
Abstract
New symmetric N,N'-bis(thioamido)thio/carbohydrazones (1–4) were prepared from thio/carbohydrazide and various isothiocyanates with high yields and characterized the structures of the compounds. The compounds were evaluated as new corrosion inhibitors for corrosion of mild steel in 0.1 M HCl solutions using polarization and electrochemical impedance spectroscopy (EIS) techniques, and all compounds showed effective inhibitory properties. Antioxidant activity values determined using the DPPH method for all compounds showed higher antioxidant activity than the standard Trolox (5.89 µM). The IC50 values of the compounds were obtained between 3.45 and 4.25 µM, and also showed a ranking of \(4>2>3>1>\mathrm{Trolox}\), with antioxidant activities better than Trolox. Compound 4 (1,5-bis((3-fluorophenyl)carbamothioyl)carbonohydrazide) exhibited the highest activity in terms of both antioxidant and anti-corrosion properties. DFT calculations were also performed to determine the spectroscopic properties of the compounds and to analyze the structure–activity relationship for their antioxidant characteristics. It was investigated how DPPH reactions are affected by the dependence of delocalization indices and charge densities of N–H bonds on intramolecular interactions. As well as the calculations of intramolecular interactions and some QTAIM parameters, Laplacian bond order and bond polarity index (BPI) analysis were also performed to examine the electronic properties of reactive N–H bonds.
Graphical abstract
New symmetric N,N'-bis(thioamido)thio/carbohydrazones were obtained. Structures of all compounds were elucidated using spectroscopic approaches. Antioxidant activities of the compounds were determined by DPPH method. QTAIM analyses were used to examine the relationship between the electronic properties and antioxidant activity. The compounds were evaluated as new corrosion inhibitors for corrosion of mild steel in 0.1 M HCl.

Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Compounds including thio/carbohydrazides are a significant class of synthetic organic chemistry. Thio/carbohydrazone compounds have a variety of biological, physical, and pharmaceutical applications [1,2,3,4]. They have revealed wide spectrum of activities or properties such as physicochemical, antioxidant, antileishmanial, carbonic anhydrase inhibitors, antimicrobial, electrochemical, antibacterial, antitubercular, corrosion inhibitor, antitumor, cell growth inhibitory, and antiviral [3, 5,6,7,8,9,10,11,12,13,14,15,16].
Many functional compounds such as thio/carbohydrazone derivatives have been the subject of research in recent years with their antioxidant properties such as reducing the damages of oxidative stress in living organisms and preventing DNA oxidation [17,18,19].
In the literature, there are studies on corrosion inhibitors that explain the good performance of the synthesized molecules by the presence of aromatic rings, heteroatoms and thioketone groups in their structures [20, 21]. Moreover, different studies have reported that this difference in inhibitory effects varies according to the type of functional group [22,23,24,25].
In this paper, we have synthesized new N,N'-bis(thioamido)thiocarbohydrazone and carbohydrazones derived from thio/carbohydrazide and various isothiocyanates for the first time. The structures of the synthesized compounds were characterized by using FTIR, 1H NMR, and 13C NMR spectroscopic techniques and elemental analysis. The experimental results were compared with the density functional theory (DFT) calculations and were found to be in good agreement. DPPH free radical scavenging method was used to determine the antioxidant properties of the compounds. The relationship between antioxidant properties and structural and electronic properties of the compounds was also examined by QTAIM, Laplacian bond order (LBO) and bond polarity index (BPI) analysis. Furthermore, the corrosion inhibition of mild steel in 0.1 M HCl solution of all compounds was investigated using potentiodynamic polarization and electrochemical impedance spectroscopy (EIS) techniques.
Materials and methods
Instruments and chemicals
All chemical materials were purchased from Sigma-Aldrich or Merck Chemical Company and were used without further purification. Eurovector EA3000 elemental analyzer was used to determine elemental analysis. A Stuart SMP30 melting point apparatus was used to record melting points. IR spectra were taken by a Bruker Alpha Fourier transform IR (FT-IR) spectrometer. NMR spectra were obtained from a JEOL ECX-400 (400 MHz) spectrophotometer in dimethyl-d6 sulfoxide (DMSO-d6). A Shimadzu UVM-1240 UV–visible spectrophotometer was used for absorption measurements. Corrosion study was performed using a Gamry Reference 3000 Potentiostat/Galvanostat/ZRA.
Synthesis of the compounds (1–4)
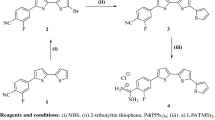
A solution of an appropriate isothiocyanates (8.0 mmol) in ethanol (25 mL) was added to a solution of thio/carbohydazide (4.0 mmol) in ethanol (25 mL) with stirring. The reaction mixture was refluxed for 3 h. The solution was cooled to room temperature, and the precipitate was filtered, washed three times with ethanol, and dried. The products, shown in Scheme 1, were obtained in good yield (80–97%).

General synthesis of new N,N'-bis(thioamido)thiocarbohydrazone and carbohydrazone
Antioxidant activity assay
DPPH free radical scavenging method [26, 27], which is used to examine the antioxidant activities of molecules depending on the hydrogen losing abilities of compounds, was used for antioxidant activity measurements of the compounds 1–4. Stock solutions of the compounds were prepared in DMSO at 200 µM. Then, it was added to the previously prepared DPPH solution with sufficient ethanol so that the final compound concentrations were in the range of 0.4–10 µM with a total of 6 mL solution [28, 29]. Then, the absorbances of the mixtures kept at room temperature in a dark room were read at 517 nm, and these absorbances were used in percentage inhibition calculations Eq. 1 [30]:
where A0 and A1 represent the absorbance of the control sample and the compound, respectively. Next, IC50 (mg/mL) values were calculated, expressing the amount of antioxidant required to reduce the initial DPPH concentration by 50% [31].
Corrosion inhibition assays
Electrochemical measurements were carried out using mild steel (composition; 0.134% C, 0.0514% Ni, 0.555% Mn, 0.0117% P, 0.200% Si, 0.0258% S and 99.022% Fe) placed inside a cylindrical teflon rod as the working electrode. Electrode surface area was prepared as 0.1963 cm2 and experiments were performed at a constant temperature of 293 K. The surface of the working electrode was grinded on water-lubricated silicon carbide papers (800, 1000, 1200 grid) and polishing with diamond paste down to 1 μm finish. A 150 mL cell assembly was prepared using platinum as counter electrode and saturated Ag/AgCl calomel electrode as reference electrode. Experiments were carried out at five different concentrations (1 × 10–5 M, 3 × 10–5 M, 5 × 10–5 M, 7 × 10–5 M, 1 × 10–4 M) by passing N2 gas from the solution in 0.1 M HCl environment. Inhibitor compound solutions were prepared as a 2 × 10–4 M stock solution in dimethylsulfoxide (DMSO). Potentiodynamic polarization experiments were performed over the potential range —250 to 250 mV at a scan rate of 1 mV/s. Electrochemical impedance spectroscopy (EIS) measurements were performed at equilibrium potential in the frequency range of 0.1 Hz–105 Hz and with a signal size of 10 mV AC.
Computational procedure
The optimization calculations were performed with the GAUSSIAN 09 software package program [32] using Kohn–Sham density functional theory (KS-DFT) [33, 34], without any geometric constraints. Becke3–Lee–Yang–Parr hybrid change-correlation functional (B3LYP) [35,36,37,38] with 6-311++g(2d,2p) basis set for optimizations and spectral data calculations was used. It was also determined that the compounds do not have imaginary frequencies, i.e., the results correspond to the global minimum at the potential energy surfaces.
Highest Molecular Orbital (HOMO) and Low Lowest Molecular Orbital (LUMO) energy eigenvalues, calculated in gas phase, were used to calculate electronic parameters such as energy gap ∆E, chemical hardness (η), and electronegativity (χ).
The proton and carbon nuclear magnetic resonance (1H and 13C NMR) shifts of the compounds were calculated by the conductor-like polarizable continuum model (CPCM) in the DMSO phase in accordance with the experiment, using the gauge-including atomic orbital (GIAO) method. Quantum theory of atom in molecule (QTAIM) [39,40,41,42,43,44] calculations were performed at the level of 6-311++g(2d,2p) to analyze the effects of substituted groups on the electronic properties of compounds, and to determine the intramolecular interactions and electron charge distribution. Non-covalent interaction (NCI), and Fukui index calculations were performed at the same level of theory in the gas phase. In order to analyze the reactive N–H bonds in DPPH reactions and the electronic properties of the bonds in intramolecular interactions, Laplacian bond order (LBO) [45], delocalization index (DI), percentage of number of electrons localized in H atom (%L), electrostatic interaction energy (EIE) between the total charge distribution of hydrogen and the nitrogen nucleus were calculated. Also, bond polarity index (BPI) [46] calculations were also performed at the 6-31g(d) level of theory.
Results and discussion
Physical properties
Physical appearances, melting points, yields, and elemental analysis data of the compounds are summarized in Tables 1 and 2.
IR spectra
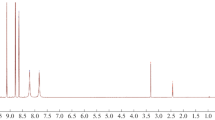
In the FT-IR spectra of the compounds, the asymmetric and symmetric stretching bands of the thio/carbohydrazide amino group (–NH2) were not observed at 3400–3200 cm−1. These results indicated a successful reaction as expected. All compounds 1–4, the amino vibrations (N–H1 and N–H2) of the thio/carbohydrazide region were observed at 3325–3249 and 3270–3147 cm−1, respectively. The amino peak (N–H3) was shown at 3225–3043 cm−1 for all compounds. The thiocarbonyl group vibrations (–C=S) of the thiocarbohydrazide region were observed at 1292–1170 cm−1. The –C–N stretching vibrations were shown at 1168–1135 cm−1. The aromatic or aliphatic –CH stretching vibrations were observed at 3067–2814 cm−1. For compounds 3 and 4, the –C=O stretching vibrations of carbohydrazide were observed at 1672 and 1684 cm−1, respectively. In compound 4, the Ar–F stretching vibration was observed at 984 cm−1 (see at Figures S10–S13 in Supplementary information). These vibrations values are highly consistent with reported results of similar compounds [5, 6, 9, 10, 14]. Experimental and theoretical vibration results are given in Table 3.
1H NMR analysis
The 1H NMR spectra of the compounds were detected in DMSO-d6 as solvent. DMSO-d6 and water in DMSO (HOD, H2O) peaks are observed at around 2.00, 2.50 (quintet), and 3.30 ppm (variable, based on the solvent and its concentration), respectively [47]. For all compounds 1–4, the amino signals (–NH) of the structures were detected at 9.78–7.63 ppm (see Figures S2–S5 in Supplementary information). The N–H1 and N–H2 signals of thio/carbohydrazide region were resonated as a singlet at around 9.78–8.97 and 9.61–8.52 ppm. The N–H3 signals were observed at around 8.93–7.63 ppm. For compound 1, the methyl (–CH3) signal coupled to the N–H3 proton and was resonated as a doublet peak at 2.84–2.83 ppm. For compounds 2 and 3, the methylene group (–CH2–N) proton signal was observed as a quartet at 3.49–3.44 and 3.51–3.46 ppm; the methyl group (–CH3) proton signal resonated as a triplet at 1.11–1.07 and 1.10–1.07 ppm, respectively. In compound 4, the aromatic protons (4H) were detected as a multiplied at 7.53–6.93 ppm. The data are consistent with values for similar compounds given in the literature [5, 6, 9, 14, 48, 49]. 1H NMR calculations were carried out in the DMSO phase in accordance with the experimental process. Experimental and theoretical proton chemical shift values of the synthesized compounds are given in Table 4.
13C NMR analysis
The 13C NMR spectra of the compounds were taken in DMSO-d6; the chemical shifts are summarized in Table 5. The –C=S (C2) peaks were observed at 182.36–181.67 ppm for all compounds. All compounds, the –C=S (C1) or –C=O (C1) peaks were detected at 184.77–157.02 ppm (see Figures S6–S9 in Supplementary information). For compounds 1–3, the –C3 peaks were observed at 31.50 (–CH3), 39.11 (–CH2), and 38.89 ppm (–CH2), respectively. The C3 carbon atoms shifted downfield (high values of δ), which was caused by the presence of the amino group (–NH). For compounds 2 and 3, the methyl carbon atoms (–CH3) resonated at 14.82 and 14.91 ppm. Furthermore, in compounds 4, the C atoms (for C3–C8) were also split into doublets caused by interacting with the atomic nucleus of F. The carbon atoms (C3–C8) were detected at 163.22–111.97 ppm. The C5 carbon atom shifted downfield (high values of δ, 163–161 ppm) which was caused by the presence of the fluorine atom. Moreover, the C3 carbon atom was resonated at around 141 ppm, shifted downfield due to the presence of the amino group (Ar–NH). These observations are in agreement with the results of reported similar compounds [5, 6, 9, 48, 49].
Evaluation of antioxidant activity
The antioxidant activities of the compounds were determined by the calculated inhibition percentages at different concentrations as shown in Fig. 1. Antioxidant inhibition percentages of four different molecules increased in direct proportion to the concentration. All compounds showed a higher percentage of inhibition against Trolox at all concentrations in the 0.4–10 µM range.

Percentage change of inhibition calculated by DPPH method for Trolox and compounds 1–4. The calculated results (expressed as vertical bars) indicate that the observed experimental effect is genuine at 95% certainty. Trolox was used as the standard antioxidant for antioxidant capacity assays
IC50 values were also calculated to evaluate antioxidant activities. The values obtained for Trolox and compounds 1–4 are summarized in Table 6. As a result of IC50 values, antioxidant activities for the synthesized compounds followed the order of 4 > 2 > 3 > 1. When compounds 2 and 3 were evaluated among themselves, it was found that antioxidant activity of the compound containing thiocarbohydrazone (C=S) was higher than in carbohydrazone group (C=O).
It was observed that the fluorine groups of compound 4, which has the highest antioxidant activity, easily provide electrons and thus contribute to the increase of the antioxidant activity of the compound, as in various studies [49, 50].
Evaluation of corrosion activity
Potentiodynamic polarization studies
Potentiodynamic polarization experiments were performed at four different inhibitor concentrations in order to determine the inhibitory effects of the compounds on the corrosion of mild steel in 0.1 M HCl solution. In Fig. 2, the E-log curves obtained for mild steel in 0.1 M HCl medium containing four different concentrations of compound 4 are given. From the E-log curves obtained by Tafel extrapolation method, anodic Tafel slope (βa), cathodic Tafel slope (βc), and icorr values were determined. Then, using Eq. 2 [51], the inhibition efficiency [IE (%)] was calculated from the icorr values:
where icorr* and icorr values indicate the corrosion current densities measured in inhibitor and non-inhibitor media, respectively. Calculated parameters are given in Table 7 collectively. The data in Table 7 and the polarization curves in Fig. 2 show that the inhibitory effect causes a significant reduction in the corrosion rate compared to the blank sample.

Potentiodynamic polarization curves obtained for mild steel in solutions containing compound 4 at different concentrations in 0.1 M HCl medium
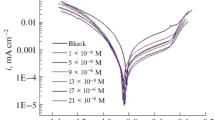
As seen in the current potential curves obtained in 0.1 M HCl solution at different concentrations of the synthesized inhibitors, a decrease in current values is observed in both anodic and cathodic regions, especially at high concentrations. The decrease in current values with the addition of inhibitor in the anodic and cathodic regions indicates that adsorption occurs in both regions [52]. It was observed that both the polarization resistance and the percent inhibitory efficiency increased as the inhibitor concentration increased for the synthesized molecules.
Experimental data revealed that as the concentration of the compounds increased, the cathodic current densities decreased and the inhibitory activity increased. Although there appears to be a positive shift in corrosion potentials (Ecorr) compared to blank, this shift is not very pronounced for all compounds. The shift in the Ecorr value of the compounds at the highest concentration is in the range of 9–23 mV. According to Ferreira et al. [53], if the displacement at the corrosion potential is more than 85 mV with respect to the corrosion potential of the cavity, the inhibitor can be viewed as cathodic or anodic type. In this study, the maximum shift is 23 mV, and it is thought that the compounds reduce the dissolution of mild steel anodicly and also delay the cathodic hydrogen formation reaction, thus showing mixed-type inhibitory properties (see Figures S14–S17 in Supplementary information). Similar results have been reported in the literature [54, 55].
Electrochemical impedance spectroscopy (EIS) measurements
In this paper, the electrochemical behaviors at the interface of mild steel in acid solution with and without inhibitor are summarized. The inhibitory effects of compounds 1–4 on the corrosion of steel in 0.1 M HCl solution were studied by EIS method. Nyquist diagrams containing four different concentrations of compound 4 are given in Fig. 3 (see Figures S18–S21 in Supplementary information).

Nyquist diagrams obtained for mild steel in solutions containing compound 4 at different concentrations in 0.1 M HCl medium
Nyquist diagrams obtained from the reactions in the environment with and without inhibitors show a capacitive loop in the form of suppressed semicircles. This capacitive loop is related to the charge transfer process that controls the dissolution of mild steel in 0.1 M HCl solution and the protective film layer formed on the surface [56].
Bode plots of impedance spectra obtained with and without the addition of synthesized inhibitors are given in Fig. 4. Accordingly, it is clearly seen that an increase in the impedance of the double layer (Fig. 4a) and in the maximum phase angle (Fig. 4b) is observed as a result of the addition of inhibitor. Findings from Bode plots for all media with and without inhibitor additives show that it consists of a depressive-capacitive cycle corresponding to a time constant. The increase within the diameter of the impedance spectra, which becomes quite evident in the presence of the compounds and with the increase in concentration, is much higher than in the uninhibited solution. Experimental data suggest that the compounds were adsorbed on the metal surface and a protective film was formed [57, 58].

a Bode and b phase plots for mild steel in 0.1 M HCl medium with or without compound 4 at different concentrations
The Nyquist diagrams were analyzed using the Framework Data Acquisition Software and the corrosion of mild steel was defined with the equivalent circuit given in Fig. 5 [59, 60]:

Electrochemical equivalent circuit used for the metal/solution interface of impedance spectra
The equivalent circuit consists of the polarization resistor (Rp) and the fixed phase element (CPE) connected in parallel and the solution resistor (Rs). The impedance of the constant phase elements (CPE) used instead of the pure double-layer capacitor for modeling the corrosion system is expressed as follows:
where Q is the CPE constant and CPE exponent, j is the imaginary unit, w is the angular frequency, and n is the phase shift, which can be used as a measure of surface inhomogeneity [61].
Using experimental data to determine system parameters, Q values can be converted to Cdl capacitance and used for comparison. Thus, Cdl values were calculated from Q and n values using Eq. 4 [62]:
In Table 8, the inhibitor efficiency values (IE) calculated by Eq. 5 and the coating fraction \(\theta\) values calculated by Eq. 6 are given using the electrochemical parameters:
where \({R}_{\mathrm{p}}^{i}\) and \({R}_{\mathrm{p}}^{^\circ }\) values show the load transfer resistance values measured in the environment with and without inhibitor, respectively. In addition, Rp, Q, and Cdl values are given in Table 8.
Table 8 clearly shows that with increasing concentration of thiocarbohydrazones and carbohydrazones derivatives, the polarization resistance exhibits an increasing trend and the Rp values are larger than the uninhibited solution, reaching a maximum value of 235.5 Ω cm2 for compound 4 at 3.20 µM concentration. This effect shows that the load transfer process is the speed determining step in the dissolution reaction of steel in the environment with and without inhibitors. The increase in Rp values showed that charge transfer was delayed on the surface of mild steel in the presence of inhibitor [63]. In addition, slowing of the dissolution on the steel surface is evidenced by the presence of inhibitor at the mild steel/electrolyte interface, increasing the double layer thickness and thus decreasing the Cdl values [64]. In other words, the charge transfer process mainly controls the dissolution of mild steel in 0.1 M HCl solution [56, 65].
Computational results
The HOMO and LUMO energy, EHOMO, and ELUMO, are indicators of a molecule's tendency to donate and accept electrons, respectively. Chemical reactivity is directly related to the interaction between the HOMO–LUMO levels of the reacting compounds. However, the frontier molecular orbital energies and other electronic parameters derived from them are sensitively affected by possible conformations of the compounds. Due to the dynamic nature of reactions, including both environmental interactions and internal properties of the compounds, it is necessary to be very careful in interpreting a reaction mechanism through these parameters. Although the parameters have more than one variable, the fact that similar results can be obtained in repetitive experiments reveals that the reaction dynamics repeat themselves within certain limits.
The HOMO–LUMO energy gap is a useful parameter for both the DPPH reaction and as a measure of the reactivity of the inhibitor molecule to adsorption on the metal surface. As ∆E decreases, the reactivity of the molecule increases, resulting in an increase of %IE while decreasing the IC50 value of the molecule. In other words, the lower energy gap provides higher inhibition efficiency since the energy required to remove an electron from the last occupied orbital is lower. In addition, molecules with a low HOMO–LUMO energy range are soft, and these soft base inhibitors are more effective for metals. As a result of the calculations, it was seen that the data of compound 4 differed significantly from those of other compounds. The results given in Table 9 show that compound 4 has the lowest energy gap (3.744 eV), suggesting that compound 4 is more reactive than other compounds and may outperform other compounds as a corrosion inhibitor. Likewise, the hardness of compound 4 (1.872 eV) was noticeably lower than the other compounds, and its electronegativity (4.518 eV) and polarizability (296.67 a.u) were calculated to be higher.
Due to the conformational freedom of the compounds and the dynamic nature of the reactions, the variable intra/intermolecular interactions cause the electronic parameters to be insufficient to determine the qualitative and quantitative results of the reactions. Reactive N–H bonds are among the most important factors determining the reactions of compounds with DPPH. Weak N–H bonds mean that the DPPH∙ + RH → DPPH + R∙ reaction is easier to perform, that is, the compound with weak N–H bonds exhibits high antioxidant properties. Although electron density by itself cannot explain every detail, it is a fundamental concept for describing chemical reactivity and contributes to the explanation of nucleophilic and electrophilic attacks in terms of electrostatic interactions. Also, the change in electron density under the influence of a reagent is significant. In Fukui analysis, the distribution of HOMO electron density is considered the most important for electrophilic attack, and LUMOs, especially, are considered in nucleophilic substitution reactions. For condensed Fukui indices defined by the finite differences method, the nucleophilicity, electrophilicity, and radical attack susceptibility of an atom in a molecule are given as follows [66]:
The weak N–H bonds in the compounds determine the nucleophilicity and thus the probability of reaction with DPPH. Therefore, condensed Fukui indices were calculated to learn about which atoms in the compounds are more likely to lose an electron. Since N–H1, N–H2, N–H1*, and N–H2* atoms exhibit intramolecular interaction (see Fig. 6; all QTAIM data and NCI surfaces are given in Supplementary file, Figure S22), their charge density varies. Intramolecular interactions do not make it very convenient to use the electronic data of those regions to determine the reactive sites of compounds. Moreover, N–H3 and N–H3* structures that do not show intramolecular interactions are more likely not to be subjected to steric effects and may therefore play a more reactive role in nucleophilic reactions. The \({f}^{-}\) indices calculated for N–H3 structures are given in Fig. 7. The \({f}^{-}\) indices of the N3 atoms in the compounds were calculated larger than those of the N3* atoms, resulting in that the N–H3 structures were more reactive than the N–H3* structures. \({f}^{-}\) indices for N3 and N3* were calculated as 0.04091 and 0.03633 a.u for compound 4; 0.03221 and 0.03063 a.u for compound 3; 0.03220 and 0.03069 a.u for compound 2; 0.03069 and 0.02915 a.u for compound 1, respectively. Moreover, the probabilities of the N-H3 structures to react with DPPH can be correlated with the mean values of the \({f}^{-}\) indices. In fact, a result consistent with the experimental data in the form of \({f}_{(4)}^{-}>{f}_{(2)}^{-}>{f}_{(3)}^{-}>{f}_{(1)}^{-}\) was observed between the average values of \({f}^{-}\) indices of N–H3 structures. In addition, the \({f}^{-}\) indices of the H3 and H3* atoms bonding with N3 and N3* atoms revealed an inverse relationship (Fig. 7).

QTAIM data of the compound 1 [Green numbers indicate Laplacian bond order (LBO), Red numbers show the electron density, ⍴ (e/Bohr3), on BCP]. (Color figure online)

Fukui (\({f}^{-}\)) indices of N–H3 and N–H3* atoms of the compounds
QTAIM data revealed that the charge density in BCPs of N–H is related to the bond order and, therefore, to the DPPH reaction. The decrease in the charge density of the more reactive N–H3 and N–H3* bonds resulted in a lower LBO (the LBO values of N–H bonds are given in Table S3 in the Supplementary file). The LBO is a parameter directly proportional to bond strength, and accordingly, compounds with reactive N–H bonds with a lower charge density showed higher antioxidant properties. Also, the delocalization of electrons on N–H bonds affects the polarity of the bond. The bond polarity index is given as
where EIA and EIB representing the energy index (average energy per valence electron of the atom) of atoms A and B, respectively (see Ref. [46] for more information). \({\mathrm{EI}}^{\mathrm{ref}}\) are the energy indexes of the homonuclear molecular structures of the related atoms. The polarity of the covalent N–H bond is related to the difference between the electronegativity of the atoms, and electrons naturally migrate toward the more electronegative nitrogen atom. It was observed that the 3-fluorophenyl group reduced the polarity of the N–H bonds, in other words, reduced the electronegativity difference between the N and H atoms and consequently weakened the bond strength. The bond polarity indices of the N–H3 and N–H3* structures of the compounds are given in Fig. 8. It was found that compounds with N–H bonds with lower BPI values exhibited better antioxidant properties in the reactions of the compounds with DPPH compared to those with higher ones.

Bond polarity index of the N–H3 and N–H3* atoms of the compounds
Percentage values of electrons on reactive hydrogen atoms (%L) are also compatible/proportional with their bond polarities. The percentages for N–H3 and N–H3* were the smallest in compound 4, which had the highest antioxidant properties, and were calculated as 25.3906 and 25.4130, respectively, while compound 1 with the lowest antioxidant properties was calculated the highest as 25.6925 and 25.6607, respectively. These relative percentages of the N–H3 structures of compounds are a quantitative indicator of electron localization on reactive H atoms and are among the quantities that also determine the N–H bond length and bond strength. It was also calculated that the electrostatic interaction energy (EIE) between the total charge distributions of the active hydrogen atoms and the nitrogen nucleus took parallel values with the polarity of the N–H bonds, and the values were found to be directly related to the antioxidant properties of the compounds (see Table S1-S2 in Supplementary file).
Conclusions
In the paper, new symmetric N,N'-bis(thioamido)thiocarbohydrazone and carbohydrazone derivatives (1–4) were obtained from thio/carbohydrazide and various isothiocyanates with very high yields (80–97%). The structures of the compounds were elucidated by FTIR, 1H NMR, and 13C NMR spectroscopic techniques and elemental analysis. All compounds exhibited stronger antioxidant properties compared to Trolox that is a standard. Furthermore, when evaluated in terms of corrosion inhibitory properties in an acidic medium, all the compounds also showed a very strong corrosion rate slowing effect compared to the blank sample. Among the synthesized molecules, compound 4 had the highest activity for both antioxidant and corrosion inhibitor properties.
Although more than one variable determines the steps of a chemical reaction, experiments have shown that the reaction dynamics repeat themselves within certain limits. It is not always easy to reveal and analyze these dynamics. Although the electronic properties of a molecular structure cannot fully account for many variables such as conformations, intra and intermolecular interactions, acidity, temperature, etc., they contain important clues about the nature of reactions. For example, the electronic properties of N–H bonds are very useful in determining the reaction of the compounds with DPPH. Calculations revealed that as the bond polarization of reactive N–H structures decreased, the antioxidant property of the compound increased in accordance with the experiments. The relationship between the electron loss abilities of the reactive N–H atoms in the compounds, that is, nucleophilic properties, and their reactions with DPPH was revealed by calculating the condensed Fukui indices. It was observed that the N–H bonds of nitrogen atoms with a larger Fukui index were weaker and reacted better with DPPH. The QTAIM data revealed that the electron density on N–H correlated with LBO and thus with the DPPH reaction. Reactive N–H bonds with a lower electron density caused a lower LBO and consequently a decrease in bond strength and a higher antioxidant property of the compound. It was also calculated that the 3-fluorophenyl group decreased the polarity of the N–H bonds, thus weakening the bond strength. Furthermore, it was calculated that the electrostatic interaction energy between the total charge distributions of active hydrogen atoms and the nitrogen nucleus took values proportional to the polarity of the N–H bonds, and it was also revealed that the values were directly related to the antioxidant properties of the compounds. In this context, QTAIM, Laplacian bond order (LBO), and bond polarity index (BPI) analysis of active hydrogen bonds have been demonstrated to be an easy and useful method to determine the antioxidant activities of compounds.
References
A.A. Aly, A.A. Hassan, E.S.S. AbdEl-latief, J. Heterocycl. Chem. 55, 2196 (2018)
K. Gangarapu, S. Manda, A. Jallapally, S. Thota, S.S. Karki, J. Balzarini, E. De Clercq, H. Tokuda, Med. Chem. Res. 23, 1046 (2014)
T. Aboul-Fadl, F.A. Bin-Jubair, O. Aboul-Wafa, Eur. J. Med. Chem. 45, 4578 (2010)
F. Kurzer, M. Wilkinson, Chem. Rev. 70, 111 (1970)
G.S. Mrđan, G.G. Vastag, D.Đ Škorić, M.M. Radanović, T.Ž Verbić, M.K. Milčić, I.N. Stojiljković, O.S. Marković, B.M. Matijević, Struct. Chem. 32, 1231 (2021)
H. Muğlu, M.S. Çavuş, T. Bakır, H. Yakan, J. Mol. Struct. 1196, 819 (2019)
M.T. Muhammad, N. Ghouri, K.M. Khan, M.I. Choudhary, S. Perveen, Med. Chem. 14, 725 (2018)
S. Iqbal, M. Saleem, M.K. Azim, M. Taha, U. Salar, K.M. Khan, S. Perveen, M.I. Choudhary, Bioorganic Chem. 72, 89 (2017)
K. El-Mahdy, A. El-Kazak, M. Abdel-Megid, M. Seada, O. Farouk, Acta Chim. Slov. 63, 18 (2016)
Y. Kaya, A. Erçağ, A. Koca, J. Mol. Struct. 1102, 117 (2015)
K. Kumar, M. Kamboj, K. Jain, D. Singh, Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 128, 243 (2014)
V. Parshutin, N. Shol’toyan, S. Sidelnikova, G. Volodina, O. Bologa, V. Shofranskii, Surf. Eng. Appl. Electrochem. 45, 212 (2009)
M. Sathisha, U.N. Shetti, V. Revankar, K. Pai, Eur. J. Med. Chem. 43, 2338 (2008)
M. Sathisha, V.K. Revankar, K. Pai, Met. Based Drugs 2008, 362105 (2008)
A. Kolocouris, K. Dimas, C. Pannecouque, M. Witvrouw, G.B. Foscolos, G. Stamatiou, G. Fytas, G. Zoidis, N. Kolocouris, G. Andrei, Bioorganic Med. Chem. Lett. 12, 723 (2002)
H. Yakan, Res. Chem. Intermed. 46, 3979 (2020)
A.A. Al-Amiery, Y.K. Al-Majedy, H.H. Ibrahim, A.A. Al-Tamimi, Org. Med. Chem. Lett. 2, 1 (2012)
T.K. Bakır, J.B. Lawag, Res. Chem. Intermed. 46, 2541 (2020)
G. Kiran, M. Sarangapani, T. Gouthami, A.R. Narsimha Reddy, Toxicol. Environ. Chem. 95, 367 (2013)
M. Lashkari, M. Arshadi, Chem. Phys. 299, 131 (2004)
A. Stoyanova, G. Petkova, S. Peyerimhoff, Chem. Phys. 279, 1 (2002)
D.S. Chauhan, K. Ansari, A. Sorour, M. Quraishi, H. Lgaz, R. Salghi, Int. J. Biol. Macromol. 107, 1747 (2018)
N. Esmaeili, J. Neshati, I. Yavari, J. Ind. Eng. Chem. 22, 159 (2015)
K.E. Mouaden, D. Chauhan, M. Quraishi, L. Bazzi, Sustain. Chem. Pharm. 15, 100213 (2020)
M. Rbaa, P. Dohare, A. Berisha, O. Dagdag, L. Lakhrissi, M. Galai, B. Lakhrissi, M.E. Touhami, I. Warad, A. Zarrouk, J. Alloys Compd. 833, 154949 (2020)
S.B. Kedare, R. Singh, J. Food Sci. Technol. 48, 412 (2011)
H. Yakan, S. Cakmak, H. Kutuk, S. Yenigun, T. Ozen, Res. Chem. Intermed. 46, 2767 (2020)
W. Brand-Williams, M.-E. Cuvelier, C. Berset, LWT-Food Sci. Technol. 28, 25 (1995)
N. Yayli, G. Kılıç, G. Celik, N. Kahriman, Ş Kanbolat, A. Bozdeveci, ŞA. Karaoğlu, R. Aliyazıcıoğlu, H.E. Sellitepe, İS. Doğan, Turk. J. Chem. 45, 788 (2021)
N. Naik, H. Vijay Kumar, P.B. Vidyashree, J. Pharm. Res. 4, 2686 (2011)
E.N. Frankel, A.S. Meyer, J. Sci. Food Agric. 80, 1925 (2000)
M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. Petersson, D. Fox, in Wallingford CT (2010)
W. Kohn, L.J. Sham, Phys. Rev. 140, A1133 (1965)
P. Hohenberg, W. Kohn, Phys. Rev. 136, B864 (1964)
A.D. Becke, J. Chem. Phys. 98, 1372 (1993)
C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37, 785 (1988)
A.A. Fesenko, A.D. Shutalev, Tetrahedron 76, 131340 (2020)
M.M. Efremova, A.P. Molchanov, A.S. Novikov, G.L. Starova, A.A. Muryleva, A.V. Slita, V.V. Zarubaev, Tetrahedron 76, 131104 (2020)
R.F. Bader, Chem. Rev. 91, 893 (1991)
R. Bader, Atoms in molecule (Oxford University Press, Oxford, 1990)
R.F. Bader, Acc. Chem. Res. 18, 9 (1985)
M.S. Çavuş, H. Yakan, H. Muğlu, T. Bakır, J. Phys. Chem. Solids 140, 109362 (2020)
H. Muğlu, H. Yakan, A.G.A. Misbah, M.S. Çavuş, T.K. Bakır, Res. Chem. Intermed. 47, 4985 (2021)
H. Muğlu, M. Akın, M.S. Çavuş, H. Yakan, N. Şaki, E. Güzel, Comput. Biol. Chem. 107618 (2021)
T. Lu, F. Chen, J. Phys. Chem. A 117, 3100 (2013)
L.C. Allen, D.A. Egolf, E.T. Knight, C. Liang, J. Phys. Chem. 94, 5602 (1990)
D. Williams, I. Fleming, (McGraw-Hill, New York, 1996)
H. Yakan, T.K. Bakır, M.S. Çavuş, H. Muğlu, Res. Chem. Intermed. 46, 5417 (2020)
H. Muğlu, H. Yakan, T.K. Bakir, Turk. J. Chem. 44, 237 (2020)
H. Yakan, El-Cezeri J. Sci. Eng. 8, 155 (2021)
H. Lgaz, R. Salghi, S. Jodeh, B. Hammouti, J. Mol. Liq. 225, 271 (2017)
M. Özcan, F. Karadağ, I. Dehri, Colloids Surf. Physicochem. Eng. Aspects 316, 55 (2008)
E. Ferreira, C. Giacomelli, F. Giacomelli, A. Spinelli, Mater. Chem. Phys. 83, 129 (2004)
A. El-Sayed, Corros. Prev. Control 43, 27 (1996)
A. Yurt, S. Ulutas, H. Dal, Appl. Surf. Sci. 253, 919 (2006)
H. Lgaz, K.S. Bhat, R. Salghi, S. Jodeh, M. Algarra, B. Hammouti, I.H. Ali, A. Essamri, J. Mol. Liq. 238, 71 (2017)
H. Lgaz, R. Salghi, I.H. Ali, Int. J. Electrochem. Sci 13, 250 (2018)
M. Messali, M. Larouj, H. Lgaz, N. Rezki, F. Al-Blewi, M. Aouad, A. Chaouiki, R. Salghi, I.-M. Chung, J. Mol. Struct. 1168, 39 (2018)
F. Bentiss, M. Lagrenee, M. Traisnel, J. Hornez, Corros. Sci. 41, 789 (1999)
N.K. Gupta, C. Verma, R. Salghi, H. Lgaz, A. Mukherjee, M. Quraishi, New J. Chem. 41, 13114 (2017)
I. Ahamad, R. Prasad, M. Quraishi, Mater. Chem. Phys. 124, 1155 (2010)
A. Chaouiki, H. Lgaz, I.-M. Chung, I. Ali, S.L. Gaonkar, K. Bhat, R. Salghi, H. Oudda, M. Khan, J. Mol. Liq. 266, 603 (2018)
S.K. Saha, A. Dutta, P. Ghosh, D. Sukul, P. Banerjee, Phys. Chem. Chem. Phys. 18, 17898 (2016)
A.O. Yüce, B.D. Mert, G. Kardaş, B. Yazıcı, Corros. Sci. 83, 310 (2014)
E. Khamis, M. Ameer, N. AlAndis, G. Al-Senani, Corrosion 56, 127 (2000)
W. Yang, W.J. Mortier, J. Am. Chem. Soc. 108, 5708 (1986)
Acknowledgements
This study was not supported by any organization. The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Contributions
M. Serdar Çavuş: Theoretical Calculations, Writing–Review. Hasan Yakan: Spectroscopic Characterization, Visualization, Writing–Review. Cihan Özorak: Antioxidant Assay, Corrosion Inhibitors Studies, Writing–Review. Halit Muğlu: Synthesis, Characterization, Writing–Review. Temel Kan Bakır: Antioxidant Assay, Corrosion Inhibitors Studies, Writing–Review.
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
All spectra (IR, 1H, and 13C NMR) of the compounds, potentiodynamic polarization curves, Nyquist diagrams, and QTAIM data are presented in the supporting information.
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Çavuş, M.S., Yakan, H., Özorak, C. et al. New N,N'-bis(thioamido)thiocarbohydrazones and carbohydrazones: synthesis, structure characterization, antioxidant activity, corrosion inhibitors and DFT studies. Res Chem Intermed 48, 1593–1613 (2022). https://doi.org/10.1007/s11164-022-04659-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-022-04659-z