
Abstract
Sustainable and renewable production of platform chemicals and fuels has been gradually rising. Formic acid is one of the important chemicals for leather, cosmetic and pharmaceutical industries as well as hydrogen source. In this study, selective oxidation of biomass-derived glucose to formic acid was investigated under base free medium at 70 °C over synthesized hydrotalcite-like catalysts using hydrogen peroxide as oxidant. Effect of Mg/Al ratio (6/1, 3/1, 1/1, 1/3 and 1/6) and heat treatment (drying and calcination) on catalyst structure and product distributions; effect of calcination temperature (450, 650 and 900 °C), solvent composition (ethanol/water) and reaction temperature (30, 50 and 70 °C) on catalytic activity and product selectivity were investigated. Reducing the Mg/Al ratio enhanced the density of metal-OH bonds, surface area and uniformity of pores up to some extent. The highest glucose conversion and formic acid selectivity were achieved over Mg–Al (1:3) catalyst as 38.7 and 99.0%, respectively. The calcined catalysts (at 450 °C) exhibited 7 times higher selectivities and 4 times higher activities than the dried ones. However, higher calcination temperatures did not show remarkable increments in activities and yields. Easily prepared, cheap Mg–Al (1:3) catalyst provided promising results even at low temperature with hydrogen peroxide at atmospheric medium in a low boiling point solvent (ethanol).
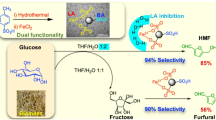
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The global market of formic acid (FA) has been steadily increasing since it is one of the efficient platform chemicals used in the food, textile and leather, pharmaceutical, and cosmetic industries [1]. Formic acid can be transported easily due to its stability at room conditions, nontoxic and nonflammable properties [2]. FA has a crucial role as a hydrogen donor [3] as decomposes easily to produce hydrogen and carbon dioxide above 140 °C [4,5,6] . This makes it favorable, safe, and economic feedstock to be used in fuel cells and many hydrogenation reactions.
Although many different methods have been applying for lab-scale production of formic acid [7,8,9,10], today two main non-renewable methods have been using for large-scale. These two methods are hydrolysis of methyl formate and carbonylation of methanol [11]. The methyl formate process is still the most widely used method since 90% of formic acid is produced industrially by this reaction. However, high process costs and the formation of undesired products along with formic acid push people to find sustainable, renewable, and cost-effective methods.
The conversion of biomass-derived molecules to fuels, especially formic acid is attractive in the last years [12,13,14,15,16,17,18,19,20,21]. Two different chemical strategies are present for glucose transformation to formic acid; dehydration-hydrolysis and oxidation. The dehydration-hydrolysis process consists of dehydration of glucose to 5-hydroxymethylfurfural [22,23,24,25,26,27] that is followed by its rehydration to formic and levulinic acids (LA) [22, 28,29,30,31]. The formation of LA together with FA limits the thermodynamic yields of these acids [28]. Nowadays, extensive efforts have been making for highly selective glucose oxidation to formic acid [14, 21, 32, 33].
Glucose oxidation to FA occurs under basic conditions or over base sites of the catalysts [14, 32]. Wide range of homogeneous and heterogeneous catalysts, different O2 sources, different reaction mediums, and conditions were investigated in the literature [12,13,14,15, 34,35,36,37]. Wang et al. (2019) achieved 91.3% formic acid yield with LiOH at room temperature for 8 h by using H2O2 as an oxidant [14]. Jin et al. (2008) investigated the fast oxidation of glucose to formic acid with H2O2 in the presence of an alkali medium (NaOH) at 250 °C. Formic acid formed only in 60 s in 75% FA yield [32]. Albert et al. (2012) reported oxidation of biomass (xylan) conversion to formic acid with Keggin-type polyoxometalate (H5PV2Mo10O40) as a homogeneous catalyst by using p-toluenesulfonic acid as an additive at 90 °C and 30 bar O2. The formic acid yield was obtained as 53% for 24 h [38]. However, it is still a great challenge to carry out the glucose oxidation process providing a high yield of formic acid under base-free and mild conditions.
Hydrotalcites (HTs), called layered double hydroxides, are natural minerals containing M+2 and M+3 metal oxides [39, 40]. They can be synthesized from basic metal salts by co-precipitation easily. HTs have a high amount of hydroxyl groups and surface basic sites which make them active and selective oxidation catalysts [40]. Promising catalytic performances of this type of catalyst were reported many times [40,41,42,43,44,45,46,47]. Sato et al. [43] brought a unique perspective to hydrotalcite type catalysts for selective formic acid production via oxidation of glucose. They calcined Mg–Al type hydrotalcite (HT) and tested in glucose oxidation in ethanol medium. Although satisfactory glucose conversion was obtained over uncalcined HT, calcination significantly improved the formic acid selectivity (upto 78.1% with complete glucose conversion). In comparison with the other common catalysts, -such as mono alkali oxides (MgO and CaO), zeolites (H-ZSM-5 and H-β) and SiO2- the highest yield was achieved over calcined HT.
The present work focused on the performance of different Mg–Al type hydrotalcite catalysts on glucose oxidation. HTs were synthesized by the co-precipitation method. These catalysts were tested in selective glucose oxidation to formic acid. Effect of Mg/Al ratio, thermal process, reaction temperature, and solvent composition on catalytic activity and formic acid selectivity were investigated under base-free mild conditions.
Experimental study
Materials
Glucose, magnesium nitrate hexahydrate (≥ 99%), aluminum nitrate nanohydrate (≥ 99%), and sodium hydroxide (97%) were purchased from Merck KGaA (Darmstadt, Germany). Sodium carbonate anhydrous (99%) and hydrogen peroxide (35%) were supplied by Isolab Chemicals (Wertheim, Germany).
Catalyst preparation
Synthesis of hydrotalcite-like catalysts with different Mg–Al ratios
Mg–Al hydrotalcites were prepared by a co-precipitation method. Solution A was prepared by dissolving the magnesium nitrate hexahydrate [Mg(NO3)2.6H2O] and the aluminum nitrate nonahydrate [Al(NO3)2.9H2O] in water. Solution B was aqueous solution of 1 M NaOH. These two solutions were simultaneously added into aqueous sodium carbonate (Na2CO3) solution at 60 °C and pH 10. After consumption of the metal solutions, the mixture was stirred for 1 h and left to mature for 18 h. The resulting precipitates were filtered, washed with deionized water, dried at 110 °C for 18 h, and calcined at 450 °C for 5 h with a heating rate of 10 °C/min. The synthesized hydrotalcites are labeled Mg–Al(x:y) (x:y = Mg/Al ratio). The Mg–Al(3:1) catalyst was also calcined at 650 and 900 °C with the same heating rate, time and labeled as Mg–Al(3:1)-650 and Mg–Al(3:1)-900, respectively.
Characterization of catalysts
The crystal phases and X-ray diffraction (XRD) patterns of the catalysts were analyzed with the Rigaku Smartlab instrument. The patterns were scanned between 2θ = 5–80° angles where the scanning speed was 3°/s. A wavelength of λ = 0.15418 nm and a power of 45 kV/40 mA of Cu Kα beam source were used. FT-IR measurements of the catalysts were made with Perkin-Elmer Brand Paragon 1000 model Fourier Transform Infrared Spectroscopy (FT-IR/ATR). Measurements were taken with a powder sample between 400 and 4000 cm−1. Nitrogen physisorption analyzes were performed with the Quantachrome Autosorb IQ2 model instrument. All samples were degassed at 120 °C for 8 h prior to adsorption. The multipoint adsorption data were obtained at 77 K and relative pressure of 760 mmHg. The surface area, pore structures, and pore volumes of the catalysts were determined by the BET method.
Catalyst screening tests
Synthesized catalysts were tested in glucose oxidation. Oxidation tests were carried out at 70 °C for 6 h in both water and ethanol medium. Hydrogen peroxide solution (35%) was used as an oxidant.
The reactions were performed in a reflux glass bubble reactor with a volume of 100 ml. A specified amount of catalyst and glucose were loaded into the reactor. This was followed by the addition of oxidant in 50 ml of solvent. The solution was stirred at 500 rpm under heating in an oil bath on the magnetic stirrer. Each sample, taken at every two hours, was cooled immediately in an ice bath, filtered with a 0.45 µm PVDF syringe filter, diluted for HPLC analysis.
Parametric studies
Parametric studies were performed with the most efficient catalyst (providing the highest conversion and FA selectivity). The effects of calcination temperatures (450 °C, 650 °C, and 900 °C), solvent compositions (ethanol, water, ethanol/water: 50/50 (v/v)), reaction temperatures (30 °C, 50 °C, and 70 °C) and catalyst/glucose ratios (1/1, 1/2 and 1/5) on glucose oxidation were investigated in order to optimize the reaction parameters.
Recyclability test
Recyclability of the catalysts was performed in the same reaction medium. After each run, used catalyst was washed, dried and calcined at 450 °C and used in the consecutive cycle. Used catalyst after the 1st run was analyzed by XRD.
Product analysis
The diluted samples were analyzed by Thermo Ultimate 3000 model high pressure liquid chromatography (HPLC) equipped with ICSep Coregel 87H3 (300 × 7.8 mm) organic acid column, UV–Visible (UV–Vis), and Refractive Index (RI) detectors. The injection volume was 20 µl. Dilute sulfuric acid (8 mM) was used as the mobile phase. The flow rate was 0.6 ml/min, the column temperature was 60 °C and the RID temperature was 45 °C. Organic acids (formic acid, acetic acid, levulinic acid) concentrations were determined by UV–Vis detector while sugar (glucose and fructose) concentrations by RID detector.
Glucose conversion (X), product carbon-yield (Y) and carbon-selectivity (S) were calculated using the following equations.
Results and discussion
Characterization of the synthesized catalysts
The XRD patterns of calcined hydrotalcite-like structures with different Mg/Al ratios are demonstrated in Fig. 1. It was observed that each catalyst was compatible with the literature in terms of Mg/Al ratios [44, 48]. Mg–Al(6:1) catalyst had three different crystal phases which were hydrotalcite (2θ = 12°, 23°, 35°), magnesium-aluminate (2θ = 65°) and magnesium oxide/periclase (2θ = 37°, 43°, 63°). The Mg–Al(3:1) had high-density hydrotalcite crystal phases (2θ = 12°, 23°, 35°, 40°, and 47°) and less magnesium oxide/periclase crystals (2θ = 43° and 63°). Pure hydrotalcite crystal structure forms when the weight ratio of Mg/Al is between 2–4% [49]. It was realized from the XRD patterns, the density of hydrotalcite crystal phases decreased outside of this range. For Mg–Al(1:1), the magnesium-aluminate (2θ = 20° and 65°) crystallinity was very low but magnesium oxide/periclase (2θ = 37°, 43°, 63°) crystallinity was high. Mg–Al(1:3) had low crystallinity or smaller crystals (< 5 nm) so it could not be detected by XRD analysis. The crystal phases of Mg–Al(1:3), were magnesium-aluminate (2θ = 44°, 63°, and 65°) and magnesium oxide/periclase (2θ = 37°). Mg–Al(1:6) catalyst had a high crystal density of magnesium-aluminate (2θ = 20°, 45°, and 65°) and low crystal density of magnesium oxide/periclase (2θ = 37°). When the Mg/Al ratio of the catalysts decreased (from the ratio Mg/Al = 3), the hydrotalcite crystals were disappeared and the density of magnesium-aluminate crystals increased.

XRD patterns of calcined hydrotalcite-like structures at different Mg/Al ratios
The high-temperature X-ray diffraction analysis was performed at different temperatures to observe the differences in the diffraction planes of uncalcined Mg–Al(3:1). The XRD patterns of this catalyst are shown in Fig. 2. Dense crystallinity was clearly visible in the patterns at room temperature and 150 °C. As seen in Fig. 2, there were hydrotalcite crystals in Mg–Al (3:1) structure at these two temperatures. When the temperature reached 250 °C, the intensity of the crystal peaks decreased. At 350 °C the crystal structure of the catalyst transformed into an amorphous structure. There was no change in crystal density up to 900 °C. Dense magnesium aluminate crystal phases were noticed at 20°, 32°, 59°, and 65° at 900 °C. As a result, when the temperature increased, the hydrotalcite structure of synthesized material changed and its crystal structure tended toward amorphous.

XRD patterns of Mg–Al (3:1) HT catalyst at different temperatures
Textural properties of the prepared hydrotalcite-like catalysts are given in Table 1. It was observed that there was a steady increase in specific surface area with the decrease in the Mg/Al ratio (up to 1:6). This increment was 20% between catalysts possessing hydrotalcite structure which was almost % 90 in catalysts having mostly magnesium–aluminum oxide structure. By increasing the calcination temperature of Mg–Al(3:1) from 450 to 650 °C, the surface area and pore diameter were improved by almost 50%, while the pore volume more than doubled. However, the structure of this catalyst did not change significantly when the calcination temperature raised to 900 °C. Effect of calcination process on textural properties is clearly noticeable in Mg–Al(1:3). Calcination highly increased the surface area of this sample.
Nitrogen adsorption–desorption isotherms and pore size distributions of the synthesized catalysts are given in Figs. 3 and 4. All of the catalysts indicated Type IV isotherm with distinct Type H3 hysteresis loop which implies mesoporous character, slit-shaped pores, wide pore size distribution, and high energy of adsorption [50]. Most of the pores are distributed in the mesopore range. Few amounts of macropores were observed.

N2 Adsorption and desorption isotherms of hydrotalcite-like catalysts a Mg-Al(1:6) b Mg-Al(1:3) c Mg-Al(1:1) d Mg-Al(1:3))

N2 Adsorption and desorption isotherms of hydrotalcite-like catalysts a Mg–Al(3:1) b Mg–Al(3:1)-650 c Mg–Al(3:1)-900 d Mg–Al(6:1))
The Mg–Al (1:3) exhibited the most narrow pore size distribution on lower mesopore region, whereas the distributions patterns of other catalysts were much broader. This suggests that Mg–Al (1:3) was more uniformly packed together compared to others (Fig. 5).

BJH Pore size distributions of catalysts with different Mg–Al ratio
The FT-IR spectra of dried hydrotalcite-like structures at different Mg/Al ratios are given in Fig. 6. Each catalyst is compatible with the literature [51, 52]. The absorption band at 3698 cm−1 was stretching vibrations of metal-OH (such as Mg-OH and Al–OH) bonds which was seen in Mg–Al(1:1), Mg–Al(1:3), and Mg–Al(1:6) materials. In these regions, Lewis acid centers formed on the aluminum side, and both basic and oxidizing centers formed on the Mg side. This band is not found in Mg–Al(3:1) and Mg–Al(6:1) catalysts [14, 34]. Absorption bands at 3400–3500 cm−1 formed by physisorption between located interphase and metal-O–H bonds. The sharp absorption bands at 2980 cm−1 were caused by the CO3−2-H2O stretching vibrations [51] formed by the hydrogen bonding of carbonate ions attached to the Mg–Al structure [53]. Also, 1350–1380 cm−1 bands were caused by CO3−2 in the structure. The band at 1646 cm−1 observed in Mg–Al(3:1) was H–O..H vibrations by the presence of crystalline water. The bands obtained at 1250 and 1066 cm−1 were the symmetric-asymmetric stretching movements of the ether groups.

FT-IR spectra of dried hydrotalcite-like catalysts
FT-IR spectra of calcined catalysts at different Mg/Al ratios are shown in Fig. 7. The intensity of metal-OH bonds (absorption band at 3698 cm−1) in Mg–Al(1:1) decreased after the calcination process, but it remained constant in Mg–Al(1:3) and Mg–Al(1:6) catalysts. In Mg–Al(3:1) and Mg–Al(6:1) catalysts, the intensity of these bonds reduced dramatically with calcination. Sharp absorption bands (CO3−2-H2O stresses) at 2901 and 2980 cm−1 maintained their intensity in Mg–Al(1:3) and Mg–Al(1:6), which decreased significantly in other catalysts as a result of calcination. For these catalysts, similar results were obtained at 1390, 1250, and 1066 cm−1 (CO3−2 in the structure). Mg–Al(1:1) also maintained its intensity at 1390 cm−1. As a result, reactive metal-OH and CO3−2 groups were preserved as crystal structures getting closer to magnesium-aluminate even if catalysts were exposed to calcination. FT-IR spectra of different calcination temperatures (650 °C and 900 °C) of Mg–Al(3:1) are also shown in Fig. 7. In the structure, the metal oxides absorption bands remained at the same level but others decreased to negligible levels.

FT-IR spectra of calcined hydrotalcite-like catalysts
Glucose conversion tests
Activity test of dried hydrotalcite-like catalysts
The glucose conversions over dried hydrotalcite-like catalysts with different Mg/Al ratios (6/1, 3/1, 1/1, 1/3, and 1/6) are given in Fig. 8. Glucose conversion was not notable in a catalyst-free environment. Changing Mg/Al ratio did not affect the glucose conversion significantly except Mg–Al(6:1). The lowest glucose conversion (8.5%) was obtained with Mg–Al(6:1). Other catalysts exhibited appx. 16–18% conversions.

Glucose conversions of dried hydrotalcite-like catalysts with different Mg–Al ratios (Solvent: Ethanol, Temperature: 70℃, Time: 6 h, Catalyst/Glucose:1/2, 2% Glucose Loading, 250% H2O2 solution)
The formic acid yields are given in Fig. 9. The highest FA yield (4.7%) and selectivity (29.4%) were obtained with Mg–Al(1:3) catalyst. The FA yield raised with the increased in the aluminum ratio (except Mg–Al(1:6)). Unexpectedly, no formic acid was produced by Mg–Al(1:6) catalyst.

Formic acid carbon yield of dried hydrotalcites with different Mg–Al ratios (Solvent: Ethanol, Temperature: 70 °C, Time: 6 h, Catalyst/Glucose:1/2, 2% Glucose Loading, 250% H2O2 Solution).
In literature, some alkaline earth metal salts (MgCl2 or CaCl2) were found very effective to oxidize the sugar but not effective alone in formic acid production [34]. The transition metals (FeCl3, CrCl3 or MnCl2 and AlCl3) were more selective for organic acids, especially formic acid [34]. The basic medium or alkali metals contribute to the conversion of glucose, but formic acid is formed by the transition metals in heterogeneously catalyzed oxidation. The results were clearly seen in Figs. 8 and 9.
Activity tests of calcined hydrotalcite-like catalysts
The glucose conversions of calcined hydrotalcite-like catalysts with different Mg/Al ratios (6/1, 3/1, 1/1, 1/3, and 1/6) are given in Fig. 10. The glucose conversion increased with reaction time in all catalysts. Increasing the Mg/Al ratio enhanced the conversion (except Mg–Al(6:1)). The highest conversion was obtained as 65.1% with Mg–Al(3:1). As compared with the dried samples, calcined ones were seen much more active (approximately 4 times) in the oxidation of glucose. Because, strong basic centers on the surfaces of hydrotalcites—formed during the calcination process- accelerated the oxidation of glucose and increased stability of catalysts [40].

Glucose conversion over calcined hydrotalcites prepared at different Mg–Al ratios (Solvent: Ethanol, Temperature: 70 °C, Catalyst/Glucose:1/2, 2% Glucose Loading, 250% H2O2 Solution, Catalyst Calcination Temperature: 450 °C).
Time-dependent formic acid carbon selectivities of the calcined catalysts are shown in Fig. 11. The selectivities of formic acid increased slightly up to 4 h with the decrease in the Mg/Al ratio. After 4th hour, FA yield significantly increased over Mg–Al(1:3) and Mg–Al(1:6) and reached to 99 and 74%, respectively. These results indicated that the breakdown of hydrogen peroxide formed O radicals; these radicals reacted with glucose molecules in virtue of Mg metal to produce intermediates [43] until the 4th hour of the reaction. These products were converted then into formic acid on the centers where the Al atom and CO3−2 ions were located [43].

Formic acid selectivity of calcined hydrotalcites with different Mg–Al ratios (Solvent: Ethanol, Temperature: 70 °C, Catalyst/Glucose:1/2, 2% Glucose Loading, 250% H2O2 Solution, Catalyst Calcination Temperature: 450 °C)
As mentioned in Fig. 1, as the Mg/Al ratio reduced, the materials moved away from the hydrotalcite structure and the crystal density shifted toward magnesium aluminates, which was very effective in formic acid production. Meanwhile, Mg–Al(1:3) and Mg–Al(1:6) catalysts preserved the Metal-OH and CO3−2 groups in their structure even after the calcination, compared to other hydrotalcites (Fig. 2). Based on these performances, the rest of the tests were continued with Mg–Al(1:3).
Effect of calcination temperature on glucose conversion and formic acid selectivity
Calcination of prepared catalysts improved the activities and formic acid selectivities. Mg-A1(3:1) was determined as a model catalyst in order to examine the effect of calcination temperature and the effect of crystal phase on catalytic activity and formic acid selectivity. This catalyst was calcined at different temperatures (450 °C, 650 °C, and 900 °C). The glucose conversion, formic acid carbon yields, and selectivities of Mg-A1(3:1)-450, Mg-A1(3:1)-650, and Mg-A1(3:1)-900 catalysts are given in Fig. 12. The highest glucose conversion (49.3%) was obtained with Mg–Al(3:1)-650 catalyst, but the highest formic acid carbon yield (7.43%) and selectivity (17.32%) were obtained with Mg–Al(3:1)-900 which was slightly higher than Mg–Al(3:1)-450. Therefore, considering the amount of energy consumption, the optimum calcination temperature was determined as 450 °C.

Glucose conversion, formic acid carbon yield, and formic acid selectivity of Mg–Al(3:1) catalyst calcined at different temperatures (Solvent: Ethanol, Temperature: 70 °C, Time: 6 h, Catalyst/Glucose:1/2, 2% Glucose Loading, 250% H2O2 Solution)
Effect of solvent type on glucose conversion and formic acid selectivity
In the literature studies, it has been observed that the type of solvent may affect the oxidation products distribution. The most suitable solvents for oxidation were determined as alcohols (ethanol) and water. For this reason, aqueous ethanol (EtOH) solutions in different proportions (100%, 50% and 0%, (EtOH-Water)) were prepared and their effects on glucose conversion and formic acid selectivities were investigated. Results are given in Figs. 13 and 14. The presence of water did not affect the catalytic activity (~ 40%) significantly where formic acid was maximized with the absence of water. The highest selectivity (99%), without any by-product, was obtained in 100% ethanol medium. It was previously reported the formation of the carboxylic acid intermediates during glucose oxidation [7]. Decarboxylation of these intermediates favors the formation of CO2, especially in aqueous mediums. When an alcohol is used as a solvent, the formation of CO2 during the reaction is prevented and formic acid yield is improved [7]. In addition, as a result of the reaction of aldehydes (glucose) with alcohols (ethanol), hemiacetal structures are formed and the conversion of glucose to non-formic acid carboxylic acids (acetic acid, glycolic acid) are prevented [54]. Thus, formic acid selectivity increases significantly in ethanol medium.

Glucose conversion of Mg–Al(3:1) catalyst calcined in the different reaction medium (Temperature: 70 °C, Time: 6 h, Catalyst/Glucose:1/2, 2% Glucose Loading, 250% H2O2 Solution)

Formic acid selectivities of Mg–Al(3:1) catalyst in different reaction mediums (Temperature: 70 °C, Time: 6 h, Catalyst/Glucose:1/2, 2% Glucose Loading, 250% H2O2 Solution)
Effect of reaction temperature on glucose conversion and formic acid selectivity
The time-dependent glucose conversions at different temperatures are given in Fig. 15. The reaction was carried out at three different temperatures (30 °C, 50 °C, and 70 °C). The highest temperature was determined as 70 °C considering decomposition of H2O2 above 70 °C and boiling point of EtOH [55]. Although the oxidation reaction was exothermic, glucose conversion (9.6%, 25.0%, and 38.7%) increased with temperature by the increase in molecular motion and surface activity. The time trends of conversions and the differences between conversion lines at each time point indicated the accuracy of the oxidation results.

Glucose conversion of Mg–Al(1:3) calcined hydrotalcites at different reaction temperatures (Solvent: Ethanol, Catalyst/Glucose:1/2, 2% Glucose Loading, 250% H2O2 Solution, Catalyst Calcination Temperature: 450 °C)
The selectivities of formic acid obtained at different temperatures are shown in Fig. 16. The highest product selectivity (99%) was obtained at the highest temperature where the lowest product selectivity (29.6%) was at the lowest reaction temperature. A huge difference in selectivities was observed between 70 and 50 °C. Although the temperature difference is only 20 °C, the slight increase in temperature could be changed the mechanism. It was noticed that the selectivity at 30 °C was higher than at 50 °C. This was due to the formation of CO2 that starts after 30 °C.

Formic acid selectivity of Mg–Al(1:3) calcined hydrotalcites at different reaction temperatures (Solvent: Ethanol, Reaction time: 6 h, Catalyst/Glucose:1/2, 2% Glucose Loading, 250% H2O2 Solution, Catalyst Calcination Temperature: 450 °C)
Effect of catalyst/glucose ratio on glucose conversion and formic acid production
Effects of catalyst/glucose (1/5, 1/2, and 1/1) ratio on glucose conversion were investigated. The time-dependent glucose conversion amounts of different catalyst/glucose ratios are given in Fig. 17. The glucose conversion increased with the amount of catalyst depending on the increase in the active sites per glucose molecule. The highest glucose conversion (71.9%) was obtained when the catalyst/glucose ratio was chosen as 1/1.

Glucose conversion of Mg–Al(1:3) calcined hydrotalcites at different catalyst/glucose ratios (Solvent: Ethanol, Reaction Temperature: 70 °C, 2% Glucose Loading, 250% H2O2 Solution, Catalyst Calcination Temperature: 450 °C)
The effect of different catalyst/glucose (1/5, 1/2, and 1/1) ratios on formic acid selectivity was examined (Fig. 18). When the amount of catalyst was increased, formic acid selectivity decreased from 99 to 64.7%. This was attributed to the formation of side reactions and the high number of active centers caused the conversion of glucose into other products. Formic acid formation at G/C = 1/5 was not remarkable. This may be due to the insufficient number of active sites per glucose molecule, instantaneous occupation of these sites, and undesired reactions between unbonded glucose molecules and solvent, products, etc.

Formic acid selectivity of Mg–Al(1:3) calcined hydrotalcites at different catalyst/glucose ratios (Solvent: Ethanol, Reaction Temperature: 70 °C, 2% Glucose Loading, 250% H2O2 Solution, Catalyst Calcination Temperature: 450 °C)
Catalyst stability
Mg–Al(1:3) catalyst were tested in a series of recycle reactions and the re-used catalyst after the 1st reaction was analyzed by XRD. XRD patterns of the used catalyst and series activity results are given in Figs. 19 and 20, respectively. Recovered catalysts after each run were washed and calcined for the consecutive reaction cycles. Glucose conversion slightly reduced after 3rd run; however it was significantly decreased after the 4th use. XRD results of the used catalyst exhibited the same crystal structure with higher intensities. This could be due to the enlarging of MgO crystals during the reaction. Dramatic drop in the activity after 4th run could be attributed to the formation of humins and deactivation of the active sites by these molecules.

XRD patterns of fresh and re-used Mg–Al (1:3) HT catalyst

Recycle reaction results of Mg- Mg–Al (1:3) HT catalyst
Conclusions
Selective oxidation of glucose to formic acid under base free mild conditions was successfully achieved over hydrotalcite-like catalysts. Synthesized catalysts exhibited high glucose conversions (~ 60% over Mg–Al (3:1)) and extremely high formic acid selectivities (~ 99% over Mg–Al (1:3)) in ethanol by using H2O2 as oxidant at 70 °C. Densities of the hydrotalcite crystals and average pore diameters of the catalysts reduced as Mg/Al ratio increased, while surface area and amount of metal-OH bonds increased. Mg–Al (1:3) had most narrow pore size distributions that indicated the more uniformly packing of this material compared to others. Mg–Al (1:3) provided the highest formic acid yield and selectivity (37% FA yield and 99% FA selectivity) among the all synthesized catalysts. The calcination process significantly improved the catalytic activities of the catalysts (~ 4X in conversion and ~ 7X in yield). The effect of water/ethanol ratio was also demonstrated. Presence of water in solvent significantly decreased the FA yield since water favored the formation of CO2. A dramatic drop in FA selectivity was observed when the reaction temperature was reduced to 50 °C due to the reaction mechanism. Doubling the catalyst/glucose ratio increased the conversion by 84% while lowering the selectivity by 30%.
References
T. Thananatthanachon, T.B. Rauchfuss, Angew. Chemie - Int. Ed. 49, 6616 (2010)
S.D. Sheet, I.C.I.S. Chem, Bus. 1, 1 (2017)
J. Zhao, M. Liu, G. Fan, L. Yang, F. Li, Ind. Eng. Chem. Res. 60, 5826 (2021)
S. Fukuzumi, T. Kobayashi, T. Suenobu, J. Am. Chem. Soc. 132, 1496 (2010)
X. Li, X. Ma, F. Shi, Y. Deng, Chemsuschem 3, 71 (2010)
B. Loges, A. Boddien, H. Junge, J.R. Noyes, W. Baumann, M. Beller, Chem. Commun. 28, 4185 (2009)
Z. Tang, W. Deng, Y. Wang, E. Zhu, X. Wan, Q. Zhang, Y. Wang, Chemsuschem 7, 1557 (2014)
F.R. Tao, C. Zhuang, Y.Z. Cui, J. Xu, Chinese Chem. Lett. 25, 757 (2014)
J. Yun, F. Jin, A. Kishita, K. Tohji, H. Enomoto, in J. Phys. Conf. Ser. (2010)
Z. Qiao, Z. Wang, C. Zhang, S. Yuan, Y. Zhu, J. Wang, AIChE J. 59, 215 (2012)
F. Joó, Chemsuschem 1, 805 (2008)
F. Wang, Y. Wang, F. Jin, G. Yao, Z. Huo, X. Zeng, Z. Jing, Ind. Eng. Chem. Res. 53, 7939 (2014)
N.V. Gromov, O.P. Taran, I.V. Delidovich, A.V. Pestunov, Y.A. Rodikova, D.A. Yatsenko, E.G. Zhizhina, V.N. Parmon, Catal. Today 278, 74 (2016)
C. Wang, X. Chen, M. Qi, J. Wu, G. Gözaydin, N. Yan, H. Zhong, F. Jin, Green Chem. 21, 6089 (2019)
K. Li, L. Bai, P.N. Amaniampong, X. Jia, J.M. Lee, Y. Yang, Chemsuschem 7, 2670 (2014)
B. Jin, G. Yao, X. Wang, K. Ding, F. Jin, A.C.S. Sustain, Chem. Eng. 5, 6377 (2017)
J. Ahlkvist, Formic and levulinic acid from cellulose via heterogeneous catalysis (2013).
T. Moreno, G. Kouzaki, M. Sasaki, M. Goto, M.J. Cocero, Carbohydr. Res. 349, 33 (2012)
J. Albert, Faraday Discuss. 202, 99 (2017)
Z. Babaei, R. Yazdanpanah Esmaeilabad, N. Orash, and A. Najafi Chermahini, Biomass Convers. Biorefinery 2, (2020).
S. Ponce, S. Wesinger, D. Ona, D. A. Streitwieser, and J. Albert, Biomass Convers. Biorefinery (2021).
Y. Qu, Y. Zhao, S. Xiong, C. Wang, S. Wang, L. Zhu, L. Ma, Energy Fuels 34, 11041 (2020)
Y. Feng, M. Zuo, T. Wang, W. Jia, X. Zhao, X. Zeng, Y. Sun, X. Tang, T. Lei, L. Lin, J. Taiwan Inst. Chem. Eng. 96, 431 (2019)
U. Tyagi and N. Anand, Biomass Convers. Biorefinery (2020).
S. Dutta, Biomass Convers. Biorefinery (2021).
F. Huang, T. Jiang, H. Dai, X. Xu, S. Jiang, L. Chen, Z. Fei, P.J. Dyson, Catal. Letters 150, 2599 (2020)
P.V. Rathod, R.B. Mujmule, W.-J. Chung, A.R. Jadhav, H. Kim, Catal. Letters 149, 672 (2019)
B. Guo, L. He, G. Tang, L. Zhang, L. Ye, B. Yue, S.C.E. Tsang, H. He, Chinese. J. Catal. 41, 1248 (2020)
V.V. Ordomsky, V.L. Sushkevich, J.C. Schouten, J. Van Der Schaaf, T.A. Nijhuis, J. Catal. 300, 37 (2013)
Y. Hou, M. Niu, W. Wu, Ind. Eng. Chem. Res. 59, 16899 (2020)
D. Garcés, E. Díaz, S. Ordóñez, Ind. Eng. Chem. Res. 56, 5221 (2017)
F. Jin, J. Yun, G. Li, A. Kishita, K. Tohji, H. Enomoto, Green Chem. 10, 612 (2008)
F. Jin, H. Enomoto, Energy Environ. Sci. 4, 382 (2011)
T. Lu, Y. Hou, W. Wu, M. Niu, S. Ren, Z. Lin, V.K. Ramani, Fuel 216, 572 (2018)
Y. Önal, S. Schimpf, P. Claus, J. Catal. 223, 122 (2004)
A. Mafra, L. Ulrich, J. Kornecki, R. Fernandez-Lafuente, P. Tardioli, M. Ribeiro, Catalysts 9(8), 657 (2019). https://doi.org/10.3390/catal9080657
S. Franz, N.D. Shcherban, I. Bezverkhyy, S.A. Sergiienko, I.L. Simakova, T. Salmi, D.Y. Murzin, Res. Chem. Intermed. 47, 2573 (2021)
J. Albert, R. Wölfel, A. Bösmann, P. Wasserscheid, Energy Environ. Sci. 5, 7956 (2012)
F. Cavani, F. Trifirò, A. Vaccari, Catal. Today 11, 173 (1991)
D. Kwon, J.Y. Kang, S. An, I. Yang, J.C. Jung, J. Energy Chem. 46, 229 (2020)
V.R. Choudhary, P.A. Chaudhari, V.S. Narkhede, Catal. Commun. 4, 171 (2003)
A.V.P. Lino, E.M. Assaf, J.M. Assaf, Catal. Today 289, 78 (2017)
R. Sato, H. Choudhary, S. Nishimura, K. Ebitani, Org. Process Res. Dev. 19, 449 (2015)
M.D. Özcan, O. Özcan, M.E. Kibar, A.N. Akin, J. Fac. Eng. Archit. Gazi Univ. 34, 1127 (2019)
O.V. Larina, P.I. Kyriienko, N.D. Shcherban, P.S. Yaremov, D.Y. Balakin, I. Khalakhan, K. Veltruská, S.O. Soloviev, S.M. Orlyk, Ind. Eng. Chem. Res. 60, 11964 (2021)
M.A. López-Martínez, I. Melián-Cabrera, Ind. Eng. Chem. Res. 60, 11848 (2021)
P. Völs, S. Hilbert, B. Störr, N. Bette, A. Lißner, J. Seidel, F. Mertens, Ind. Eng. Chem. Res. 60, 5114 (2021)
C.E. Daza, C.R. Cabrera, S. Moreno, R. Molina, Appl. Catal. A Gen. 378, 125 (2010)
W.T. Reichle, Solid State Ionics 22, 135 (1986)
G. Zi, Z. Yan, Y. Wang, Y. Chen, Y. Guo, F. Yuan, W. Gao, Y. Wang, J. Wang, Carbohydr. Polym. 115, 146 (2015)
R.C. Zeng, X.T. Li, Z.G. Liu, F. Zhang, S.Q. Li, H.Z. Cui, Mater. Sci. 9, 355 (2015)
C.L. Caldeira, V.S.T. Ciminelli, K. Osseo-Asare, Geochim. Cosmochim. Acta 74, 1777 (2010)
B. Kostura, D. Matýsek, J. Kukutschová, J. Leško, Ann. Chim. Sci. Des Mater. 37, 11 (2012)
J. Liu, Z. Du, Y. Yang, T. Lu, F. Lu, J. Xu, Chemsuschem 5, 2151 (2012)
B. Sarkar, C. Pendem, L.N. Sivakumar Konathala, R. Tiwari, T. Sasaki, R. Bal, Chem. Commun. 50, 9707 (2014)
Acknowledgements
This work was mainly financially supported by Research Fund of Usak University (2019/DTS005) and Regional Development Program. All of the synthesized catalysts were characterized in Recep Tayyip Erdoğan University Central Research Laboratory Application and Research Center, Rize, Turkey. We are grateful to all team staff for their support. Hülya A. Kılıç is grateful for the support provided by Higher Education Institution (YOK 100/2000, Fuels (fossil and bio-) and Combustion) PhD funding program.
Funding
This work was mainly financially supported by Research Fund of Usak University (2019/DTS005) and Regional Development Program.
Author information
Authors and Affiliations
Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kılıç, H.A., Kılıç, E., Erden, L. et al. Highly selective oxidation of glucose to formic acid over synthesized hydrotalcite-like catalysts under base free mild conditions. Res Chem Intermed 48, 4079–4103 (2022). https://doi.org/10.1007/s11164-022-04811-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-022-04811-9