1), 733–768 (2014)
A.D. Anbar, A.H. Knoll, Proterozoic ocean chemistry and evolution: a bioinorganic bridge? Science 297(5584), 1137–1142 (2002)
A.D. Anbar, Y. Duan, T.W. Lyons, G.L. Arnold, B. Kendall, R.A. Creaser, A.J. Kaufman, G.W. Gordon, C. Scott, J. Garvin, R. Buick, A whiff of oxygen before the great oxidation event? Science 317(5846), 1903–1906 (2007)
G. Antler, A. Pellerin, A critical look at the combined use of sulfur and oxygen isotopes to study microbial metabolisms in methane-rich environments. Front. Microbiol. 9, 519 (2018)
G. Antler, A.V. Turchyn, V. Rennie, B. Herut, O. Sivan, Coupled sulfur and oxygen isotope insight into bacterial sulfate reduction in the natural environment. Geochim. Cosmochim. Acta 118, 98–117 (2013)
G. Antler, A.V. Turchyn, B. Herut, A. Davies, V.C.F. Rennie, O. Sivan, Sulfur and oxygen isotope tracing of sulfate driven anaerobic methane oxidation in estuarine sediments. Estuar. Coast. Shelf Sci. 142, 4–11 (2014)
G. Antler, A.V. Turchyn, S. Ono, O. Sivan, T. Bosak, Combined 34S, 33S and 18O isotope fractionations record different intracellular steps of microbial sulfate reduction. Geochim. Cosmochim. Acta 203, 364–380 (2017)
D. Archer, B. Buffett, V. Brovkin, Ocean methane hydrates as a slow tipping point in the global carbon cycle. Proc. Natl. Acad. Sci. USA 106(49), 20596–20601 (2009)
A.R. Babbin, R.G. Keil, A.H. Devol, B.B. Ward, Organic matter stoichiometry, flux, and oxygen control nitrogen loss in the ocean. Science 344(6182), 406–408 (2014)
D. Babikov, Recombination reactions as a possible mechanism of mass-independent fractionation of sulfur isotopes in the Archean atmosphere of Earth. Proc. Natl. Acad. Sci. 114(12), 3062–3067 (2017)
A. Bachan, K.V. Lau, M.R. Saltzman, E. Thomas, L.R. Kump, J.L. Payne, American journal of science: a model for the decrease in amplitude of carbon isotope excursions across the Phanerozoic. Am. J. Sci. 317(6), 641–676 (2017)
M.P. Badger, T.B. Chalk, G.L. Foster, P.R. Bown, S.J. Gibbs, P.F. Sexton, D.N. Schmidt, H. Pälike, A. Mackensen, R.D. Pancost, Insensitivity of alkenone carbon isotopes to atmospheric CO2 at low to moderate CO2 levels. Clim. Past 15(2), 539–554 (2019)
F. Bak, N. Pfennig, Chemolithotrophic growth of Desulfovibrio sulfodismutans sp. nov. by disproportionation of inorganic sulfur compounds. Arch. Microbiol. 147(2), 184–189 (1987)
N. Balci, W.C. Shanks, B. Mayer, K.W. Mandernack, Oxygen and sulfur isotope systematics of sulfate produced by bacterial and abiotic oxidation of pyrite. Geochim. Cosmochim. Acta 71(15), 3796–3811 (2007)
J.L. Banner, G.N. Hanson, Calculation of simultaneous isotopic and trace element variations during water-rock interaction with applications to carbonate diagenesis. Geochim. Cosmochim. Acta 54(11), 3123–3137 (1990)
H. Bao, Sulfate: a time capsule for Earth’s O2, O3, and H2O. Chem. Geol. 395, 108–118 (2015)
H. Bao, D.R. Marchant, Quantifying sulfate components and their variations in soils of the McMurdo Dry Valleys, Antarctica. J. Geophys. Res., Atmos. 111(D16), D16301 (2006)
H. Bao, M.H. Thiemens, J. Farquhar, D.A. Campbell, C.C.-W. Lee, K. Heine, D.B. Loope, Anomalous 17O compositions in massive sulphate deposits on the Earth. Nature 406(6792), 176–178 (2000)
H. Bao, G.M. Michalski, M.H. Thiemens, Sulfate oxygen-17 anomalies in desert varnishes. Geochim. Cosmochim. Acta 65(13), 2029–2036 (2001a)
H. Bao, M.H. Thiemens, K. Heine, Oxygen-17 excesses of the central namib gypcretes: spatial distribution. Earth Planet. Sci. Lett. 192(2), 125–135 (2001b)
H. Bao, J.R. Lyons, C. Zhou, Triple oxygen isotope evidence for elevated CO2 levels after a neoproterozoic glaciation. Nature 453(7194), 504–506 (2008)
H. Bao, I.J. Fairchild, P.M. Wynn, C. Spötl, Stretching the envelope of past surface environments: neoproterozoic Glacial Lakes from Svalbard. Science 323(5910), 119–122 (2009)
H. Bao, S. Yu, D.Q. Tong, Massive volcanic SO2 oxidation and sulphate aerosol deposition in Cenozoic North America. Nature 465(7300), 909–912 (2010)
Y.M. Bar-On, R. Phillips, R. Milo, The biomass distribution on Earth. Proc. Natl. Acad. Sci. USA 115(25), 6506–6511 (2018)
B. Batchelor, A.W. Lawrence, A kinetic model for autotrophic denitrification using elemental sulfur. Water Res. 12(12), 1075–1084 (1978)
N.H. Batjes, Total carbon and nitrogen in the soils of the world. Eur. J. Soil Sci. 65(1), 10–21 (2014)
T. Bauersachs, S. Schouten, J. Compaore, U. Wollenzien, L.J. Stal, J.S. Sinninghe Damste, Nitrogen isotopic fractionation associated with growth on dinitrogen gas and nitrate by cyanobacteria. Limnol. Oceanogr. 54(4), 1403–1411 (2009)
D.A. Beard, H. Qian, Relationship between thermodynamic driving force and one-way fluxes in reversible processes. PLoS ONE 2(1), 1–4 (2007)
V. Beaumont, F. Robert, Nitrogen isotope ratios of kerogens in precambrian cherts: a record of the evolution of atmosphere chemistry? Precambrian Res. 96(1–2), 63–82 (1999)
G.E. Bebout, M.L. Fogel, Nitrogen-isotope compositions of metasedimentary rocks in the Catalina Schist, California: implications for metamorphic devolatilization history. Geochim. Cosmochim. Acta 56(7), 2839–2849 (1992)
G.E. Bebout, M.L. Fogel, P. Cartigny, Nitrogen: highly volatile yet surprisingly compatible. Elements 9(5), 333–338 (2013)
A. Bekker, H.D. Holland, Oxygen overshoot and recovery during the early paleoproterozoic. Earth Planet. Sci. Lett. 317(318), 295–304 (2012)
A. Bekker, H.D. Holland, P.L. Wang, D. Rumble, H.J. Stein, J.L. Hannah, L.L. Coetzee, N.J. Beukes, Dating the rise of atmospheric oxygen. Nature 427(6970), 117–120 (2004)
I.A. Berg, D. Kockelkorn, W.H. Ramos-Vera, R.F. Say, J. Zarzycki, M. Hügler, B.E. Alber, G. Fuchs, Autotrophic carbon fixation in archaea. Nat. Rev. Microbiol. 8(6), 447–460 (2010)
R.A. Berner, Models for carbon and sulfur cycles and atmospheric oxygen; application to Paleozoic geologic history. Am. J. Sci. 287(3), 177–196 (1987)
R.A. Berner, D.E. Canfield, A new model for atmospheric oxygen over Phanerozoic time. Am. J. Sci. 289(4), 333–361 (1989)
R.A. Berner, Z. Kothavala, Geocarb III: a revised model of atmospheric CO2 over Phanerozoic time. Am. J. Sci. 301(2), 182–204 (2001)
V.J. Bertics, J.A. Sohm, T. Treude, C.E.T. Chow, D.G. Capone, J.A. Fuhrman, W. Ziebis, Burrowing deeper into benthic nitrogen cycling: the impact of bioturbation on nitrogen fixation coupled to sulfate reduction. Mar. Ecol. Prog. Ser. 409, 1–15 (2010)
E. Bertran, A. Waldeck, B.A. Wing, I. Halevy, W.D. Leavitt, A.S. Bradley, D.T. Johnston, Oxygen isotope effects during microbial sulfate reduction: applications to sediment cell abundances. ISME J. 14(6), 1508–1519 (2020)
R.I. Bickley, V. Vishwanathan, Photocatalytically induced fixation of molecular nitrogen by near UV radiation. Nature 280(5720), 306–308 (1979)
I.N. Bindeman, J.M. Eiler, B.A. Wing, J. Farquhar, Rare sulfur and triple oxygen isotope geochemistry of volcanogenic sulfate aerosols. Geochim. Cosmochim. Acta 71(9), 2326–2343 (2007)
L.R. Bird, K.S. Dawson, G.L. Chadwick, J.M. Fulton, V.J. Orphan, K.H. Freeman, Carbon isotopic heterogeneity of coenzyme F430 and membrane lipids in methane-oxidizing archaea. Geobiology 17(6), 611–627 (2019)
N. Blair, A. Leu, E. Munoz, J. Olsen, E. Kwong, D. Des Marais, Carbon isotopic fractionation in heterotrophic microbial metabolism. Appl. Environ. Microbiol. 50(4), 996–1001 (1985)
C.C. Blair, S. D’Hondt, A.J. Spivack, R.H. Kingsley, Radiolytic hydrogen and microbial respiration in subsurface sediments. Astrobiology 7(6), 951–970 (2007)
R.E. Blankenship, M.T. Madigan, C.E. Bauer (eds.), Anoxygenic Photosynthetic Bacteria, Advances in Photosynthesis and Respiration (Springer, Dordrecht, 2004)
M.B. Blaser, L.K. Dreisbach, R. Conrad, Carbon isotope fractionation of 11 acetogenic strains grown on H2 and CO2. Appl. Environ. Microbiol. 79(6), 1787–1794 (2013)
A.J. Boller, P.J. Thomas, C.M. Cavanaugh, K.M. Scott, Low stable carbon isotope fractionation by coccolithophore RubisCO. Geochim. Cosmochim. Acta 75(22), 7200–7207 (2011)
A.J. Boller, P.J. Thomas, C.M. Cavanaugh, K.M. Scott, Isotopic discrimination and kinetic parameters of RubisCO from the marine bloom-forming diatom, Skeletonema costatum. Geobiology 13(1), 33–43 (2015)
M.E. Böttcher, B. Oelschläger, T. Höpner, H.-J. Brumsack, J. Rullkötter, Sulfate reduction related to the early diagenetic degradation of organic matter and “black spot” formation in tidal sandflats of the German Wadden Sea (southern North Sea): stable isotope (13C, 34S, 18O) and other geochemical results. Org. Geochem. 29(5–7), 1517–1530 (1998)
M.E. Böttcher, B. Thamdrup, T. Vennemann, Oxygen and sulfur isotope fractionation during anaerobic bacterial disproportionation of elemental sulfur. Geochim. Cosmochim. Acta 65(10), 1601–1609 (2001)
M.E. Böttcher, B. Thamdrup, M. Gehre, A. Theune, 34S/32S and 18O/16O fractionation during sulfur disproportionation by desulfobulbus propionicus. Geomicrobiol. J. 22(5), 219–226 (2005)
S.R. Boyd, P. Philippot, Precambrian ammonium biogeochemistry: a study of the Moine metasediments, Scotland. Chem. Geol. 144(3–4), 257–268 (1998)
A.S. Bradley, W.D. Leavitt, D.T. Johnston, Revisiting the dissimilatory sulfate reduction pathway. Geobiology 9(5), 446–457 (2011)
A.S. Bradley, W.D. Leavitt, M. Schmidt, A.H. Knoll, P.R. Girguis, D.T. Johnston, Patterns of sulfur isotope fractionation during microbial sulfate reduction. Geobiology 14(1), 91–101 (2016)
S.D. Bridgham, J.P. Megonigal, J.K. Keller, N.B. Bliss, C. Trettin, The carbon balance of North American wetlands. Wetlands 26(4), 889–916 (2006)
P. Brimblecombe, The global sulfur cycle, in Treatise on Geochemistry, vol. 10, 2nd edn. (Elsevier, Amsterdam, 2013), pp. 559–591
H.G. Britton, The Ussing relationship and chemical reactions: possible application to enzymatic investigations. Nature 205(4978), 1323–1324 (1965)
J.J. Brocks, G.D. Love, R.E. Summons, A.H. Knoll, G.A. Logan, S.A. Bowden, Biomarker evidence for green and purple sulphur bacteria in a stratified palaeoproterozoic sea. Nature 437(7060), 866–870 (2005)
J.J. Brocks, A.J. Jarrett, E. Sirantoine, C. Hallmann, Y. Hoshino, T. Liyanage, The rise of algae in cryogenian oceans and the emergence of animals. Nature 548(7669), 578–581 (2017)
W.S. Broecker, J. van Donk, Insolation Changes, Ice Volumes, and the O18 Record in Deep-Sea Cores Rev. Geophys. 8(1), 169–198 (1970)
B. Brunner, S.M. Bernasconi, A revised isotope fractionation model for dissimilatory sulfate reduction in sulfate reducing bacteria. Geochim. Cosmochim. Acta 69(20), 4759–4771 (2005)
B. Brunner, S. Contreras, M.F. Lehmann, O. Matantseva, M. Rollog, T. Kalvelage, G. Klockgether, G. Lavik, M.S.M. Jetten, B. Kartal, M.M.M. Kuypers, Nitrogen isotope effects induced by anammox bacteria. Proc. Natl. Acad. Sci. 110(47), 18994–18999 (2013)
R.N. Bryant, C. Jones, M.R. Raven, M.L. Gomes, W.M. Berelson, A.S. Bradley, D.A. Fike, Sulfur isotope analysis of microcrystalline iron sulfides using secondary ion mass spectrometry imaging: extracting local paleo-environmental information from modern and ancient sediments. Rapid Commun. Mass Spectrom. 33(5), 491–502 (2019)
C. Buchwald, K. Grabb, C.M. Hansel, S.D. Wankel, Constraining the role of iron in environmental nitrogen transformations: dual stable isotope systematics of abiotic NO2- reduction by Fe(II) and its production of N2O. Geochim. Cosmochim. Acta 186, 1–12 (2016)
R. Buick, D.J. Des Marais, A.H. Knoll, Stable isotopic compositions of carbonates from the mesoproterozoic bangemall group, northwestern Australia. Chem. Geol. 123(1–4), 153–171 (1995)
R.J. Buresh, J.T. Moraghan, Chemical reduction of nitrate by ferrous iron. J. Environ. Qual. 5(3), 320–325 (1976)
A. Burke, K.A. Moore, M. Sigl, D.C. Nita, J.R. McConnell, J.F. Adkins, Stratospheric eruptions from tropical and extra-tropical volcanoes constrained using high-resolution sulfur isotopes in ice cores. Earth Planet. Sci. Lett. 521, 113–119 (2019)
V. Busigny, O. Lebeau, M. Ader, B. Krapež, A. Bekker, Nitrogen cycle in the late Archean ferruginous ocean. Chem. Geol. 362, 115–130 (2013)
N.J. Butterfield, Macroevolution and macroecology through deep time. Palaeontology 50(1), 41–55 (2007)
D.E. Canfield, A new model for proterozoic ocean chemistry. Nature 396(6710), 450–453 (1998)
D.E. Canfield, Biogeochemistry of sulfur isotopes. Rev. Mineral. Geochem. 43, 606–636 (2001)
D.E. Canfield, The evolution of the Earth surface sulfur reservoir. Am. J. Sci. 304(10), 839–861 (2004)
D.E. Canfield, J. Farquhar, Animal evolution, bioturbation, and the sulfate concentration of the oceans. Proc. Natl. Acad. Sci. USA 106(20), 8123–8127 (2009)
D.E. Canfield, R. Raiswell, The evolution of the sulfur cycle. Am. J. Sci. 299(7–9), 697–723 (1999)
D.E. Canfield, A. Teske, Late proterozoic rise in atmospheric oxygen concentration inferred from phylogenetic and sulphur-isotope studies. Nature 382(6587), 127–132 (1996)
D.E. Canfield, M.T. Rosing, C. Bjerrum, Early anaerobic metabolisms. Philos. Trans. R. Soc. Lond. B, Biol. Sci. 361(1474), 1819–1834 (2006)
D.E. Canfield, J. Farquhar, A.L. Zerkle, High isotope fractionations during sulfate reduction in a low-sulfate euxinic ocean analog. Geology 38(5), 415–418 (2010)
J.D. Caranto, K.M. Lancaster, Nitric oxide is an obligate bacterial nitrification intermediate produced by hydroxylamine oxidoreductase. Proc. Natl. Acad. Sci. USA 114(31), 8217–8222 (2017)
J.D. Caranto, A.C. Vilbert, K.M. Lancaster, Nitrosomonas europaea cytochrome P460 is a direct link between nitrification and nitrous oxide emission. Proc. Natl. Acad. Sci. USA 113(51), 14704–14709 (2016)
E.J. Carpenter, H.R. Harvey, F. Brian, D.G. Capone, Biogeochemical tracers of the marine cyanobacterium trichodesmium. Deep-Sea Res., Part 1, Oceanogr. Res. Pap. 44(1), 27–38 (1997)
P. Cartigny, B. Marty, Nitrogen isotopes and mantle geodynamics: the emergence of life and the atmosphere–crust–mantle connection. Elements 9(5), 359–366 (2013)
K.L. Casciotti, Inverse kinetic isotope fractionation during bacterial nitrite oxidation. Geochim. Cosmochim. Acta 73(7), 2061–2076 (2009)
D.C. Catling, M.W. Claire, How Earth’s atmosphere evolved to an oxic state: a status report. Earth Planet. Sci. Lett. 237(1–2), 1–20 (2005)
D.C. Catling, K.J. Zahnle, The Archean atmosphere. Sci. Adv. 6(9), eaax1420 (2020)
A.R. Cavazos, M. Taillefert, Y. Tang, J.B. Glass, Kinetics of nitrous oxide production from hydroxylamine oxidation by birnessite in seawater. Mar. Chem. 202, 49–57 (2018)
T.E. Cerling, J.M. Harris, B.J. MacFadden, M.G. Leakey, J. Quade, V. Eisenmann, J.R. Ehieringer, Global vegetation change through the Miocene/Pliocene boundary. Nature 389(6647), 153–158 (1997)
L.A. Chambers, P.A. Trudinger, Are thiosulfate and trithionate intermediates in dissimilatory sulfate reduction? J. Bacteriol. 123(1), 36–40 (1975)
S. Chen, A.C. Gagnon, J.F. Adkins, Carbonic anhydrase, coral calcification and a new model of stable isotope vital effects. Geochim. Cosmochim. Acta 236, 179–197 (2018)
M.I. Chicarelli, J.M. Hayes, B.N. Popp, C.B. Eckardt, J.R. Maxwell, Carbon and nitrogen isotopic compositions of alkyl porphyrins from the Triassic Serpiano oil shale. Geochim. Cosmochim. Acta 57(6), 1307–1311 (1993)
P. Ciais, C. Sabine, G. Bala, L. Bopp, V. Brovkin, J. Canadell, A. Chhabra, R. DeFries, J. Galloway, M. Heimann, C. Jones, C.L. Quéré, R. Myneni, S. Piao, P. Thornton, Carbon and other biogeochemical cycles, in Climate Change 2013 the Physical Science Basis: Working Group i Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, vol. 9781107057 (2013), pp. 465–570
M.W. Claire, J.F. Kasting, S.D. Domagal-Goldman, E.E. Stüeken, R. Buick, V.S. Meadows, Modeling the signature of sulfur mass-independent fractionation produced in the Archean atmosphere. Geochim. Cosmochim. Acta 141, 365–380 (2014)
G.E. Claypool, in Ventilation of Marine Sediments Indicated by Depth Profiles of Pore Water Sulfate and \(\delta^{34}\)S. The Geochemical Society Special Publications, vol. 9 (Elsevier, Amsterdam, 2004), pp. 59–65
H.G. Close, R. Bovee, A. Pearson, Inverse carbon isotope patterns of lipids and kerogen record heterogeneous primary biomass. Geobiology 9(3), 250–265 (2011)
A.J. Coby, F.W. Picardal, Inhibition of NO3- and NO2- reduction by microbial Fe(III) reduction: evidence of a reaction between NO2- and cell surface-bound Fe2+. Appl. Environ. Microbiol. 71(9), 5267–5274 (2005)
R.E. Criss, Principles of Stable Isotope Distribution (Oxford University Press, London, 1999)
P.W. Crockford, J.A. Hayles, H. Bao, N.J. Planavsky, A. Bekker, P.W. Fralick, G.P. Halverson, T.H. Bui, Y. Peng, B.A. Wing, Triple oxygen isotope evidence for limited mid-Proterozoic primary productivity. Nature 559(7715), 613–616 (2018)
P.W. Crockford, M. Kunzmann, A. Bekker, J. Hayles, H. Bao, G.P. Halverson, Y. Peng, T.H. Bui, G.M. Cox, T.M. Gibson, S. Wörndle, R. Rainbird, A. Lepland, N.L. Swanson-Hysell, S. Master, B. Sreenivas, A. Kuznetsov, V. Krupenik, B.A. Wing, Claypool Continued: Extending the Isotopic Record of Sedimentary Sulfate (2019)
S.A. Crowe, G. Paris, S. Katsev, C. Jones, S.-T. Kim, A.L. Zerkle, S. Nomosatryo, D.A. Fowle, J.F. Adkins, A.L. Sessions, J. Farquhar, D.E. Canfield, Sulfate was a trace constituent of Archean seawater. Science 346(6210), 735–739 (2014)
H. Daims, E.V. Lebedeva, P. Pjevac, P. Han, C. Herbold, M. Albertsen, N. Jehmlich, M. Palatinszky, J. Vierheilig, A. Bulaev, R.H. Kirkegaard, M. Von Bergen, T. Rattei, B. Bendinger, P.H. Nielsen, M. Wagner, Complete nitrification by nitrospira bacteria. Nature 528(7583), 504–509 (2015)
T. Dalsgaard, B. Thamdrup, L. Farías, N.P. Revsbech, Anammox and denitrification in the oxygen minimum zone of the eastern South Pacific. Limnol. Oceanogr. 57(5), 1331–1346 (2012)
S.O. Danielache, C. Eskebjerg, M.S. Johnson, Y. Ueno, N. Yoshida, High-precision spectroscopy of 32S, 33S and 34S sulfur dioxide: ultraviolet absorption cross sections and isotope effects. J. Geophys. Res., Atmos. 113(D17), D17314 (2008)
S.O. Danielache, S. Hattori, M.S. Johnson, Y. Ueno, S. Nanbu, N. Yoshida, Photoabsorption cross-section measurements of 32S, 33S, 34S, and 36S sulfur dioxide for the B1B1-X1A1 absorption band. J. Geophys. Res., Atmos. 117(D24), D24301 (2012)
S.O. Danielache, S. Tomoya, A. Kondorsky, I. Tokue, S. Nanbu, Nonadiabatic calculations of ultraviolet absorption cross section of sulfur monoxide: isotopic effects on the photodissociation reaction. J. Chem. Phys. 140(4), 044319 (2014)
A.E. Dekas, R.S. Poretsky, V.J. Orphan, Deep-sea archaea fix and share nitrogen in methane-consuming microbial consortia. Science 326(5951), 422–426 (2009)
C.C. Delwiche, P.L. Steyn, Nitrogen isotope fractionation in soils and microbial reactions. Environ. Sci. Technol. 4(11), 929–935 (1970)
B. Demirel, P. Scherer, The roles of acetotrophic and hydrogenotrophic methanogens during anaerobic conversion of biomass to methane: a review. Rev. Environ. Sci. Biotechnol. 7(2), 173–190 (2008)
M.J. DeNiro, S. Epstein, Mechanism of carbon isotope fractionation associated with lipid synthesis. Science 197(4300), 261–263 (1977)
M.J. DeNiro, S. Epstein, Influence of diet on the distribution of carbon isotopes in animals. Geochim. Cosmochim. Acta 42(5), 495–506 (1978)
D.J. Des Marais, Isotopic evolution of the biogeochemical carbon cycle during the Proterozoic Eon. Org. Geochem. 27(5–6), 185–193 (1997)
D.J. Des Marais, Isotopic evolution of the biogeochemical carbon cycle during the precambrian. Rev. Mineral. Geochem. 43(1), 555–578 (2001)
P. Dhakal, C.J. Matocha, F.E. Huggins, M.M. Vandiviere, Nitrite reactivity with magnetite. Environ. Sci. Technol. 47(12), 6206–6213 (2013)
G.R. Dickens, Rethinking the global carbon cycle with a large, dynamic and microbially mediated gas hydrate capacitor. Earth Planet. Sci. Lett. 213(3–4), 169–183 (2003)
A.G. Dickson, C. Goyet, Handbook of methods for the analysis of the various parameters of the carbon dioxide system in sea water. U. S. Department of Energy 1994(September):176, 187 (1994)
T.A. Doane, The abiotic nitrogen cycle. ACS Earth Space Chem. 1(7), 411–421 (2017)
S.D. Domagal-Goldman, J.F. Kasting, D.T. Johnston, J. Farquhar, Organic haze, glaciations and multiple sulfur isotopes in the mid-Archean era. Earth Planet. Sci. Lett. 269(1–2), 29–40 (2008)
H.L. Drake, J.M. Akagi, Characterization of a novel thiosulfate-forming enzyme isolated from Desulfovibrio vulgaris. J. Bacteriol. 132(1), 132–138 (1977)
J.L. Eigenbrode, K.H. Freeman, Late Archean rise of aerobic microbial ecosystems. Proc. Natl. Acad. Sci. USA 103(43), 15759–15764 (2006)
D.L. Eldridge, J. Farquhar, Rates and multiple sulfur isotope fractionations associated with the oxidation of sulfide by oxygen in aqueous solution. Geochim. Cosmochim. Acta 237, 240–260 (2018)
Y. Endo, S.O. Danielache, Y. Ueno, S. Hattori, M.S. Johnson, N. Yoshida, H.G. Kjaergaard, Photoabsorption cross-section measurements of 32S, 33S, 34S and 36S sulfur dioxide from 190 to 220 nm. J. Geophys. Res., Atmos. 120(6), 2546–2557 (2015)
Y. Endo, Y. Ueno, S. Aoyama, S.O. Danielache, Sulfur isotope fractionation by broadband uv radiation to optically thin so2 under reducing atmosphere. Earth Planet. Sci. Lett. 453, 9–22 (2016)
G. Etiope, K.R. Lassey, R.W. Klusman, E. Boschi, Reappraisal of the fossil methane budget and related emission from geologic sources. Geophys. Res. Lett. 35(9), L09307 (2008)
P.G. Falkowski, The Biological and Geological Contingencies for the Rise of Oxygen on Earth (2011)
P. Falkowski, R.J. Scholes, E. Boyle, J. Canadell, D. Canfield, J. Elser, N. Gruber, K. Hibbard, P. Hogberg, S. Linder, F.T. Mackenzie, B. Moore, T. Pedersen, Y. Rosental, S. Seitzinger, V. Smetacek, W. Steffen, The global carbon cycle: a test of our knowledge of Earth as a system. Science 290(5490), 291–296 (2000)
J. Farquhar, M.H. Thiemens, Oxygen cycle of the Martian atmosphere-regolith system: \(\Delta^{17}\)O of secondary phases in Nakhla and Lafayette. J. Geophys. Res., Planets 105(E5), 11991–11997 (2000)
G.D. Farquhar, M.H. O’Leary, J.A. Berry, On the relationship between carbon isotope discrimination and the intercellular carbon dioxide concentration in leaves. Aust. J. Plant Physiol. 9(2), 121–137 (1982)
G.D. Farquhar, J.R. Ehleringer, K.T. Hubick, Carbon isotope discrimination and photosynthesis. Annu. Rev. Plant Physiol. Plant Mol. Biol. 40(1), 503–537 (1989)
J. Farquhar, H. Bao, M. Thiemens, Atmospheric influence of Earth’s earliest sulfur cycle. Science 289(5480), 756–758 (2000)
J. Farquhar, J. Savarino, S. Airieau, M.H. Thiemens, Observation of wavelength-sensitive mass-independent sulfur isotope effects during SO2 photolysis: implications for the early atmosphere. J. Geophys. Res., Planets 106(E12), 32829–32839 (2001)
J. Farquhar, D.T. Johnston, B.A. Wing, K.S. Habicht, D.E. Canfield, S. Airieau, M.H. Thiemens, Multiple sulphur isotopic interpretations of biosynthetic pathways: implications for biological signatures in the sulphur isotope record. Geobiology 1(1), 27–36 (2003)
J. Farquhar, D.T. Johnston, B.A. Wing, Implications of conservation of mass effects on mass-dependent isotope fractionations: influence of network structure on sulfur isotope phase space of dissimilatory sulfate reduction. Geochim. Cosmochim. Acta 71(24), 5862–5875 (2007)
H.A. Favre, W.H. Powell, Nomenclature of Organic Chemistry (Royal Chem. Soc., London, 2014)
K. Fennel, M. Follows, P.G. Falkowski, The co-evolution of the nitrogen, carbon and oxygen cycles in the proterozoic ocean. Am. J. Sci. 305(6-8 SPEC. ISS.), 526–545 (2005)
J.G. Ferry, How to make a living by exhaling methane. Annu. Rev. Microbiol. 64(1), 453–473 (2010)
V. Fichtner, H. Strauss, A. Immenhauser, D. Buhl, R.D. Neuser, A. Niedermayr, Diagenesis of carbonate associated sulfate. Chem. Geol. 463, 61–75 (2017)
D.A. Fike, A.S. Bradley, C.V. Rose, Rethinking the ancient sulfur cycle. Annu. Rev. Earth Planet. Sci. 43(1), 593–622 (2015)
K. Finster, Microbiological disproportionation of inorganic sulfur compounds. J. Sulfur Chem. 29(3–4), 281–292 (2008)
W.W. Fischer, J. Hemp, J.E. Johnson, Evolution of oxygenic photosynthesis. Annu. Rev. Earth Planet. Sci. 44(1), 647–683 (2016)
D. Fowler, M. Coyle, U. Skiba, M.A. Sutton, J.N. Cape, S. Reis, L.J. Sheppard, A. Jenkins, B. Grizzetti, J.N. Galloway, P. Vitousek, A. Leach, A.F. Bouwman, K. Butterbach-Bahl, F. Dentener, D. Stevenson, M. Amann, M. Voss, The global nitrogen cycle in the twenty-first century. Philos. Trans. R. Soc. Lond. B, Biol. Sci. 368(1621), 20130164 (2013)
H.B. Franz, A.C. McAdam, D.W. Ming, C. Freissinet, P.R. Mahaffy, D.L. Eldridge, W.W. Fischer, J.P. Grotzinger, C.H. House, J.A. Hurowitz, S.M. McLennan, S.P. Schwenzer, D.T. Vaniman, P.D. Archer, S.K. Atreya, P.G. Conrad, J.W. Dottin, J.L. Eigenbrode, K.A. Farley, D.P. Glavin, S.S. Johnson, C.A. Knudson, R.V. Morris, R. Navarro-González, A.A. Pavlov, R. Plummer, E.B. Rampe, J.C. Stern, A. Steele, R.E. Summons, B. Sutter, Large sulfur isotope fractionations in Martian sediments at Gale crater. Nat. Geosci. 10(9), 658–662 (2017)
T. Freudenthal, T. Wagner, F. Wenzhöfer, M. Zabel, G. Wefer, Early diagenesis of organic matter from sediments of the Eastern subtropical Atlantic: evidence from stable nitrogen and carbon isotopes. Geochim. Cosmochim. Acta 65(11), 1795–1808 (2001)
C. Frey, J.W. Dippner, M. Voss, Close coupling of N-cycling processes expressed in stable isotope data at the redoxcline of the Baltic Sea. Glob. Biogeochem. Cycles 28(9), 974–991 (2014)
N.U. Frigaard, C. Dahl, Sulfur metabolism in phototrophic sulfur bacteria. Adv. Microb. Physiol. 54, 103–200 (2008)
E.D. Galbraith, M. Kienast, A.L. Albuquerque, M.A. Altabet, F. Batista, D. Bianchi, S.E. Calvert, S. Contreras, X. Crosta, R. De Pol-Holz, N. Dubois, J. Etourneau, R. Francois, T.C. Hsu, T. Ivanochko, S.L. Jaccard, S.J. Kao, T. Kiefer, S. Kienast, M.F. Lehmann, P. Martinez, M. McCarthy, A.N. Meckler, A. Mix, J. Möbius, T.F. Pedersen, L. Pichevin, T.M. Quan, R.S. Robinson, E. Ryabenko, A. Schmittner, R. Schneider, A. Schneider-Mor, M. Shigemitsu, D. Sinclair, C. Somes, A.S. Studer, J.E. Tesdal, R. Thunell, J.Y. Terence Yang, The acceleration of oceanic denitrification during deglacial warming. Nat. Geosci. 6(7), 579–584 (2013)
J.N. Galloway, D.G. Capone, E.W. Boyer, R.W. Howarth, S.P. Seitzinger, G.P. Asner, C.C. Cleveland, P.A. Green, E.A. Holland, D.M. Karl, A.F. Michales, J.H. Porter, A.R. Townsend, C.J. Vorosmarty, Nitrogen cycles: past, present, and future. Biogeochemistry 70(153), 226 (2004)
R.M. Garrels, A. Lerman, Coupling of the sedimentary sulfur and carbon cycles; an improved model. Am. J. Sci. 284(9), 989–1007 (1984)
J. Garvin, R. Buick, A.D. Anbar, G.L. Arnold, A.J. Kaufman, Isotopic evidence for an aerobic nitrogen cycle in the latest Archean. Science 323(5917), 1045–1048 (2009)
B.C. Gill, T.W. Lyons, M.R. Saltzman, Parallel, high-resolution carbon and sulfur isotope records of the evolving Paleozoic marine sulfur reservoir. Palaeogeogr. Palaeoclimatol. Palaeoecol. 256(3–4), 156–173 (2007)
B.C. Gill, T.W. Lyons, T.D. Frank, Behavior of carbonate-associated sulfate during meteoric diagenesis and implications for the sulfur isotope paleoproxy. Geochim. Cosmochim. Acta 72(19), 4699–4711 (2008)
L.V. Godfrey, P.G. Falkowski, The cycling and redox state of nitrogen in the archaean ocean. Nat. Geosci. 2(10), 725–729 (2009)
T.P. Goldstein, Z. Aizenshtat, Thermochemical sulfate reduction a review. J. Therm. Anal. 42(1), 241–290 (1994)
K.C. Grabb, C. Buchwald, C.M. Hansel, S.D. Wankel, A dual nitrite isotopic investigation of chemodenitrification by mineral-associated Fe(II) and its production of nitrous oxide. Geochim. Cosmochim. Acta 196, 388–402 (2017)
J. Granger, D.M. Sigman, M.F. Lehmann, P.D. Tortell, Nitrogen and oxygen isotope fractionation during dissimilatory nitrate reduction by denitrifying bacteria. Limnol. Oceanogr. 53(6), 2533–2545 (2008)
E.M. Griffith, A. Paytan, Barite in the ocean - occurrence, geochemistry and palaeoceanographic applications. Sedimentology 59(6), 1817–1835 (2012)
N. Gruber, The marine nitrogen cycle: overview and challenges, in Nitrogen in the Marine Environment (Elsevier, Amsterdam, 2008), pp. 1–50
A.P. Gumsley, K.R. Chamberlain, W. Bleeker, U. Söderlund, M.O. De Kock, E.R. Larsson, A. Bekker, Timing and tempo of the great oxidation event. Proc. Natl. Acad. Sci. USA 114(8), 1811–1816 (2017)
K.S. Habicht, D.E. Canfield, J. Rethmeier, Sulfur isotope fractionation during bacterial reduction and disproportionation of thiosulfate and sulfite. Geochim. Cosmochim. Acta 62(15), 2585–2595 (1998)
K.S. Habicht, M. Gade, B. Thamdrup, P. Berg, D.E. Canfield, Calibration of sulfate levels in the Archean ocean. Science 298(5602), 2372–2374 (2002)
I. Halevy, Production, preservation, and biological processing of mass-independent sulfur isotope fractionation in the Archean surface environment. Proc. Natl. Acad. Sci. 110(44), 17644–17649 (2013)
I. Halevy, D.T. Johnston, D.P. Schrag, Explaining the structure of the Archean mass-independent sulfur isotope record. Science 329(5988), 204–207 (2010)
I. Halevy, S.E. Peters, W.W. Fischer, Sulfate burial constraints on the Phanerozoic sulfur cycle. Science 337(6092), 331–334 (2012)
H.C.B. Hansen, C.B. Koch, H. Nancke-Krogh, O.K. Borggaard, J. Sørensen, Abiotic nitrate reduction to ammonium: key role of green rust. Environ. Sci. Technol. 30(6), 2053–2056 (1996)
A.K. Hardison, C.K. Algar, A.E. Giblin, J.J. Rich, Influence of organic carbon and nitrate loading on partitioning between dissimilatory nitrate reduction to ammonium (DNRA) and N2 production. Geochim. Cosmochim. Acta 164, 146–160 (2015)
C.E. Harman, A.A. Pavlov, D. Babikov, J.F. Kasting, Chain formation as a mechanism for mass-independent fractionation of sulfur isotopes in the Archean atmosphere. Earth Planet. Sci. Lett. 496, 238–247 (2018)
D.L. Hartmann, A.M. Klein Tank, M. Rusticucci, L.V. Alexander, S. Brönnimann, Y.A.R. Charabi, F.J. Dentener, E.J. Dlugokencky, D.R. Easterling, A. Kaplan, B.J. Soden, P.W. Thorne, M. Wild, P. Zhai, Observations: atmosphere and surface, in Climate Change 2013 the Physical Science Basis: Working Group I Contribution to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change (2013), pp. 159–254
J.M. Hayes, Global methanotrophy of the Archean-proterozoic transition, in Early Life on Earth, Nobel Symposium No. 84 (Columbia University Press, New York, 1994), pp. 220–235
J.M. Hayes, Fractionation of carbon and hydrogen isotopes in biosynthetic processes, in Stable Isotope Geochemistry, vol. 43 (2001), pp. 225–277
J.M. Hayes, J.R. Waldbauer, The carbon cycle and associated redox processes through time. Philos. Trans. R. Soc. Lond. B, Biol. Sci. 361(1470), 931–950 (2006)
J.M. Hayes, H. Strauss, A.J. Kaufman, The abundance of 13C in marine organic matter and isotopic fractionation in the global biogeochemical cycle of carbon during the past 800 Ma. Chem. Geol. 161(1), 103–125 (1999)
J.D. Hays, J. Imbrie, N.J. Shackleton, Variations in the Earth’s Orbit: Pacemaker of the Ice Ages (1976)
J. Heil, H. Vereecken, N. Brüggemann, A review of chemical reactions of nitrification intermediates and their role in nitrogen cycling and nitrogen trace gas formation in soil. Eur. J. Soil Sci. 67(1), 23–39 (2016)
S.P. Hesselbo, D.R. Gröcke, H.C. Jenkyns, C.J. Bjerrum, P. Farrimond, H.S. Morgans Bell, O.R. Green, Massive dissociation of gas hydrate during a Jurassic oceanic anoxic event. Nature 406(6794), 392–395 (2000)
M.B. Higgins, R.S. Robinson, J.M. Husson, S.J. Carter, A. Pearson, Dominant eukaryotic export production during ocean anoxic events reflects the importance of recycled NH4+. Proc. Natl. Acad. Sci. USA 109(7), 2269–2274 (2012)
K.U. Hinrichs, R.E. Summons, V. Orphan, S.P. Sylva, J.M. Hayes, Molecular and isotopic analysis of anaerobic methaneoxidizing communities in marine sediments. Org. Geochem. 31(12), 1685–1701 (2000)
K.U. Hinrichs, J.M. Hayes, W. Bach, A.J. Spivackl, L.R. Hmelo, N.G. Holm, C.G. Johnson, S.P. Sylva, Biological formation of ethane and propane in the deep marine subsurface. Proc. Natl. Acad. Sci. USA 103(40), 14684–14689 (2006)
M.P. Hoch, M.L. Fogel, D.L. Kirchman, Isotope fractionation associated with ammonium uptake by a marine bacterium. Limnol. Oceanogr. 37(7), 1447–1459 (1992)
M.S. Hodgskiss, P.W. Crockford, Y. Peng, B.A. Wing, T.J. Horner, A productivity collapse to end Earth’s great oxidation. Proc. Natl. Acad. Sci. USA 116(35), 17207–17212 (2019)
P.F. Hoffman, The great oxidation and a siderian snowball Earth: MIF-S based correlation of paleoproterozoic glacial epochs. Chem. Geol. 362, 143–156 (2013)
P.F. Hoffman, A.J. Kaufman, G.P. Halverson, D.P. Schrag, A neoproterozoic snowball Earth. Science 281(5381), 1342–1346 (1998)
B.M. Hoffman, D. Lukoyanov, Z.Y. Yang, D.R. Dean, L.C. Seefeldt, Mechanism of nitrogen fixation by nitrogenase: the next stage. Chem. Rev. 114(8), 4041–4062 (2014)
T. Höink, A. Lenardic, A.M. Jellinek, Earth’s thermal evolution with multiple convection modes: a Monte-Carlo approach. Phys. Earth Planet. Inter. 221, 22–26 (2013)
H.D. Holland, The Chemical Evolution of the Atmosphere and Oceans (Princeton University Press, Princeton, 1984)
H.D. Holland, The oxygenation of the atmosphere and oceans. Philos. Trans. R. Soc. Lond. B, Biol. Sci. 361(1470), 903–915 (2006)
B.D. Holt, R. Kumar, P.T. Cunningham, Primary sulfates in atmospheric sulfates: estimation by oxygen isotope ratio measurements. Science 217(4554), 51–53 (1982)
B. Hönisch, N.G. Hemming, D. Archer, M. Siddall, J.F. McManus, Atmospheric carbon dioxide concentration across the mid-Pleistocene transition. Science 324(5934), 1551–1554 (2009)
C.H. House, J.W. Schopf, K.D. McKeegan, C.D. Coath, Carbon Isotopic Composition of Individual Precambrian Microfossils. Geology 28(8), 707–710 (2000)
E. Ingall, R. Jahnke, Evidence for enhanced phosphorus regeneration from marine sediments overlain by oxygen depleted waters. Geochim. Cosmochim. Acta 58(11), 2571–2575 (1994)
G. Izon, A.L. Zerkle, I. Zhelezinskaia, J. Farquhar, R.J. Newton, S.W. Poulton, J.L. Eigenbrode, M.W. Claire, Multiple oscillations in Neoarchaean atmospheric chemistry. Earth Planet. Sci. Lett. 431, 264–273 (2015)
G. Izon, A.L. Zerkle, K.H. Williford, J. Farquhar, S.W. Poulton, M.W. Claire, Biological regulation of atmospheric chemistry en route to planetary oxygenation. Proc. Natl. Acad. Sci. 114(13), E2571–E2579 (2017)
O. Jagoutz, F.A. Macdonald, L. Royden, Low-latitude arc-continent collision as a driver for global cooling. Proc. Natl. Acad. Sci. USA 113(18), 4935–4940 (2016)
J.P. Jasper, J.M. Hayes, A carbon isotope record of CO2 levels during the late quaternary. Nature 347(6292), 462–464 (1990)
H.C. Jenkyns, Geochemistry of oceanic anoxic events. Geochem. Geophys. Geosyst. 11(3), Q03004 (2010)
B. Johnson, C. Goldblatt, The nitrogen budget of Earth. Earth-Sci. Rev. 148, 150–173 (2015)
D.T. Johnston, Multiple sulfur isotopes and the evolution of Earth’s surface sulfur cycle. Earth-Sci. Rev. 106(1–2), 161–183 (2011)
D.T. Johnston, J. Farquhar, B.A. Wing, A.J. Kaufman, D.E. Canfield, K.S. Habicht, Multiple sulfur isotope fractionations in biological systems: a case study with sulfate reducers and sulfur disproportionators. Am. J. Sci. 305(6-8 SPEC. ISS.), 645–660 (2005)
D.T. Johnston, B.C. Gill, A. Masterson, E. Beirne, K.L. Casciotti, A.N. Knapp, W. Berelson, Placing an upper limit on cryptic marine sulphur cycling. Nature 513(7519), 530–533 (2014)
B.B. Jørgensen, Mineralization of organic matter in the sea bed - the role of sulphate reduction. Nature 296(5858), 643–645 (1982)
B.B. Jørgensen, A thiosulfate shunt in the sulfur cycle of marine sediments. Science 249(4965), 152–154 (1990)
B.B. Jørgensen, D.J. Des Marais, Competition for sulfide among colorless and purple sulfur bacteria in cyanobacterial mats. FEMS Microbiol. Lett. 38(3), 179–186 (1986)
B.B. Jørgensen, A.J. Findlay, A. Pellerin, The biogeochemical sulfur cycle of marine sediments. Front. Microbiol. 10, 849 (2019)
C.K. Junium, M.A. Arthur, Nitrogen cycling during the Cretaceous, Cenomanian- Turonian Oceanic Anoxic Event II. Geochem. Geophys. Geosyst. 8(3), Q03002 (2007)
C.K. Junium, M.A. Arthur, K.H. Freeman, Compound-specific \(\delta{15}\)N and chlorin preservation in surface sediments of the Peru Margin with implications for ancient bulk \(\delta^{15}\)N records. Geochim. Cosmochim. Acta 160, 306–318 (2015)
C.K. Junium, A.J. Dickson, B.T. Uveges, Perturbation to the nitrogen cycle during rapid Early Eocene global warming. Nat. Commun. 9, 1 (2018)
S. Kadoya, D.C. Catling, Constraints on hydrogen levels in the Archean atmosphere based on detrital magnetite. Geochim. Cosmochim. Acta 262, 207–219 (2019)
M.J. Kampschreur, R. Kleerebezem, W.W. de Vet, M.C. Van Loosdrecht, Reduced iron induced nitric oxide and nitrous oxide emission. Water Res. 45(18), 5945–5952 (2011)
A. Kampschulte, H. Strauss, The sulfur isotopic evolution of Phanerozoic seawater based on the analysis of structurally substituted sulfate in carbonates. Chem. Geol. 204(3–4), 255–286 (2004)
A. Kampschulte, P. Bruckschen, H. Strauss, The sulphur isotopic composition of trace sulphates in carboniferous brachiopods: implications for coeval seawater, correlation with other geochemical cyles and isotope stratigraphy. Chem. Geol. 175(1–2), 149–173 (2001)
J.A. Karhu, H.D. Holland, Carbon isotopes and the rise of atmospheric oxygen. Geology 24(10), 867–870 (1996)
A.J. Kaufman, A.H. Knoll, Neoproterozoic variations in the C-isotopic composition of seawater: stratigraphic and biogeochemical implications. Precambrian Res. 73(1–4), 27–49 (1995)
C.D. Keeling, The Suess effect: 13Carbon-14Carbon interrelations. Environ. Int. 2(4–6), 229–300 (1979)
R.F. Keeling, S.C. Piper, M. Heimann, Global and hemispheric CO2 sinks deduced from changes in atmospheric O2 concentration. Nature 381(6579), 218–221 (1996)
S. Kirschke, P. Bousquet, P. Ciais, M. Saunois, J.G. Canadell, E.J. Dlugokencky, P. Bergamaschi, D. Bergmann, D.R. Blake, L. Bruhwiler, P. Cameron-Smith, S. Castaldi, F. Chevallier, L. Feng, A. Fraser, M. Heimann, E.L. Hodson, S. Houweling, B. Josse, P.J. Fraser, P.B. Krummel, J.F. Lamarque, R.L. Langenfelds, C. Le Quéré, V. Naik, S. O’doherty, P.I. Palmer, I. Pison, D. Plummer, B. Poulter, R.G. Prinn, M. Rigby, B. Ringeval, M. Santini, M. Schmidt, D.T. Shindell, I.J. Simpson, R. Spahni, L.P. Steele, S.A. Strode, K. Sudo, S. Szopa, G.R. Van Der Werf, A. Voulgarakis, M. Van Weele, R.F. Weiss, J.E. Williams, G. Zeng, Three Decades of Global Methane Sources and Sinks (2013)
J.L. Kirschvink, Late proterozoic low-latitude global glaciation: the snowball Earth, in The Proterozoic Biosphere: A Multidisciplinary Study, vol. 52, ed. by J.W. Schopf, C. Klein (1992), pp. 51–52, Chap. 2.3
J.L. Kirschvink, R.E. Kopp, Palaeoproterozoic ice houses and the evolution of oxygen-mediating enzymes: the case for a late origin of photosystem II. Philos. Trans. R. Soc. Lond. B, Biol. Sci. 363(1504), 2755–2765 (2008)
J.L. Kirschvink, E.J. Gaidos, L.E. Bertani, N.J. Beukes, J. Gutzmer, L.N. Maepa, R.E. Steinberger, Paleoproterozoic snowball Earth: extreme climatic and geochemical global change and its biological consequences. Proc. Natl. Acad. Sci. USA 97(4), 1400–1405 (2000)
N. Klueglein, A. Kappler, Abiotic oxidation of Fe(II) by reactive nitrogen species in cultures of the nitrate-reducing Fe(II) oxidizer Acidovorax sp. BoFeN1 - questioning the existence of enzymatic Fe(II) oxidation. Geobiology 11(2), 180–190 (2013)
A.N. Knapp, The sensitivity of marine N2 fixation to dissolved inorganic nitrogen. Front. Microbiol. 3, 374 (2012)
K. Knittel, A. Boetius, Anaerobic oxidation of methane: progress with an unknown process. Annu. Rev. Microbiol. 63(1), 311–334 (2009)
M.C. Koehler, E.E. Stüeken, M.A. Kipp, R. Buick, A.H. Knoll, Spatial and temporal trends in precambrian nitrogen cycling: a mesoproterozoic offshore nitrate minimum. Geochim. Cosmochim. Acta 198, 315–337 (2017)
K. Koop-Jakobsen, A.E. Giblin, The effect of increased nitrate loading on nitrate reduction via denitrification and DNRA in salt marsh sediments. Limnol. Oceanogr. 55(2), 789–802 (2010)
R.E. Kopp, J.L. Kirschvink, I.A. Hilburn, C.Z. Nash, The paleoproterozoic snowball Earth: a climate disaster triggered by the evolution of oxygenic photosynthesis. Proc. Natl. Acad. Sci. USA 102(32), 11131–11136 (2005)
J. Korenaga, in Archean Geodynamics and the Thermal Evolution of Earth. Geophysical Monograph Series, vol. 164 (Blackwell Publishing Ltd., Oxford, 2006), pp. 7–32
S. Kotelnikova, Microbial production and oxidation of methane in deep subsurface. Earth-Sci. Rev. 58(3–4), 367–395 (2002)
B. Kraft, H.E. Tegetmeyer, R. Sharma, M.G. Klotz, T.G. Ferdelman, R.L. Hettich, J.S. Geelhoed, M. Strous, The environmental controls that govern the end product of bacterial nitrate respiration. Science 345(6197), 676–679 (2014)
J. Krissansen-Totton, R. Buick, D.C. Catling, A statistical analysis of the carbon isotope record from the Archean to Phanerozoic and implications for the rise of oxygen. Am. J. Sci. 315(4), 275–316 (2015)
J. Krissansen-Totton, S. Olson, D.C. Catling, Disequilibrium biosignatures over Earth history and implications for detecting exoplanet life. Sci. Adv. 4(1), eaao5747 (2018)
P.P. Kumar, G.K. Manohar, S.S. Kandalgaonkar, Global distribution of nitric oxide produced by lightning and its seasonal variation. J. Geophys. Res. 100(D6), 11203–11208 (1995)
L.R. Kump, M.E. Barley, Increased subaerial volcanism and the rise of atmospheric oxygen 2.5 billion years ago. Nature 448(7157), 1033–1036 (2007)
L.R. Kump, R.M. Garrels, Modeling atmospheric O2 in the global sedimentary redox cycle. Am. J. Sci. 286(5), 337–360 (1986)
L.R. Kump, C. Junium, M.A. Arthur, A. Brasier, A. Fallick, V. Melezhik, A. Lepland, A.E. Črne, G. Luo, Isotopic evidence for massive oxidation of organic matter following the great oxidation event. Science 334(6063), 1694–1696 (2011)
M. Kusakabe, B.W. Robinson, Oxygen and sulfur isotope equilibria in the BaSO4-HSO4-H2O system from 110 to 350C and applications. Geochim. Cosmochim. Acta 41(8), 1033–1040 (1977)
M.M. Kuypers, Y. van Breugel, S. Schouten, E. Erba, J.S. Damsté, N2-fixing cyanobacteria supplied nutrient N for Cretaceous oceanic anoxic events. Geology 32(10), 853–856 (2004)
T.A. Laakso, D.P. Schrag, A small marine biosphere in the proterozoic. Geobiology 17(2), 161–171 (2019)
H. Lammer, A.L. Zerkle, S. Gebauer, N. Tosi, L. Noack, M. Scherf, E. Pilat-Lohinger, M. Güdel, J.L. Grenfell, M. Godolt, A. Nikolaou, Origin and evolution of the atmospheres of early Venus, Earth and Mars. Astron. Astrophys. Rev. 26(1), 2 (2018)
M. Laneuville, M. Kameya, H.J. Cleaves, Earth without life: a systems model of a global abiotic nitrogen cycle. Astrobiology 18(7), 897–914 (2018)
W.D. Leavitt, I. Halevy, A.S. Bradley, D.T. Johnston, Influence of sulfate reduction rates on the Phanerozoic sulfur isotope record. Proc. Natl. Acad. Sci. USA 110(28), 11244–11249 (2013)
C.C.-W. Lee, M.H. Thiemens, The \(\delta^{17}\)O and \(\delta^{18}\)O measurements of atmospheric sulfate from a coastal and high Alpine region: a mass-independent isotopic anomaly. J. Geophys. Res., Atmos. 106(D15), 17359–17373 (2001)
H. Lee, J.D. Muirhead, T.P. Fischer, C.J. Ebinger, S.A. Kattenhorn, Z.D. Sharp, G. Kianji, Massive and prolonged deep carbon emissions associated with continental rifting. Nat. Geosci. 9(2), 145–149 (2016)
C.-T.A. Lee, H. Jiang, R. Dasgupta, M. Torres, A framework for understanding whole-Earth carbon cycling, in Deep Carbon (Cambridge University Press, Cambridge, 2019), pp. 313–357
N. Lehnert, H.T. Dong, J.B. Harland, A.P. Hunt, C.J. White, Reversing nitrogen fixation. Nat. Rev. Chem. 2(10), 278–289 (2018)
S.L. Lewis, M.A. Maslin, Defining the Anthropocene. Nature 519(7542), 171–180 (2015)
G.N. Lewis, M. Randall, Thermodynamics and the Free Energy of Chemical Substances (McGraw-Hill, New York, 1923)
L.E. Lisiecki, M.E. Raymo, A Pliocene-Pleistocene stack of 57 globally distributed benthic \(\delta \)18O records. Paleoceanography 20(1), 1–17 (2005)
P. Liu, C.E. Harman, J.F. Kasting, Y. Hu, J. Wang, Can organic haze and O2 plumes explain patterns of sulfur mass-independent fractionation during the Archean? Earth Planet. Sci. Lett. 526, 115767 (2019)
R.M. Lloyd, Oxygen isotope behavior in the sulfate-water system. J. Geophys. Res. 73(18), 6099–6110 (1968)
G. Luo, S. Ono, N.J. Beukes, D.T. Wang, S. Xie, R.E. Summons, Rapid oxygenation of Earth’s atmosphere 2.33 billion years ago. Sci. Adv. 2(5), 1–10 (2016)
M. Luo, Z.Q. Chen, G.R. Shi, X. Feng, H. Yang, Y. Fang, Y. Li, Microbially induced sedimentary structures (MISSs) from the Lower Triassic Kockatea Formation, northern Perth Basin, Western Australia: Palaeoenvironmental implications. Palaeogeogr. Palaeoclimatol. Palaeoecol. 519, 236–247 (2019)
G.W. Luther, B. Sundby, B.L. Lewis, P.J. Brendel, N. Silverberg, Interactions of manganese with the nitrogen cycle: alternative pathways to dinitrogen. Geochim. Cosmochim. Acta 61(19), 4043–4052 (1997)
D. Lüthi, M. Le Floch, B. Bereiter, T. Blunier, J.M. Barnola, U. Siegenthaler, D. Raynaud, J. Jouzel, H. Fischer, K. Kawamura, T.F. Stocker, High-resolution carbon dioxide concentration record 650,000-800,000 years before present. Nature 453(7193), 379–382 (2008)
J.R. Lyons, Mass-independent fractionation of sulfur isotopes by isotope-selective photodissociation of so2. Geophys. Res. Lett. 34(22), L22811 (2007)
T.W. Lyons, C.T. Reinhard, N.J. Planavsky, The rise of oxygen in Earth’s early ocean and atmosphere. Nature 506(7488), 307–315 (2014)
F.A. Macdonald, N.L. Swanson-Hysell, Y. Park, L. Lisiecki, O. Jagoutz, Arc-continent collisions in the tropics set Earth’s climate state. Science 364(6436), 181–184 (2019)
S.A. Macko, M.L. Fogel, P. Hare, T. Hoering, Isotopic fractionation of nitrogen and carbon in the synthesis of amino acids by microorganisms. Chem. Geol., Isot. Geosci. Sect. 65(1), 79–92 (1987)
C. Magnabosco, L.H. Lin, H. Dong, M. Bomberg, W. Ghiorse, H. Stan-Lotter, K. Pedersen, T.L. Kieft, E. van Heerden, T.C. Onstott, The biomass and biodiversity of the continental subsurface. Nat. Geosci. 11(10), 707–717 (2018)
N. Mahmoudi, S.R. Beaupré, A.D. Steen, A. Pearson, Sequential bioavailability of sedimentary organic matter to heterotrophic bacteria. Environ. Microbiol. 19(7), 2629–2644 (2017)
B.D. Marino, M.B. McElroy, R.J. Salawitch, W.G. Spaulding, Glacial-to-interglacial variations in the carbon isotopic composition of atmospheric CO2. Nature 357(6378), 461–466 (1992)
W. Martens-Habbena, W. Qin, R.E. Horak, H. Urakawa, A.J. Schauer, J.W. Moffett, E.V. Armbrust, A.E. Ingalls, A.H. Devol, D.A. Stahl, The production of nitric oxide by marine ammonia-oxidizing archaea and inhibition of archaeal ammonia oxidation by a nitric oxide scavenger. Environ. Microbiol. 17(7), 2261–2274 (2015)
E. Martin, I. Bindeman, Mass-independent isotopic signatures of volcanic sulfate from three supereruption ash deposits in Lake Tecopa, California. Earth Planet. Sci. Lett. 282(1), 102–114 (2009)
A.P. Martin, D.J. Condon, A.R. Prave, A. Lepland, A Review of Temporal Constraints for the Palaeoproterozoic Large, Positive Carbonate Carbon Isotope Excursion (the Lomagundi-Jatuli Event). Earth-Sci. Rev. 127, 242–261 (2013)
E. Mason, M. Edmonds, A.V. Turchyn, Remobilization of crustal carbon may dominate volcanic arc emissions. Science 357(6348), 290–294 (2017)
A.L. Masterson, J. Farquhar, B.A. Wing, Sulfur mass-independent fractionation patterns in the broadband uv photolysis of sulfur dioxide: pressure and third body effects. Earth Planet. Sci. Lett. 306(3–4), 253–260 (2011)
A.L. Masterson, B.A. Wing, A. Paytan, J. Farquhar, D.T. Johnston, The minor sulfur isotope composition of Cretaceous and Cenozoic seawater sulfate. Paleoceanography 31(6), 779–788 (2016)
H.L. McClelland, J. Bruggeman, M. Hermoso, R.E. Rickaby, The origin of carbon isotope vital effects in coccolith calcite. Nat. Commun. 8, 1–16 (2017)
T.M. McCollom, E.L. Shock, Geochemical constraints on chemolithoautotrophic metabolism by microorganisms in seafloor hydrothermal systems. Geochim. Cosmochim. Acta 61(20), 4375–4391 (1997)
W.F. McDonough, S.s. Sun, The composition of the Earth. Chem. Geol. 120(3–4), 223–253 (1995)
N.R. McKenzie, B.K. Horton, S.E. Loomis, D.F. Stockli, N.J. Planavsky, C.T.A. Lee, Continental arc volcanism as the principal driver of icehouse-greenhouse variability. Science 352(6284), 444–447 (2016)
D.L. McRose, X. Zhang, A.M. Kraepiel, F.M. Morel, Diversity and activity of alternative nitrogenases in sequenced genomes and coastal environments. Front. Microbiol. 8, 267 (2017)
A.J. Medford, M.C. Hatzell, Photon-driven nitrogen fixation: current progress, thermodynamic considerations, and future outlook. ACS Catal. 7(4), 2624–2643 (2017)
M.P. Mehta, J.A. Baross, Nitrogen fixation at 92∘C by a hydrothermal vent archaeon. Science 314(5806), 1783–1786 (2006)
E.D. Melton, E.D. Swanner, S. Behrens, C. Schmidt, A. Kappler, The interplay of microbially mediated and abiotic reactions in the biogeochemical Fe cycle. Nat. Rev. Microbiol. 12(12), 797–808 (2014)
K.M. Meyer, L.R. Kump, Oceanic Euxinia in Earth history: causes and consequences. Annu. Rev. Earth Planet. Sci. 36(1), 251–288 (2008)
P.J. Mickler, L.A. Stern, J.L. Banner, Large kinetic isotope effects in modern speleothems. Bull. Geol. Soc. Am. 118(1–2), 65–81 (2006)
J.J. Middelburg, Marine Carbon Biogeochemistry. Springer Briefs in Earth System Sciences (Springer, Cham, 2019)
J.V. Mills, G. Antler, A.V. Turchyn, Geochemical evidence for cryptic sulfur cycling in salt Marsh sediments. Earth Planet. Sci. Lett. 453, 23–32 (2016)
M. Minagawa, E. Wada, Nitrogen isotope ratios of red tide organisms in the East China Sea: a characterization of biological nitrogen fixation. Mar. Chem. 19(3), 245–259 (1986)
K.D. Monson, J.M. Hayes, Biosynthetic control of the natural abundance of carbon 13 at specific positions within fatty acids in Saccharomyces cerevisiae. Isotopic fractionations in lipid synthesis as evidence for peroxisomal regulation. J. Biol. Chem. 257(10), 5568–5575 (1982)
M.A. Moran, B.P. Durham, Sulfur metabolites in the pelagic ocean. Nat. Rev. Microbiol. 17(11), 665–678 (2019)
R.D. Müller, M. Sdrolias, C. Gaina, W.R. Roest, Age, spreading rates, and spreading asymmetry of the world’s ocean crust. Geochem. Geophys. Geosyst. 9(4), 1–19 (2008)
T. Murakami, B. Sreenivas, S.D. Sharma, H. Sugimori, Quantification of atmospheric oxygen levels during the paleoproterozoic using paleosol compositions and iron oxidation kinetics. Geochim. Cosmochim. Acta 75(14), 3982–4004 (2011)
R. Navarro-González, M.J. Molina, L.T. Molina, Nitrogen fixation by volcanic lightning in the early Earth. Geophys. Res. Lett. 25(16), 3123–3126 (1998)
R. Navarro-González, C.P. McKay, D.N. Mvondo, A possible nitrogen crisis for archaean life due to reduced nitrogen fixation by lightning. Nature 412(6842), 61–64 (2001)
A. Neftel, H. Oeschger, J. Schwander, B. Stauffer, R. Zumbrunn, Ice core sample measurements give atmospheric CO2 content during the past 40,000 yr. Nature 295(5846), 220–223 (1982)
D.W. Nelson, J.M. Bremner, Gaseous products of nitrite decomposition in soils. Soil Biol. Biochem. 2(3), 203–204 (1970)
E. Noor, A. Flamholz, W. Liebermeister, A. Bar-Even, R. Milo, A note on the kinetics of enzyme action: a decomposition that highlights thermodynamic effects. FEBS Lett. 587(17), 2772–2777 (2013)
T.F. Oliveira, C. Vonrhein, P.M. Matias, S.S. Venceslau, I.A. Pereira, M. Archer, The crystal structure of Desulfovibrio vulgaris dissimilatory sulfite reductase bound to DsrC provides novel insights into the mechanism of sulfate respiration. J. Biol. Chem. 283(49), 34141–34149 (2008)
S. Ono, Photochemistry of sulfur dioxide and the origin of mass-independent isotope fractionation in Earth’s atmosphere. Annu. Rev. Earth Planet. Sci. 45, 301–329 (2017)
S. Ono, W.C. Shanks, O.J. Rouxel, D. Rumble, S-33 constraints on the seawater sulfate contribution in modern seafloor hydrothermal vent sulfides. Geochim. Cosmochim. Acta 71(5), 1170–1182 (2007)
S. Ono, A.R. Whitehill, J.R. Lyons, Contribution of isotopologue self-shielding to sulfur mass-independent fractionation during sulfur dioxide photolysis. J. Geophys. Res., Atmos. 118(5), 2444–2454 (2013)
V.J. Orphan, C.H. House, K.U. Hinrichs, K.D. McKeegan, E.F. DeLong, Multiple archaeal groups mediate methane oxidation in anoxic cold seep sediments. Proc. Natl. Acad. Sci. USA 99(11), 7663–7668 (2002)
C.J. Ottley, W. Davison, W.M. Edmunds, Chemical catalysis of nitrate reduction by iron(II). Geochim. Cosmochim. Acta 61(9), 1819–1828 (1997)
M. Pagani, K.H. Freeman, M.A. Arthur, Late miocene atmospheric CO2 concentrations and the expansion of C4 grasses. Science 285(5429), 876–879 (1999)
M. Pagani, J.C. Zachos, K.H. Freeman, B. Tipple, S. Bohaty, Atmospheric science: marked decline in atmospheric carbon dioxide concentrations during the Paleogene. Science 309(5734), 600–603 (2005)
M. Pagani, K. Caldeira, D. Archer, J.C. Zachos An Ancient Carbon Mystery. Science 341(5805), 1556–1557 (2006)
M. Pagani, M. Huber, Z. Liu, S.M. Bohaty, J. Henderiks, W. Sijp, S. Krishnan, R.M. DeConto, The role of carbon dioxide during the onset of Antarctic glaciation. Science 334(6060), 1261–1264 (2011)
H. Palme, H. O’Neill, Cosmochemical estimates of mantle composition, in Treatise on Geochemistry, vol. 3, 2nd edn. (2013), pp. 1–39
C.A. Partin, P.M. Sadler, Slow net sediment accumulation sets snowball Earth apart from all younger glacial episodes. Geology 44(12), 1019–1022 (2016)
V. Pasquier, P. Sansjofre, M. Rabineau, S. Revillon, J. Houghton, D.A. Fike, Pyrite sulfur isotopes reveal glacial-interglacial environmental changes. Proc. Natl. Acad. Sci. USA 114(23), 5941–5945 (2017)
A.A. Pavlov, J.F. Kasting, Mass-independent fractionation of sulfur isotopes in Archean sediments: strong evidence for an anoxic Archean atmosphere. Astrobiology 2(1), 27–41 (2002)
A.A. Pavlov, J.F. Kasting, L.L. Brown, K.A. Rages, R. Freedman, Greenhouse warming by CH4 in the atmosphere of early Earth. J. Geophys. Res., Planets 105(E5), 11981–11990 (2000)
A. Paytan, M. Kastner, D. Campbell, M.H. Thiemens, Sulfur isotopic composition of Cenozoic seawater sulfate. Science 282(5393), 1459–1462 (1998)
A. Pellerin, G. Antler, S.A. Holm, A.J. Findlay, P.W. Crockford, A.V. Turchyn, B.B. Jørgensen, K. Finster, Large sulfur isotope fractionation by bacterial sulfide oxidation. Sci. Adv. 5(7), eaaw1480 (2019)
I.A. Pereira, A.R. Ramos, F. Grein, M.C. Marques, S.M. da Silva, S.S. Venceslau, A comparative genomic analysis of energy metabolism in sulfate reducing bacteria and archaea. Front. Microbiol. 2, 69 (2011)
K.E. Peters, R.E. Sweeney, I.R. Kaplan, Correlation of carbon and nitrogen stable isotope ratios in sedimentary organic matter. Limnol. Oceanogr. 23(4), 598–604 (1978)
N.J. Planavsky, P. McGoldrick, C.T. Scott, C. Li, C.T. Reinhard, A.E. Kelly, X. Chu, A. Bekker, G.D. Love, T.W. Lyons, Widespread iron-rich conditions in the mid-Proterozoic ocean. Nature 477(7365), 448–451 (2011)
N.J. Planavsky, A. Bekker, A. Hofmann, J.D. Owens, T.W. Lyons, Sulfur record of rising and falling marine oxygen and sulfate levels during the Lomagundi event. Proc. Natl. Acad. Sci. USA 109(45), 18300–18305 (2012)
B.N. Popp, E.A. Laws, R.R. Bidigare, J.E. Dore, K.L. Hanson, S.G. Wakeham, Effect of phytoplankton cell geometry on carbon isotopic fractionation. Geochim. Cosmochim. Acta 62(1), 69–77 (1998)
D. Postma, Kinetics of nitrate reduction by detrital Fe(II)-silicates. Geochim. Cosmochim. Acta 54(3), 903–908 (1990)
S.W. Poulton, P.W. Fralick, D.E. Canfield, Spatial variability in oceanic redox structure 1.8 billion years ago. Nat. Geosci. 3(7), 486–490 (2010)
M.J. Prather, C.D. Holmes, J. Hsu, Reactive greenhouse gas scenarios: systematic exploration of uncertainties and the role of atmospheric chemistry. Geophys. Res. Lett. 39(9), L09803 (2012)
T.M. Present, G. Paris, A. Burke, W.W. Fischer, J.F. Adkins, Large carbonate associated sulfate isotopic variability between brachiopods, micrite, and other sedimentary components in Late Ordovician strata. Earth Planet. Sci. Lett. 432, 187–198 (2015)
R. Rabus, T.A. Hansen, F. Widdel, Dissimilatory sulfate- and sulfur-reducing prokaryotes, in The Prokaryotes: Prokaryotic Physiology and Biochemistry (Springer, Berlin, 2013), pp. 309–404
S.W. Ragsdale, Stealth reactions driving carbon fixation new twists to bacterial metabolic pathways that contribute to the global carbon cycle. Science 359(6375), 517–518 (2018)
V.A. Rakov, M.A. Uman, Lightning: Physics and Effects (Cambridge University Press, Cambridge, 2003)
S. Rakshit, C.J. Matocha, M.S. Coyne, Nitrite reduction by siderite. Soil Sci. Soc. Am. J. 72(4), 1070–1077 (2008)
B. Rasmussen, A. Bekker, I.R. Fletcher, Correlation of Paleoproterozoic glaciations based on U-Pb zircon ages for tuff beds in the Transvaal and Huronian Supergroups. Earth Planet. Sci. Lett. 382, 173–180 (2013)
G.H. Rau, M.A. Arthur, W.E. Dean, 15N/14N variations in Cretaceous Atlantic sedimentary sequences: implication for past changes in marine nitrogen biogeochemistry. Earth Planet. Sci. Lett. 82(3–4), 269–279 (1987)
G.H. Rau, U. Riebesell, D. Wolf-Gladrow, A model of photosynthetic 13C fractionation by marine phytoplankton based on diffusive molecular CO2 uptake. Mar. Ecol. Prog. Ser. 133(1–3), 275–285 (1996)
M.R. Raven, A.L. Sessions, J.F. Adkins, R.C. Thunell, Rapid organic matter sulfurization in sinking particles from the Cariaco Basin water column. Geochim. Cosmochim. Acta 190, 175–190 (2016)
M.R. Raven, D.A. Fike, M.L. Gomes, S.M. Webb, A.S. Bradley, H.L.O. McClelland, Organic carbon burial during OAE2 driven by changes in the locus of organic matter sulfurization. Nat. Commun. 9, 3409 (2018)
W.S. Reeburgh, Oceanic Methane Biogeochemistry (2007)
H. Ren, D.M. Sigman, A.N. Meckler, B. Plessen, R.S. Robinson, Y. Rosenthal, G.H. Haug, Foraminiferal isotope evidence of reduced nitrogen fixation in the ice age Atlantic Ocean. Science 323(5911), 244–248 (2009)
V.C. Rennie, A.V. Turchyn, The preservation of \(\delta^{34}\mathrm{S}_{\mathrm{SO}_{4}}\) and \(\delta^{18}\mathrm{O}_{\mathrm{SO}_{4}}\) in carbonate-associated sulfate during marine diagenesis: a 25 Myr test case using marine sediments. Earth Planet. Sci. Lett. 395, 13–23 (2014)
V.C. Rennie, G. Paris, A.L. Sessions, S. Abramovich, A.V. Turchyn, J.F. Adkins, Cenozoic record of \(\delta^{34}\)S in foraminiferal calcite implies an early Eocene shift to deep-ocean sulfide burial. Nat. Geosci. 11(10), 761–765 (2018)
D. Rickard, G.W. Luther, Chemistry of iron sulfides. Chem. Rev. 107(2), 514–562 (2007)
B.A. Ridley, J.E. Dye, J.G. Walega, J. Zheng, F.E. Grahek, W. Rison, On the production of active nitrogen by thunderstorms over New Mexico. J. Geophys. Res., Atmos. 101(15), 20985–21005 (1996)
A.L. Robertson, J. Roadt, I. Halevy, J.F. Kasting, Greenhouse warming by nitrous oxide and methane in the Proterozoic Eon. Geobiology 9(4), 313–320 (2011)
A. Robock, Volcanic eruptions and climate. Rev. Geophys. 38(2), 191–219 (2000)
A.B. Romero, M.H. Thiemens, Mass-independent sulfur isotopic compositions in present-day sulfate aerosols. J. Geophys. Res., Atmos. 108(D16), 4524 (2003)
C.V. Rose, S.M. Webb, M. Newville, A. Lanzirotti, J.A. Richardson, N.J. Tosca, J.G. Catalano, A.S. Bradley, D.A. Fike, Insights into past ocean proxies from micron-scale mapping of sulfur species in carbonates. Geology 47(9), 833–837 (2019)
D.H. Rothman, J.M. Hayes, R.E. Summons, Dynamics of the Neoproterozoic carbon cycle. Proc. Natl. Acad. Sci. USA 100(14), 8124–8129 (2003)
M.J. Russell, A.J. Hall, W. Martin, Serpentinization as a source of energy at the origin of life. Geobiology 8(5), 355–371 (2010)
Z. Sade, I. Halevy, New constraints on kinetic isotope effects during CO2(aq) hydration and hydroxylation: revisiting theoretical and experimental data. Geochim. Cosmochim. Acta 214, 246–265 (2017)
G. Saleh, A.R. Oganov, Novel stable compounds in the C-H-O ternary system at high pressure. Sci. Rep. 6, 32486 (2016)
V.A. Samarkin, M.T. Madigan, M.W. Bowles, K.L. Casciotti, J.C. Priscu, C.P. McKay, S.B. Joye, Abiotic nitrous oxide emission from the hypersaline Don Juan Pond in Antarctica. Nat. Geosci. 3(5), 341–344 (2010)
A.A. Santos, S.S. Venceslau, F. Grein, W.D. Leavitt, C. Dahl, D.T. Johnston, I.A.C. Pereira, A protein trisulfide couples dissimilatory sulfate reduction to energy conservation. Science 350(6267), 1541–1545 (2015)
M. Saunois, A.R. Stavert, B. Poulter, P. Bousquet, J.G. Canadell, R.B. Jackson, P.A. Raymond, E.J. Dlugokencky, S. Houweling, P.K. Patra, P. Ciais, V.K. Arora, D. Bastviken, P. Bergamaschi, D.R. Blake, G. Brailsford, L. Bruhwiler, K.M. Carlson, M. Carrol, S. Castaldi, N. Chandra, C. Crevoisier, P.M. Crill, K. Covey, C.L. Curry, G. Etiope, C. Frankenberg, N. Gedney, M.I. Hegglin, L. Höglund-Isaksson, G. Hugelius, M. Ishizawa, A. Ito, G. Janssens-Maenhout, K.M. Jensen, F. Joos, T. Kleinen, P.B. Krummel, R.L. Langenfelds, G.G. Laruelle, L. Liu, T. Machida, S. Maksyutov, K.C. McDonald, J. McNorton, P.A. Miller, J.R. Melton, I. Morino, J. Müller, F. Murguia-Flores, V. Naik, Y. Niwa, S. Noce, S. O’Doherty, R.J. Parker, C. Peng, S. Peng, G.P. Peters, C. Prigent, R. Prinn, M. Ramonet, P. Regnier, W.J. Riley, J.A. Rosentreter, A. Segers, I.J. Simpson, H. Shi, S.J. Smith, L.P. Steele, B.F. Thornton, H. Tian, Y. Tohjima, F.N. Tubiello, A. Tsuruta, N. Viovy, A. Voulgarakis, T.S. Weber, M. van Weele, G.R. van der Werf, R.F. Weiss, D. Worthy, D. Wunch, Y. Yin, Y. Yoshida, W. Zhang, Z. Zhang, Y. Zhao, B. Zheng, Q. Zhu, Q. Zhu, Q. Zhuang, The global methane budget 2000–2017. Earth Syst. Sci. Data 12(3), 1561–1623 (2020)
J. Savarino, A. Romero, J. Cole-Dai, S. Bekki, M. Thiemens, UV-induced mass-independent sulfur isotope fractionation in stratospheric volcanic sulfate. Geophys. Res. Lett. 30(21), 2131 (2003)
E.A. Schauble, Applying stable isotope fractionation theory to new systems. Rev. Mineral. Geochem. 55, 65–111 (2004)
S.O. Schlanger, H.C. Jenkyns, Cretaceous oceanic anoxic events: causes and consequences. Geol. Mijnb. 55(3–4), 179–184 (1976)
M. Schobben, B. van de Schootbrugge, Increased stability in carbon isotope records reflects emerging complexity of the biosphere. Front. Earth Sci. 7, 87 (2019)
S. Schouten, H.M. Van Kaam-Peters, W.I.C. Rijpstra, M. Schoell, J.S. Sinninghe Damste, Effects of an oceanic anoxic event on the stable carbon isotopic composition of early Toarcian carbon. Am. J. Sci. 300(1), 1–22 (2000)
D.P. Schrag, J.A. Higgins, F.A. Macdonald, D.T. Johnston, Authigenic carbonate and the history of the global carbon cycle. Science 339(6119), 540–543 (2013)
S. Schwietzke, O.A. Sherwood, L.M. Bruhwiler, J.B. Miller, G. Etiope, E.J. Dlugokencky, S.E. Michel, V.A. Arling, B.H. Vaughn, J.W. White, P.P. Tans, Upward revision of global fossil fuel methane emissions based on isotope database. Nature 538(7623), 88–91 (2016)
A. Sclafani, L. Palmisano, M. Schiavello, N2 photoreduction and phenol and nitrophenol isomers photooxidation as examples of heterogeneous photocatalytic reactions. Res. Chem. Intermed. 18(2), 211–226 (1993)
S.P. Seitzinger, J.A. Harrison, E. Dumont, A.H.W. Beusen, A.F. Bouwman, Sources and delivery of carbon, nitrogen, and phosphorus to the coastal zone: an overview of global nutrient export from watersheds (NEWS) models and their application. Glob. Biogeochem. Cycles 19(4), GB4S05 (2005)
N.J. Shackleton, Oxygen isotopes, ice volume and sea level. Quat. Sci. Rev. 6(3–4), 183–190 (1987)
L. Shawar, I. Halevy, W. Said-Ahmad, S. Feinstein, V. Boyko, A. Kamyshny, A. Amrani, Dynamics of pyrite formation and organic matter sulfurization in organic-rich carbonate sediments. Geochim. Cosmochim. Acta 241, 219–239 (2018)
Y. Shen, R. Buick, D.E. Canfield, Isotopic evidence for microbial sulphate reduction in the early archaean era. Nature 410(6824), 77–81 (2001)
Y. Shen, J. Farquhar, A. Masterson, A.J. Kaufman, R. Buick, Evaluating the role of microbial sulfate reduction in the early Archean using quadruple isotope systematics. Earth Planet. Sci. Lett. 279(3–4), 383–391 (2009)
S.M. Sievert, C. Vetriani, Chemoautotrophy at deep-sea vents, past, present, and future. Oceanography 25(1), 218–233 (2012)
D.M. Sigman, E.A. Boyle, Glacial/interglacial variations in atmospheric carbon dioxide. Nature 407(6806), 859–869 (2000)
D.M. Sigman, K.L. Karsh, K.L. Casciotti, Ocean process tracers: nitrogen isotopes in the ocean, in Encyclopedia of Ocean Sciences, ed. by S. Edition, J.H. Steele (Academic Press, Oxford, 2009), pp. 40–54
M.S. Sim, T. Bosak, S. Ono, Large sulfur isotope fractionation does not require disproportionation. Science 333(6038), 74–77 (2011)
M.S. Sim, S. Ono, T. Bosak, Effects of iron and nitrogen limitation on sulfur isotope fractionation during microbial sulfate reduction. Appl. Environ. Microbiol. 78(23), 8368–8376 (2012)
J.S. Sinninghe Damste, J.W. De Leeuw, Analysis, structure and geochemical significance of organically-bound sulphur in the geosphere: state of the art and future research. Org. Geochem. 16(4–6), 1077–1101 (1990)
J.S. Sinninghe Damsté, M.D. Kok, J. Köster, S. Schouten, Sulfurized carbohydrates: an important sedimentary sink for organic carbon? Earth Planet. Sci. Lett. 164(1–2), 7–13 (1998)
R. Sirevåg, B.B. Buchanan, J.A. Berry, J.H. Troughton, Mechanisms of CO2 fixation in bacterial photosynthesis studied by the carbon isotope fractionation technique. Arch. Microbiol. 112(1), 35–38 (1977)
S.P. Slotznick, W.W. Fischer, Examining Archean methanotrophy. Earth Planet. Sci. Lett. 441, 52–59 (2016)
L.E. Snyder, D. Buhl, B. Zuckerman, P. Palmer, Microwave detection of interstellar formaldehyde. Phys. Rev. Lett. 22(13), 679–681 (1969)
E. Sofen, B. Alexander, S. Kunasek, The impact of anthropogenic emissions on atmospheric sulfate production pathways, oxidants, and ice core \(\Delta\)17O(SO42-). Atmos. Chem. Phys. 11, 3565–3578 (2011)
E.D. Sofen, B. Alexander, E.J. Steig, M.H. Thiemens, S.A. Kunasek, H.M. Amos, A.J. Schauer, M.G. Hastings, J. Bautista, T.L. Jackson, L.E. Vogel, J.R. Mcconnell, D.R. Pasteris, E.S. Saltzman, WAIS Divide ice core suggests sustained changes in the atmospheric formation pathways of sulfate and nitrate since the 19th century in the extratropical Southern Hemisphere. Atmos. Chem. Phys. 14(11), 5749–5769 (2014)
R.M. Soo, J. Hemp, P. Hugenholtz, Evolution of photosynthesis and aerobic respiration in the cyanobacteria. Free Radic. Biol. Med. 140, 200–205 (2019)
J. Sørensen, L. Thorling, Stimulation by lepidocrocite (7-FeOOH) of Fe(II)-dependent nitrite reduction. Geochim. Cosmochim. Acta 55(5), 1289–1294 (1991)
R. Spahni, R. Wania, L. Neef, M. Van Weele, I. Pison, P. Bousquet, C. Frankenberg, P.N. Foster, F. Joos, I.C. Prentice, P. Van Velthoven, Constraining global methane emissions and uptake by ecosystems. Biogeosciences 8(6), 1643–1665 (2011)
C.L. Stanton, C.T. Reinhard, J.F. Kasting, N.E. Ostrom, J.A. Haslun, T.W. Lyons, J.B. Glass, Nitrous oxide from chemodenitrification: a possible missing link in the proterozoic greenhouse and the evolution of aerobic respiration. Geobiology 16(6), 597–609 (2018)
C.J. Still, J.A. Berry, G.J. Collatz, R.S. DeFries, Global distribution of C3 and C4 vegetation: carbon cycle implications. Glob. Biogeochem. Cycles 17(1), 6-1–6-14 (2003)
D.A. Stolper, C.B. Keller, A record of deep-ocean dissolved O2 from the oxidation state of iron in submarine basalts. Nature 553(7688), 323–327 (2018)
K.L. Straub, M. Benz, B. Schink, F. Widdel, Anaerobic, nitrate-dependent microbial oxidation of ferrous iron. Appl. Environ. Microbiol. 62(4), 1458–1460 (1996)
H. Strauss, D.J. Des Marais, J.M. Hayers, R.E. Summons, Concentrations of organic carbon and maturities and elemental compositions of kerogens, in The Proterozoic Biosphere, a Multidisciplinary Study (1992), pp. 95–101
T.O. Strohm, B. Griffin, W.G. Zumft, B. Schink, Growth yields in bacterial denitrification and nitrate ammonification. Appl. Environ. Microbiol. 73(5), 1420–1424 (2007)
E.E. Stüeken, R. Buick, B.M. Guy, M.C. Koehler, Isotopic evidence for biological nitrogen fixation by molybdenum-nitrogenase from 3.2 Gyr. Nature 520(7549), 666–669 (2015a)
E.E. Stüeken, R. Buick, A.J. Schauer, Nitrogen isotope evidence for alkaline lakes on late Archean continents. Earth Planet. Sci. Lett. 411, 1–10 (2015b)
E.E. Stüeken, M.A. Kipp, M.C. Koehler, R. Buick, The evolution of Earth’s biogeochemical nitrogen cycle. Earth-Sci. Rev. 160, 220–239 (2016)
E.E. Stüeken, J. Zaloumis, J. Meixnerová, R. Buick, Differential metamorphic effects on nitrogen isotopes in kerogen extracts and bulk rocks. Geochim. Cosmochim. Acta 217, 80–94 (2017)
E.E. Stüeken, S.M. Som, M. Claire, S. Rugheimer, M. Scherf, L. Sproß, N. Tosi, Y. Ueno, H. Lammer, Mission to planet Earth: the first two billion years. Space Sci. Rev. 216(2), 31 (2020)
R.E. Summons, P.D. Franzmann, P.D. Nichols, Carbon isotopic fractionation associated with methylotrophic methanogenesis. Org. Geochem. 28(7–8), 465–475 (1998)
P.K. Swart, Global synchronous changes in the carbon isotopic composition of carbonate sediments unrelated to changes in the global carbon cycle. Proc. Natl. Acad. Sci. USA 105(37), 13741–13745 (2008)
P.K. Swart, The geochemistry of carbonate diagenesis: the past, present and future. Sedimentology 62(5), 1233–1304 (2015)
F. Tabataba-Vakili, J.L. Grenfell, J.-M. Grießmeier, H. Rauer, Atmospheric effects of stellar cosmic rays on Earth-like exoplanets orbiting M-dwarfs. Astron. Astrophys. 585, A96 (2016)
C. Tamocai, J.G. Canadell, E.A. Schuur, P. Kuhry, G. Mazhitova, S. Zimov, Soil organic carbon pools in the northern circumpolar permafrost region. Glob. Biogeochem. Cycles 23(2), GB2023 (2009)
G.G. Tcherkez, G.D. Farquhar, T.J. Andrews, Despite slow catalysis and confused substrate specificity, all ribulose bisphosphate carboxylases may be nearly perfectly optimized. Proc. Natl. Acad. Sci. USA 103(19), 7246–7251 (2006)
K. Tennakone, J.M. Bandara, C.T. Thaminimulla, W.D. Jayatilake, U.S. Ketipearachchi, O.A. Ileperuma, M.K. Priyadarshana, Photoreduction of dinitrogen to ammonia by ultrafine particles of Fe(O)OH formed by photohydrolysis of Iron(II) bicarbonate. Langmuir 7(10), 2166–2168 (1991)
B. Thamdrup, Bacterial manganese and iron reduction in aquatic sediments, in Advances in Microbial Ecology, ed. by B. Schink (Springer, Berlin, 2000), pp. 41–84
B. Thamdrup, K. Finster, J.W. Hansen, F. Bak, Bacterial disproportionation of elemental sulfur coupled to chemical reduction of iron or manganese. Appl. Environ. Microbiol. 59(1), 101–108 (1993)
B. Thamdrup, K. Finster, H. Fossing, J.W. Hansen, B.B. Jørgensen, Thiosulfate and sulfite distributions in porewater of marine sediments related to manganese, iron, and sulfur geochemistry. Geochim. Cosmochim. Acta 58(1), 67–73 (1994)
M.H. Thiemens, History and applications of mass-independent isotope effects. Annu. Rev. Earth Planet. Sci. 34, 217–262 (2006)
C. Thomazo, M. Ader, P. Philippot, Extreme 15N-enrichments in 2.72-Gyr-old sediments: evidence for a turning point in the nitrogen cycle. Geobiology 9(2), 107–120 (2011)
M.M. Tice, D.R. Lowe, Photosynthetic microbial mats in the 3,416-Myr-old ocean. Nature 431(7008), 549–552 (2004)
B. Tissot, D. Welte, Principles of Stable Isotope Distribution (Springer, Berlin, 1984)
C. Tornabene, R.C. Martindale, X.T. Wang, M.F. Schaller, Detecting photosymbiosis in fossil scleractinian corals. Sci. Rep. 7(1), 9465 (2017)
A.V. Turchyn, O. Sivan, D. Schrag, Oxygen isotopic composition of sulfate in deep sea pore fluid: evidence for rapid sulfur cycling. Geochim. Cosmochim. Acta 70(18), A660 (2006)
A.V. Turchyn, V. Brüchert, T.W. Lyons, G.S. Engel, N. Balci, D.P. Schrag, B. Brunner, Kinetic oxygen isotope effects during dissimilatory sulfate reduction: a combined theoretical and experimental approach. Geochim. Cosmochim. Acta 74(7), 2011–2024 (2010)
P. Van Cappellen, E.D. Ingall, Redox stabilization of the atmosphere and oceans by phosphorus-limited marine productivity. Science 271(5248), 493–496 (1996)
E.M. van den Berg, J.L. Rombouts, J.G. Kuenen, R. Kleerebezem, M.C. van Loosdrecht, Role of nitrite in the competition between denitrification and DNRA in a chemostat enrichment culture. AMB Express 7(1), 91 (2017)
M.T. Van Der Meer, S. Schouten, J.S. Sinninghe Damsté, The effect of the reversed tricarboxylic acid cycle on the 13C contents of bacterial lipids. Org. Geochem. 28(9–10), 527–533 (1998)
M.A. Van Kessel, D.R. Speth, M. Albertsen, P.H. Nielsen, H.J. Op Den Camp, B. Kartal, M.S. Jetten, S. Lücker, Complete nitrification by a single microorganism. Nature 528(7583), 555–559 (2015)
M. Vandenbroucke, C. Largeau, Kerogen origin, evolution and structure. Org. Geochem. 38(5), 719–833 (2007)
M. Vargas, K. Kashefi, E.L. Blunt-harris, D.R. Lovley, Fe (III) reduction on early Earth. Nature 395, 65–67 (1998)
J. Veizer, D. Ala, K. Azmy, P. Bruckschen, D. Buhl, F. Bruhn, G.A. Garden, A. Diener, S. Ebneth, Y. Godderis, T. Jasper, C. Korte, F. Pawellek, O.G. Podlaha, H. Strauss, 87Sr/86Sr, \(\delta\)13C and \(\delta\)18O evolution of Phanerozoic seawater. Chem. Geol. 161(1), 59–88 (1999)
S.S. Venceslau, Y. Stockdreher, C. Dahl, I.A. Pereira, The “bacterial heterodisulfide” DsrC is a key protein in dissimilatory sulfur metabolism. Biochim. Biophys. Acta, Bioenerg. 1837(7), 1148–1164 (2014)
A.R. Waldeck, B.R. Cowie, E. Bertran, B.A. Wing, I. Halevy, D.T. Johnston, Deciphering the atmospheric signal in marine sulfate oxygen isotope composition. Earth Planet. Sci. Lett. 522, 12–19 (2019)
J.C. Walker, P.B. Hays, J.F. Kasting, A negative feedback mechanism for the long-term stabilization of Earth’s surface temperature. J. Geophys. Res. 86(C10), 9776–9782 (1981)
X.T. Wang, D.M. Sigman, A.L. Cohen, D.J. Sinclair, R.M. Sherrell, M.A. Weigand, D.V. Erler, H. Ren, Isotopic composition of skeleton-bound organic nitrogen in reef-building symbiotic corals: a new method and proxy evaluation at Bermuda. Geochim. Cosmochim. Acta 148, 179–190 (2015)
S.D. Wankel, A.S. Bradley, D.L. Eldridge, D.T. Johnston, Determination and application of the equilibrium oxygen isotope effect between water and sulfite. Geochim. Cosmochim. Acta 125, 694–711 (2014)
L.M. Ward, P.M. Shih, The evolution and productivity of carbon fixation pathways in response to changes in oxygen concentration over geological time. Free Radic. Biol. Med. 140, 188–199 (2019)
L.M. Ward, B. Rasmussen, W.W. Fischer, Primary productivity was limited by electron donors prior to the advent of oxygenic photosynthesis. J. Geophys. Res., Biogeosci. 124(2), 211–226 (2019)
M.R. Warke, T.D. Rocco, A.L. Zerkle, A. Lepland, A.R. Prave, A.P. Martin, Y. Ueno, D.J. Condon, M.W. Claire, The great oxidation event preceded a paleoproterozoic “snowball Earth”. Proc. Natl. Acad. Sci. 117(24), 13314–13320 (2020)
M.C. Weiss, F.L. Sousa, N. Mrnjavac, S. Neukirchen, M. Roettger, S. Nelson-Sathi, W.F. Martin, The physiology and habitat of the last universal common ancestor. Nat. Microbiol. 1(9), 16116 (2016)
C.B. Wenk, B.A. Wing, I. Halevy, Electron carriers in microbial sulfate reduction inferred from experimental and environmental sulfur isotope fractionations. ISME J. 12(2), 495–507 (2018)
J.P. Werne, D.J. Hollander, A. Behrens, P. Schaeffer, P. Albrecht, J.S. Sinninghe Damsté, Timing of early diagenetic sulfurization of organic matter: a precursor-product relationship in Holocene sediments of the anoxic Cariaco Basin, Venezuela. Geochim. Cosmochim. Acta 64(10), 1741–1751 (2000)
A.R. Whitehill, S. Ono, Excitation band dependence of sulfur isotope mass-independent fractionation during photochemistry of sulfur dioxide using broadband light sources. Geochim. Cosmochim. Acta 94, 238–253 (2012)
A.R. Whitehill, C. Xie, X. Hu, D. Xie, H. Guo, S. Ono, Vibronic origin of sulfur mass-independent isotope effect in photoexcitation of SO2 and the implications to the early Earth’s atmosphere. Proc. Natl. Acad. Sci. 110(44), 17697–17702 (2013)
A.R. Whitehill, B. Jiang, H. Guo, S. Ono, SO2 photolysis as a source for sulfur mass-independent isotope signatures in stratospheric aerosols. Atmos. Chem. Phys. Discuss. 14(16), 23499–23554 (2015)
E.B. Wilkes, A. Pearson, A general model for carbon isotopes in red-lineage phytoplankton: interplay between unidirectional processes and fractionation by RubisCO. Geochim. Cosmochim. Acta 265, 163–181 (2019)
K.H. Williford, T. Ushikubo, K. Lepot, K. Kitajima, C. Hallmann, M.J. Spicuzza, R. Kozdon, J.L. Eigenbrode, R.E. Summons, J.W. Valley, Carbon and sulfur isotopic signatures of ancient life and environment at the microbial scale: neoArchean shales and carbonates. Geobiology 14(2), 105–128 (2016)
B.A. Wing, I. Halevy, Intracellular metabolite levels shape sulfur isotope fractionation during microbial sulfate respiration. Proc. Natl. Acad. Sci. USA 111(51), 18116–18125 (2014)
C.R. Witkowski, J.W. Weijers, B. Blais, S. Schouten, J.S. Sinninghe Damsté, Molecular fossils from phytoplankton reveal secular PCO2 trend over the Phanerozoic. Sci. Adv. 4(11), eaat4556 (2018)
U.G. Wortmann, B. Chernyavsky, S.M. Bernasconi, B. Brunner, M.E. Böttcher, P.K. Swart, Oxygen isotope biogeochemistry of pore water sulfate in the deep biosphere: Dominance of isotope exchange reactions with ambient water during microbial sulfate reduction (ODP Site 1130). Geochim. Cosmochim. Acta 71(17), 4221–4232 (2007)
T. Wotte, G.A. Shields-Zhou, H. Strauss, Carbonate-associated sulfate: experimental comparisons of common extraction methods and recommendations toward a standard analytical protocol. Chem. Geol. 326(327), 132–144 (2012)
N. Wu, J. Farquhar, H. Strauss, S.-T. Kim, D.E. Canfield, Evaluating the S-isotope fractionation associated with Phanerozoic pyrite burial. Geochim. Cosmochim. Acta 74(7), 2053–2071 (2010)
N. Wu, J. Farquhar, H. Strauss, \(\delta\)34S and \(\delta\)33S records of Paleozoic seawater sulfate based on the analysis of carbonate associated sulfate. Earth Planet. Sci. Lett. 399, 44–51 (2014)
J. Yang, C.K. Junium, N.V. Grassineau, E.G. Nisbet, G. Izon, C. Mettam, A. Martin, A.L. Zerkle, Ammonium availability in the late archaean nitrogen cycle. Nat. Geosci. 12(7), 553–557 (2019)
S. Zaarur, D.T. Wang, S. Ono, T. Bosak, Influence of phosphorus and cell geometry on the fractionation of sulfur isotopes by several species of Desulfovibrio during microbial sulfate reduction. Front. Microbiol. 8, 890 (2017)
J. Zachos, H. Pagani, L. Sloan, E. Thomas, K. Billups, Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 292(5517), 686–693 (2001)
K. Zahnle, M. Claire, D. Catling, The loss of mass-independent fractionation in sulfur due to a palaeoproterozoic collapse of atmospheric methane. Geobiology 4(4), 271–283 (2006)
R.E. Zeebe, Kinetic fractionation of carbon and oxygen isotopes during hydration of carbon dioxide. Geochim. Cosmochim. Acta 139, 540–552 (2014)
R.E. Zeebe, D. Wolf-Gladrow, in CO2 in Seawater: Equilibrium, Kinerics, Isotopes. Elsevier Oceanography Series, vol. 65 (2001), pp. 1–341
R.E. Zeebe, J. Bijma, D.A. Wolf-Gladrow, A diffusion-reaction model of carbon isotope fractionation in foraminifera. Mar. Chem. 64(3), 199–227 (1999a)
R.E. Zeebe, D.A. Wolf-Gladrow, H. Jansen, On the time required to establish chemical and isotopic equilibrium in the carbon dioxide system in seawater. Mar. Chem. 65(3–4), 135–153 (1999b)
R.E. Zeebe, J.C. Zachos, G.R. Dickens, Carbon dioxide forcing alone insufficient to explain palaeocene-Eocene thermal maximum warming. Nat. Geosci. 2(8), 576–580 (2009)
A.L. Zerkle, C.K. Junium, D.E. Canfield, C.H. House, Production of 15N-depleted biomass during cyanobacterial N2-fixation at high Fe concentrations. J. Geophys. Res., Biogeosci. 113(3), G03014 (2008)
A.L. Zerkle, J. Farquhar, D.T. Johnston, R.P. Cox, D.E. Canfield, Fractionation of multiple sulfur isotopes during phototrophic oxidation of sulfide and elemental sulfur by a green sulfur bacterium. Geochim. Cosmochim. Acta 73(2), 291–306 (2009)
A.L. Zerkle, A. Kamyshny, L.R. Kump, J. Farquhar, H. Oduro, M.A. Arthur, Sulfur cycling in a stratified euxinic lake with moderately high sulfate: constraints from quadruple S isotopes. Geochim. Cosmochim. Acta 74(17), 4953–4970 (2010)
A.L. Zerkle, M.W. Claire, S.D. Domagal-Goldman, J. Farquhar, S.W. Poulton, A bistable organic-rich atmosphere on the neoarchaean Earth. Nat. Geosci. 5(5), 359–363 (2012)
A.L. Zerkle, D.S. Jones, J. Farquhar, J.L. Macalady, Sulfur isotope values in the sulfidic Frasassi cave system, central Italy: a case study of a chemolithotrophic S-based ecosystem. Geochim. Cosmochim. Acta 173, 373–386 (2016)
A.L. Zerkle, S.W. Poulton, R.J. Newton, C. Mettam, M.W. Claire, A. Bekker, C.K. Junium, Onset of the aerobic nitrogen cycle during the Great Oxidation Event. Nature 542(7642), 465–467 (2017)
J. Zhang, P.D. Quay, D.O. Wilbur, Carbon isotope fractionation during gas-water exchange and dissolution of CO2. Geochim. Cosmochim. Acta 59(1), 107–114 (1995)
X. Zhang, A.L. Gillespie, A.L. Sessions, Large D/H variations in bacterial lipids reflect central metabolic pathways. Proc. Natl. Acad. Sci. USA 106(31), 12580–12586 (2009)
X. Zhang, D.M. Sigman, F.M. Morel, A.M. Kraepiel, Nitrogen isotope fractionation by alternative nitrogenases and past ocean anoxia. Proc. Natl. Acad. Sci. USA 111(13), 4782–4787 (2014)
I. Zhelezinskaia, A.J. Kaufman, J. Farquhar, J. Cliff, Large sulfur isotope fractionations associated with neoArchean microbial sulfate reduction. Science 346(6210), 742–744 (2014)
X. Zhu-Barker, A.R. Cavazos, N.E. Ostrom, W.R. Horwath, J.B. Glass, The importance of abiotic reactions for nitrous oxide production. Biogeochemistry 126(3), 251–267 (2015)
Acknowledgements
This paper is the product of the geobiology working group within the International Space Science Institute (ISSI) workshop ‘Reading Terrestrial Planet Evolution in Isotopes and Element Measurements’. We would like to thank ISSI and Europlanet for their support. Thanks to Peter Crockford, who provided invaluable feedback on the manuscript. We would also like to thank Guest Editor Helmut Lammer and three anonymous reviewers for careful and constructive comments.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
M.K. Lloyd and H.L.O. McClelland contributed equally to this work
Reading Terrestrial Planet Evolution in Isotopes and Element Measurements
Edited by Helmut Lammer, Bernard Marty, Aubrey L. Zerkle, Michel Blanc, Hugh O’Neill and Thorsten Kleine
Rights and permissions
About this article

Cite this article
Lloyd, M.K., McClelland, H.L.O., Antler, G. et al. The Isotopic Imprint of Life on an Evolving Planet. Space Sci Rev 216, 112 (2020). https://doi.org/10.1007/s11214-020-00730-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11214-020-00730-6