
Abstract
The AHA coupling of amines, haloalkane and alkynes under UV visible light was achieved with a higher yield in the presence of Au/Fe2O3. The catalyst was prepared by two methods using different gold content and then characterized by XRD, UV–vis, BET, TEM, ICP-OES and TPR spectroscopies. A comparative study of the ordinary and photocatalytic conditions, showed that the UV visible light could activate the gold nanoparticles and lead to the formation of CH2Cl• and Cl• radicals through CH2Cl2 fragmentation. The propargylamine was afforded at low temperature and a short time using 2% Au/Fe2O3. The catalyst was stable for five cycles with good photoactivity.
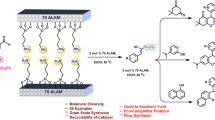
Graphical abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In organic synthesis, the irradiation by UV light has attracted many researchers, so several reactions were performed using the photocatalytic route as the conversion of alcohol to aldehyde [1] and oxidation of Benzylamine into Imines [2]. It was also used in the C–C coupling reaction as cross-coupling [3], and Suzuki–Miyaura reaction [4]. UV visible light is considered an energy source that can reduce the reaction temperature and modify the metal capacity by the excitation of its electrons, leading to a strong interaction between molecules and metal [5]. Propargylamines are interesting intermediates to synthesize built and heterocyclic molecules [6] such as pyrrole, oxazoles, and pyrazoles [7]. Furthermore, several propargylamines derivatives were used to treat Parkinson’s and Alzheimer’s disease, such as rasagiline [8]. Their preparation method has drawn the attention of a significant number of researchers. In view of the synthetic procedure, the three coupling component of alkyne, aldehyde, and amine (A3) was used in the presence of catalysts [9]. Moreover, the aldehyde is replaced by haloalkane (AHA) [10] or ketones (KA2) [11], where various heterogeneous and homogeneous transition metals catalysts have been used, such as Fe, Au, Cu, and Co [12]. Under UV light irradiation, propargylamines were prepared through copper catalyst via the coupling of arylamine, alcohol and alkynes and with the reaction carried out at room temperature [13]. Panwar et al. [14] used TiO2-PANI-AuNPs catalyst for the A3 coupling at 25 °C for 48 h. They reported that the photocatalytic method activates the catalyst.
The AHA coupling is also a simple route to propargylamines synthesis using CH2X2 as methylene moiety. This coupling involves the activation of C-X and C-H bonds by the metal. The use of heterogeneous catalyst in AHA coupling stays limited. In 2014, Sharma et al. [15] used the silica nanosphere supported iron catalyst in the AHA coupling for 10 h at 70 °C. Then, monometallic and bimetallic gold catalysts were used with good activity for 24 h at 65 °C [16, 17]. This activity is due to the small gold size nanoparticles. In the AHA coupling reaction, all catalysts were used with the ordinary condition without activation by UV light or other source.
However, different transition catalytic systems were used in the propargylamines synthesis, the Fe2O3 has mostly been used as h-Fe2O3@SiO2-CD/Ag [18], which catalyzed the A3 and KA2 coupling under ultrasonic irradiation, with high reusability of 6 runs. Recently, Magnetic AgNPs/Fe3O4/ chitosan/PVA nanocatalyst showed a high activity in the A3 coupling reaction [19]. Various ways exist to synthesize Fe2O3 NPs like sol–gel [20], precipitation and hydrothermal methods [21]. The hard template is a famous method that leads to mesoporous material formation[22]. Gold nanoparticles were prepared through different methods such as deposition precipitation with urea (DPU) [23], DP-NaOH [24, 25], and impregnation [26]. Also, the Au/Fe2O3 was prepared by co-precipitation [27], impregnation using lysine, where Fe2O3 was synthesized by the hydrothermal method. The nanoparticles were deposed with an average size of 3–6 nm [28]. The Au@Fe2O3 Nanocomposite was prepared using Fruit extract. The catalyst was used in photo-degradation [29].
In the present study, two mesoporous materials were synthesized: the mesoporous Fe2O3 was prepared with the hard template using SBA-15, while the mesoporous Au/Fe2O3 was prepared by DP-NaOH, and DPU. With the last one, different content of gold were used. Then, the effect of preparation method and nanoparticles size on the catalyst activity was demonstrated. The catalyst which gives the best yield was used also to synthesize propargylamines via AHA coupling under ordinary condition and UV light irradiation (Scheme 1).

Synthesis of propagylamines via AHA coupling reaction
Experimental section
Materials and instrumentation
All reagents were purchased from Sigma-Aldrich with high purity (99.9%).
XRD patterns of samples were recorded on a Bruker AXS D5005 diffractometer equipped with monochromatized Cu Ka radiation (l = 1.5418 Å) at 40 kV with a scanning rate of 1°/min.
UV–vis spectroscopy measurements were carried out at room temperature on a Lambda 800 UV–vis spectrometer, operating in 200–800 nm. All spectra were deconvoluted.
The inductively Coupled Plasma Optical Emission Spectrometry (ICP-OES) was used to identify the real gold content; the samples were analyzed by Perkin Elmer Optima 2000 DV instrument.
The BET surface areas and pore sizes analysis was determined from N2 adsorption–desorption of nitrogen at 77 K using a Quantachrom NOVA 1000 instrument. The samples were out gazed under vacuum at 250 °C for at least 6 h.
The Temperature Programmed Reduction experiments (TPR) were carried out in an Altamira AMI-200 apparatus. 70 mg of samples passed through 5% H2/Ar (with a flow of 30 mL/min) and heated at 800 °C with 5 °C/min.
Transmission Electron Microscopy (TEM) of samples was analyzed by a JEOL JEM-1230 electron microscope, under sonication for 20 min. Ethanol was used for dispersion of samples.
For the excitation by UV–visible irradiation, the AHA coupling reaction was monitored in a MULTIRAYS photo reactor fitted with UV lamps: 254 nm and 350 nm.
Synthesis of mesoporous Fe2O3
Firstly, Mesoporous SBA15 was synthesized by Bailiche method [30]; 4 g of amphiphilic triblock copolymer poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) was dissolved with 8.64 g of HCl (2 M) in 120 g of water under stirring at 40 °C, then 8.54 g of tetraethyl-ortho-silicate was added. The mixture was stirred again at 40 °C for 24 h. The product was crystallized in a Teflon-lined autoclave at 100 °C for 2 days. After cooling at room temperature the solid was filtered and dried at room temperature. The solid was then calcined at 500 °C for 4 h to remove the Template.
Secondly, the mesoporous iron oxide was synthesized by the mixing of Fe(NO3)3.9H2O with 0.15 g of mesoporous SBA-15, The mixture was transferred into a crucible, was placed in a muffle furnace, increasing the temperature to 500 °C for 4 h. Then, after cooling the product at room temperature, the silica template was removed by NaOH solution at 80 °C; the porous iron was recovered by centrifugation and washed with distilled water.
Synthesis of mesoporous Au/Fe2O3
Synthesis of Au/Fe2O3 by deposition precipitation with urea (DPU)
Firstly, the tetrachloroauric acid with a concentration of 3.65 × 10−4 M and a suspension of Fe2O3, and urea solution (3.6 × 10−3) were combined under stirring. The solution is heated at 80 °C. After 4 h, the mixture was filtered and washed with deionised water three times to eliminate the chloride ions. Finally, the product was dried for 24 h at 80 °C and calcined for 4 h at 400 °C under air.
Synthesis of Au/Fe2O3 by deposition with NaOH (DP-NaOH)
Under stirring, the tetrachloroauric acid, a suspension of Fe2O3 and NaOH (2 M) solution were mixed. The solution was heated at 80 °C and the same steps were used.
General procedure for the synthesis of Propargylamine
The propargylamines were prepared by the AHA coupling of phenylacetylene (2 mmol), amine (2.2 mmol), CH2Cl2 and the catalyst using 3 mL of solvent. After stirring at 60 °C for the appropriate time, the reaction mixture is extracted with H2O/CH2Cl2, dried over Na2SO4 and concentrated. This product was purified using column chromatography on silica gel. All products were characterized in our previous studies [17]. The same reaction was monitored under UV light at 254 nm and 350 nm.
Results and discussion
The real gold content in the supported catalysts was measured by ICP-OES analysis. The results are shown in Table 1. All catalyst exceeds 50% of deposit gold. The high gold deposit rate was achieved with the 2%wt Au/Fe2O3 catalyst was prepared by DPU. The increase in gold content leads to lower deposition. Also, with the catalyst prepared by DPU and DP-NaOH, the same gold content was deposited.
Figure 1 shows the XRD patterns of Fe2O3 and the x% Au/Fe2O3 catalysts. All patterns exhibit one sharp diffraction, associated with the hexagonal symmetry of mesoporous SBA-15 and corresponding to (100) reflection. This result shows that the ordered structure is maintained by solid–liquid route used in the preparation. Furthermore, the diffraction at 2 thetas equal to 49, 55, and 64° characterizes the hematite phase of the Fe2O3 [31].

XRD patterns of a Fe2O3, b 1% Au/Fe2O3, c 1% Au/Fe2O3-DP NaOH, d 2% Au/Fe2O3, e 4% Au/Fe2O3
In the Au/Fe2O3 spectra (Fig. 1b), the increase in gold content does not modify the catalyst structure; also, the gold diffraction is not detected, this suggests that gold was deposited with high dispersion and small sizes [10]. The XRD patterns of the prepared catalysts with different methods are presented in Fig. 1b and c. No difference was seen, which indicates that the method of preparation has not changed the catalyst structure.
The N2 adsorption–desorption isotherms of catalysts are illustrated in Fig. 2. All isotherms are meaningful to type IV as reported by the IUPAC classification, the mesoporous materials prototypal [32]. Table 2 displays the textural properties of samples. The increase in gold content in gold catalyst prepared by DPU results the decrease in the pore volume and the BET surface. This demonstrates that smaller gold particles are dispersed in the pore channels, which alter to narrow [18]. Samples prepared with DP-NaOH presented the same surface proprieties as 4% Au/Fe2O3 and a lower surface area than 1% Au/Fe2O3-DPU.

N2 adsorption–desorption isotherms and BJH pore size distribution of Fe2O3 and Au/Fe2O3
The deconvoluted DRUV–vis spectra of Fe2O3 and x% Au/Fe2O3 are plotted in Fig. 3. For the catalyst prepared by DP-NaOH, the gold plasmonic peak (SPR) is observed at 531 nm. This peak has shifted to higher wavelength for the catalyst prepared by DPU (560 nm, 562 nm, and 557 nm) (Table 3). Also, new bands appeared in the case of 1% Au/Fe2O3-DP-NaOH, 2% Au/Fe2O3 and 4% Au/Fe2O3 situated at 392 nm, 379 nm, and 392 nm. These bands are assigned to charged gold species Au+ [33]. Also, the bands at 246 nm, 445 nm in the Fe2O3 spectra have shifted in the supported catalyst. This is due to the d-d electronic transition of Fe3+ and iron hydroxide clusters, respectively [34]. The adsorption peak at 284 nm is pictured the metal charge transfer. It is also present in the supported catalyst with minor mobility. Another peak revealed at 732 nm and 691 nm in the 2% Au/Fe2O3 and 4% Au/Fe2O3, is defined as the d-d transition of Fe3+ [35].

DRUV–vis spectra of a Fe2O3, b 1% Au/Fe2O3-DP NaOH, c 1% Au/Fe2O3, d 2% Au/Fe2O3, e 4% Au/Fe2O3
Temperature Programmed Reduction (TPR) of Fe2O3 and Au/Fe2O3 catalysts are reported in Fig. 4. In all spectra, a broad signal situated around 360 °C, could be assigned to the reduction in Fe2O3 into Fe3O4 (magnetite) and the transformation of magnetite to FeO [36, 37]. This signal was slightly modified in its aspect in the catalyst, because of the interaction of gold with support [38]. Furthermore, in the catalyst, other peaks appear in lower temperatures. The spectra of 2% Au/Fe2O3 prepared by DPU and 1% Au/Fe2O3 prepared by DP-NaOH, present the same signal at 280 °C, 276 °C, and 235 °C, that can be attributed to the reduction in Au3+ to metallic Au0 [39]. This result is in accordance with the UV–visible result, where all catalysts present the SPR band. The spectra of 1% Au/Fe2O3 (Fig. 4c) presents a signal at 203 °C, which was shifted to 186 °C in the case of 4% Au/Fe2O3 (Fig. 4e) and it is assigned to the reduction in Au2O3 to AuO and Au3+ to Au0 [40]. Although, with high metal loading (4 wt%), a signal appears at 638 °C, is attributed to the transformation of FeO to Fe. This is probably due to the polarization of the Fe–O bonds by gold charged species.

H2-TPR profile of a Fe2O3, b 1% Au/Fe2O3DP-NaOH, c 1% Au/Fe2O3, d 2% Au/Fe2O3, e 4% Au/Fe2O3
Figure 5a shows the TEM images of the Fe2O3. The hexagonal structure is well-structured using SBA-15 as a structuring agent. The corresponding images of 1% Au/Fe2O3 prepared by DPU and DP-NaOH are presented in Fig. 5b and c, respectively. The dark contrast indicates the deposit of gold on the support surface with the homogeneous distribution. The histogram of gold particles of the 1% Au/Fe2O3 catalysts prepared by DPU (Fig. 5b) shows that the gold nanoparticles size was in average of 1–10 nm. Furthermore, for 1% Au/Fe2O3 catalysts prepared by DP-NaOH (Fig. 5e) presents larger nanoparticles with size of 1–15 nm. For the catalysts 2% Au/Fe2O3 and 4% Au/Fe2O3, prepared by DPU, the particle size depends on the gold loading (Fig. 5c, d). With 2% Au particles being less than 5 nm, increasing of the gold content to 4% the gold NPs become larger with a size of 1–15 nm. These results suggest that the preparation method of gold catalyst and gold loading affect the particle size and the decrease in gold content causes the gold NPs agglomeration.

TEM image and size distribution histogram of a Fe2O3, b 1% Au/Fe2O3, c 2% Au/Fe2O3, d 4% Au/Fe2O3, e 1% Au/Fe2O3 DP-NaOH
AHA coupling reaction
Initially, all prepared catalysts were used in the AHA coupling of phenylacetylene, CH2Cl2 and diethylamine without the base. The results are shown in Table 4. In the absence of a catalyst or using HAuCl4, Fe2 (NO3)0.9H2O and the support Fe2O3, the reaction was not carried out. Also, it is the same result using the supported catalyst 1% Au/Fe2O3. Although, using the last catalyst prepared by DP-NaOH, the yield was 30%. The increase in gold content to 2 and 4% leads to an increase in the yield of propargylamine to 45% and 35%, respectively. This result indicated that the nature of gold species and the NPs size affect the catalytic activity. Moreover, the UV–vis and TPR show that the charged and metallic species were present in the 1% Au/Fe2O3DP-NaOH, 2% Au/Fe2O3 and 4% Au/Fe2O3 catalyst, but in the 1% Au/Fe2O3 except metallic gold are supported. Also, the 2% Au/Fe2O3 has a good activity, where the gold nanoparticles size is lower than 5 nm. The same activity was achieved with the catalyst having the small gold nanoparticles [17].
To optimize the AHA coupling condition, three amounts of 2% Au/Fe2O3 catalyst were used in the coupling of phenylacetylene, CH2Cl2 and diethylamine at different temperatures and reaction time (Fig. 6). As a remark, in water, the yield does not exceed 40% at 40 °C and 60 °C. Increasing the catalyst amount, the yield increases to 40%. In the case of CH3CN, good yields were obtained by increasing the catalyst amount to 80 mg during 10 h. Adopting CH2Cl2 as a solvent, the yield rises to 60% for 10 h. In addition, without solvent, lower yields were afforded. This results due to the phenylacetylene reactivity in different solvent. As result of this part, the higher yield (80%) was obtained in CH3CN at 60 °C for 10 h using 80 mg of catalyst.

Optimization of AHA coupling reaction
Until now, the AHA coupling was afforded at 10 h by the silica nanosphere supported iron catalyst [15]. The work aims is to reduce the reaction time or temperature for high yield. So for that purpose, the reaction was carried under UV light, using two wavelengths (Table 5). We have noticed that propargylamine can’t be produced at 60 °C under 350 nm, but after four hours, another product is formed by the homocoupling of phenylacetylene. This was due to the evaporation of CH2Cl2 at 60 °C since the reaction was monitored without reflux. Decreasing the temperature to 20 °C (room temperature), the propargylamine was afforded at 2 h, 6 h, and 10 h with yield yields of 10, 60, and 80%, respectively (entries 3, 4, 5). After that, the reaction was monitored at 254 nm, and no product was formed at 2, 4, and 6 h. The reaction proceeds smoothly and gives 20% at 10 h.
The result in Table 5 indicates that during the photocatalytic reaction, the catalyst becomes more active and CH2Cl2 more reactive. To visualize this effect, the reaction of CH2Cl2 with the catalyst in CH3CN was monitored and the solution was characterized by UV–visible (Fig. 7a). The UV–visible spectra of CH2Cl2 exhibit a strong band situated around 290 nm. After irradiation at 350 nm (3.54 eV) for 2 h, the band shift to 280 nm and a new band appears at 340 nm. Continuing to irradiate to 4 h, the bands become broad and intense. The second band is due to the charged species CH2Cl+ and Cl− [41]. After 6 h, these bands shift to 320 nm and 380 nm. The CH2Cl and Cl radicals were formed by photo dissociation, which leads to the fragmentation of CH2Cl2 [42]. Alcantara et al. [43] reported that the initial excitation provokes the C–Cl bond breaking to give excited or ionized chlorine ion and CH2Cl+. These radicals are usually formed by the methylene chloride radiolyse, excitation by vacuum ultra-violet (VUV) spectroscopy, or ultrafast fragmentation channels [44, 45]. Using the irradiation at 254 nm (4.875 eV), radicals are not produced, so there is no fragmentation of CH2Cl2, and as consequence, propargylamine cannot be formed at 2, 4, and 6 h. This confirms the low yield of propargylamine obtained at 10 h. Under irradiation at 350 nm, at room temperature, the solution of the phenylacetylene, CH2Cl2 and diethylamine reaction was also characterized by UV–vis at 6 h (Fig. 7b, e). This spectrum was compared with the phenylacetylene, diethylamine and pure propargylamine spectra. The solution spectrum shows that new bands appear. Their deconvolution (Fig. 7c) reveals the presence of bands at 288 nm, 295 nm, and 345 nm, which are assigned to phenylacetylene and diethylamine, respectively. The propargylamine band is seen around 314 nm. Other bands at 275 nm, 427 nm, 440 nm, 484 nm, and 530 nm are assigned to the CH2Cl2 radicals [41].

UV–vis spectra of a CH2Cl2 under irradiation at 250 and 350 nm, b AHA coupling reaction solution, and c deconvoluted spectra of AHA coupling reaction solution
The UV–vis result shows that during the propargylamine formation, radicals are present in the solution, indicating that the fragmentation has accrued by UV light irradiation.
Several propargylamines were prepared using secondary amines (Table 6): aliphatic secondary amines such as dibutylamine and diethylamine were very reactive and gave good yield (92, 80%). In addition to that, the cyclic amines also provide a high yield of 80–90%. Moreover, the use of cyclic and aliphatic primary amine in the propargylamine synthesis does not give the desired product. As a result, the UV light activates the AHA coupling in the presence of 2% Au/Fe2O3 at room temperature, where other researchers used high temperature [15] and long-time reaction [16, 17].
After completing the reaction of phenylacetylene, CH2Cl2 and morpholine, the photocatalyst was separated by centrifugation and washed with water and CH2Cl2 to eliminate organic residuals, then, dried all night. This catalyst was used for a new reaction with the same reactants under identical conditions. Figure 8 shows that the 2% Au/Fe2O3 photocatalyst was reused for five runs without activity loss.

The reusability of 2% Au/Fe2O3 in the AHA coupling reaction under UV light
Scheme 2 shows the plausible mechanism of the AHA coupling reaction catalyzed by 2% Au/Fe2O3 under UV light irradiation. In the first step, the phenylacetylene was activated by gold nanoparticles to absorb the UV light, that excited the Plasmon gold resonance (NPs + hʋ \(\to \) h+ + e−). Then, the CH2Cl2 was fragmented under irradiation to give CH2Cl• and Cl• radicals. The chloride ion reacts with the hydrogen of secondary amine to give HCl. The CH2Cl• radical reacts with diethylamine to form the chloro-N,NR1R2-methenamine. Finally, the activation of C–Cl band of the last intermediate by gold and UV light leads to the propargylamine formation.

Possible mechanism of the photocatalytic coupling reaction
Conclusion
In the present study, we have demonstrated for the first time the photocatalytic AHA coupling of alkyne, CH2Cl2 and amine under UV irradiation. The 2% Au/Fe2O3 shows a good activity than other catalysts under ordinary conditions. This was due to the gold nanoparticles size. We have also found that the CH2Cl2 gives radicals under UV irradiation in the presence of the catalyst, which accelerates the propargylamine formation at room temperature. Moreover, the advanced photocatalyst displays a high activity mainly due to the mobility and the electron transfer at the catalyst surface. Our result offers a new application of CH2Cl2 and UV irradiation in the coupling reactions.
References
R. Zhang, Y. Liu, Z. Wang, P. Wang, Z. Zheng, X. Qin, X. Zhang, Y. Dai, M. Whangbo, B. Huang, Appl. Catal. B 254, 463 (2019)
S. Sarina, H. Zhu, E. Jaatinen, Q. Xiao, H. Liu, J. Jia, C. Chen, J. Zhao, J. Am. Chem. Soc. 135, 5793 (2013)
J.C. Tellis, D.N. Primer, G.A. Molander, Science 345, 433 (2014)
K. Mori, M. Kawashima, H. Yamashita, Chem. Commun. 50, 14501 (2014)
Z. Jiao, Z. Zhai, X. Guo, J. Phys. Chem. C 119, 3238 (2015)
S. Srivastava, New J. Chem. 43, 6469 (2019)
K. Lauder, A. Toscani, N. Scalacci, D. Castagnolo, Chem. Rev. 117, 14091 (2017)
M. Sharma, J. Mangas-Sanchez, N.J. Turner, G. Grogan, Adv. Synth. Catal. 359, 2011 (2017)
S. Sadjadi, M.M. Heravi, M. Ebrahimizadeh, J. Porous Mater. 25, 779 (2018)
A. Berrichi, R. Bachir, M. Benabdallah, N. Choukchou-Braham, Tetrahedron Lett. 56, 1302 (2015)
A. Moaddeli, M. Abdollahi-Alibeik, J. Porous Mater. 25, 147 (2018)
T.K. Saha, R. Das, ChemistrySelect 3, 147 (2018)
A. Sagadevan, V.K.K. Pampana, K.C. Hwang, Angew. Chem. Int. Ed. 58, 3838 (2019)
V. Panwar, S.L. Jain, Mater. Sci. Eng. C 99, 191 (2019)
R.K. Sharma, S. Sharma, G. Gaba, RSC. Adv. 4, 49198 (2014)
A. Berrichi, R. Bachir, S. Bedrane, N. Choukchou-Braham, K. Belkacemi, Res. Chem. Intermed. 45, 3481 (2019)
M. Bensaad, A. Berrichi, R. Bachir, S. Bedrane, Catal. Lett. 151, 1068 (2021)
S. Sadjadi, M. Malmir, M.M. Heravi, RSC. Adv. 7, 36807 (2017)
K. Ghasemi, M. Darroudi, M. Rahimi, H. Rouh, A.R. Gupta, C. Cheng, A. Amini, New J. Chem. 45, 16119 (2021)
K. Raja, M.M. Jaculine, M. Jose, S. Verma, A. Prince, K. Ilangovan, K. Sethusankar, S.J. Das, Superlattices Microstruct. 86, 306 (2015)
A. Lassoued, M.S. Lassoued, B. Dkhil, S. Ammar, A. Gadri, Phys. E: Low-Dimens. Syst. Nanostruct. 101, 212 (2018)
E. Delahaye, V. Escax, N. El Hassan, A. Davidson, R. Aquino, V. Dupuis, R. Perzynski, Y.L. Raikher, J. Phys. Chem. B 110, 26001 (2006)
N. Mehiaoui, Z. Kibou, A. Berrichi, R. Bachir, N. Choukchou-Braham, Res. Chem. Intermed. 46, 5263 (2020)
C. Santra, A. Auroux, B. Chowdhury, RSC Adv. 6, 45330 (2016)
C. Campos, M. Jofré, C.C. Torres, B. Pawelec, J. Fierro, P. Reyes, Appl. Catal. A: Gen. 482, 127 (2014)
C. Rodríguez-Martínez, Á.E. García-Domínguez, F. Guerrero-Robles, R.O. Saavedra-Díaz, G. Torres-Torres, C. Felipe, R. Ojeda-López, A. Silahua-Pavón, A. Cervantes-Uribe, J. Compos. Sci. 4, 89 (2020)
B.A.A. Silberova, G. Mul, M. Makkee, J. Moulijn, J. Catal. 243, 171 (2006)
J. Zhang, X. Liu, X. Guo, S. Wu, S. Wang, Chem. Eur. J. 16, 8108 (2010)
S. Shams, A.U. Khan, Q. Yuan, W. Ahmad, Y. Wei, Z.U.H. Khan, S. Shams, A. Ahmad, A.U. Rahman, S. Ullah, J. Photochem. Photobiol. B: Biol. 199, 111632 (2019)
Z. Bailiche, L. Chérif-Aouali, R. Cousin, S. Royer, B. Bengueddach, S. Pronier, S. Siffert, Int. J. Chem. 34, 1131 (2013)
E. Darezereshki, Mater. Lett. 65, 642 (2011)
G. Leofanti, M. Padovan, G. Tozzola, B. Venturelli, Catal. Today 41, 207 (1998)
I. Tuzovskaya, N. Bogdanchikova, A. Simakov, V. Gurin, A. Pestryakov, M. Avalos, M. Farías, Chem. Phys. 338, 23 (2007)
W. Deng, C. Carpenter, N. Yi, M. Flytzani-Stephanopoulos, Top. Catal. 44, 199 (2007)
T. Sabari Girisun, M. Saravanan, V.R. Soma, ACS Appl Nano Mater. 1, 6337 (2018)
M. Liang, W. Kang, K. Xie, J. Nat. Gas Chem. 18, 110 (2009)
. N. Salamun, H.X. Ni, S. Triwahyono, A.A. Jalil, A.H. Karim, Mal. J. Fund. Appl. Sci. 7, (2011)
Y.-X. Miao, L.-H. Ren, L. Shi, W.-C. Li, RSC Adv. 5, 62732 (2015)
A. Sandoval, R. Zanella, T.E. Klimova, Catal. Today 282, 140 (2017)
A. Venugopal, M.S. Scurrell, Appl. Catal. A: Gen. 258, 241 (2004)
S. Emmi, G. Beggiato, G. Casalbore-Miceli, Int. J. Radiat. Appl. Instrum. C: Radiat. Phys. Chem. 33, 29 (1989)
K. Kesper, F. Diehl, J.G.G. Simon, H. Specht, A. Schweig, Chem. Phys. 153, 511 (1991)
K. Alcantara, A. Gomes, W. Wolff, L. Sigaud, A. Santos, J. Phys. Chem. A 119, 8822 (2015)
B.N.C. Tenorio, R.R. Oliveira, S. Coriani, Chem. Phys. 548, 111226 (2021)
Z. Alfassi, S. Mosseri, P. Neta, J. Phys. Chem. 93, 1380 (1989)
Acknowledgements
We thank DGRDST and the University of Tlemcen for the financial of this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Berrichi, A., Bailiche, Z. & Bachir, R. Mesoporous Au/Fe2O3 catalyst for propargylamines synthesis via CH2Cl2 under visible light irradiation. Res Chem Intermed 48, 4119–4134 (2022). https://doi.org/10.1007/s11164-022-04796-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-022-04796-5