
Abstract
In this work, a series of six organic dyes-sensitized solar cells (DSSCs) combining various π-bridges with a fixed donor (triphenylamine) and a fixed electron acceptor (cyanoacrylic acid), namely D1-6, were studied. The geometrical structure, electronic and optical properties of these dyes have been investigated with the density functional theory and TD-BHandHLYP hybrid functional (time-dependent Becke-Half-and-Half-Lee–Yang–Parr’s) methods. The effects of π-bridging of the dyes have shown that the rings with a sulfur atom reduce the energy gaps and provide a redshift of the absorption spectra. Similarly, we focus on the description of the ground and excited state properties. On the other hand, the pyrrole group improves the open-circuit voltage (VOC) and the light-harvesting efficiency parameters leading to greater power conversion efficiency. Furthermore, the results revealed lowest total reorganization energy λ (λ+ and λ−) for the dye D3 with pyrrole linkage, which reflects its most favorable charge-transport properties, implying a lower charge recombination rate, faster charge injection and dye regeneration processes. Therefore, this study would provide a new path to design novel conjugated organic molecules as dyes for high-performance DSSCs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A new generation of photovoltaic cells, namely dyes-sensitized solar cells (DSSCs), also known as Grätzel cells, has attracted significant attention as an alternative to silicon solar cells due to its low-cost, flexibility, environmentally friendly and highly efficient conversion of sunlight into electricity [1,2,3]. To improve the performance of DSSCs, extensive researches have been conducted on semiconductor electrodes [4], electrolytes [5], counter-electrodes [6] and photosensitive dyes [7]. A greater effort was focused on the development of new photosensitizers for the production of high-performance organic photovoltaic cells [8]. Compared to the rare and expensive ruthenium complex dyes, the organic dyes have the advantage of being ecological and having various molecular structures with their high absorption at low cost [7, 9, 10].
The most studied dyes adopt generally a structure type of Donor-π spacer-Acceptor (D-π-A), allowing intramolecular charge transfer by photoinduction, and have high molar extinction coefficients and a building block configuration, which can absorb a wide range of photons and efficiently separate charge across the dye [11, 12]. Thus, the properties of dyes with the D-π-A structure can be adjusted by the modification of functional group (electron-donating group, π-conjugated linker and acceptor group) which make it possible to design and synthesize high-efficiency sensitizers for DSSC application [11, 12].
In the current paper, the theoretical study of structural and optoelectronic properties of D-π-A sensitizers is based on triphenylamine group, and the choice of this donor is due to its electron-donating property, high hole-transporting capability and its three-dimensional geometry which can lead to amorphous materials with interesting optoelectronic and charge-transport properties as provided by many research groups [7, 9, 13, 14]. On the other hand, different electron acceptors were designed and synthesized for dye-sensitized solar cells. However, cyanoacrylic acids have been chosen for their promising characteristics as a stable acceptor group, and this is explained by the existence of the cyanide group, which has a strong electron-withdrawing capability and strong binding ability to the TiO2 surface [13]. Thus, the choice of a suitable π-spacer is very crucial for the design of highly efficient D-π-A organic dyes as it plays a significant role in adapting HOMO and LUMO energy levels. For that, different π-conjugated spacers have been investigated: thiophene, furan, pyrrole, thiazole, oxazole and imidazole.
In this work, we aim to perform the theoretical investigation how different π-spacers groups affect the structural, electronic and optical features of organic dyes by using density functional theory (DFT) and TD-BHandHLYP hybrid functional (time-dependent Becke-Half-and-Half-Lee–Yang–Parr’s). Firstly, we designed the six new dyes by introducing different electron π-spacers. After, we estimated the parameters affecting dye performance, such as energy gap (Egap), light-harvesting efficiency (LHE), the free energy of injection (ΔGinject) and open-circuit photovoltage (VOC). The generation and transport of free holes and electrons are also studied by evaluating ionization potentials (IPs), electron affinities (EAs) and reorganization energies λ (λ+ and λ−).
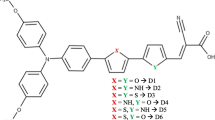
Finally, we compared the maximum absorption for the compound D1 measured experimentally with that obtained theoretically with two methods, in order to valid the appropriate functional for reproducing the optical properties of the studied compounds based on triphenylamine (Di, i = 1–6) (see Fig. 1).

Molecular structure of studied dyes
The theoretical ground-state geometry and electronic structure of the studied molecules were investigating to elucidate the relationship between molecular structure and optoelectronic properties. Furthermore, this work could facilitate the future experimental studies to design and fast screen new efficient organic dyes.
Computational methods
Density functional theory (DFT) is one of the most used methods in quantum chemistry, because it gives a result similar to the experimental data and can be used to study performing organic dyes for application in DSSCs [15]. Thereby, the geometric and electronic properties of all systems (neutral and doped) were studied by DFT method using the B3LYP [16] correlation exchange functional (Becke's three-parameter hybrid functional, and Lee–Yang–Parr’s correlation functional) combined with the 6-31G (d, p) basis set [17] for all atoms which reproduced the experimental data successfully [13].
With the aim of checking the reliability, the absorption spectra of the studied compounds have been performed using two different functional, including B3LYP in time-dependent density functional theory (TDDFT) and BHandHLYP hybrid functional (Becke-Half-and-Half-LYP) which include 50% fraction of Hartree–Fock (HF) exchange and 50% of Becke 88 exchange [18]. The purpose of using this hybrid functional is to mix the exchange energies calculated in an exact manner (Hartree–Fock) with those obtained by DFT methods in order to improve performance and to obtain results close to those obtained experimentally. The inclusion of the solvent effect in theoretical calculations is important when seeking to reproduce or predict the experimental spectra with a reasonable accuracy. Hence, the use of the tetrahydrofuran solvent (THF) was used in experimental studies [13], by using the "Conductor-Polarizable Continuum" model (C-PCM) [19].
Besides this, the choice of the exchange–correlation functional is very important to describe precisely the charge transfer properties. For that, many hybrid functionals have been used [20, 21]. In further, the B3LYP functional was successfully used to calculate the charge-transport parameters in previous studies related to DSSCs [22,23,24]. Therefore, the charge transfer properties of studied compounds were determined with the same functional.
All calculations were performed using the Gaussian 09 program package [25], and the simulation of spectra and density of orbital are doing by GaussView 5.0.
Results and discussion
Structural properties
The geometries of the studied dyes have been optimized using B3LYP/6-31G(d, p), and the optimized structures are shown in Fig. 2. Different π-spacers have been employed in order to investigate the structure–property of the performance on the variation of bridge.

Optimized ground-state geometries with B3LYP/6-31G (d, p) of studied dyes
From the optimized structures, we determined the bond lengths and dihedral angles of the six dyes (Di, i = 1–6). We define L1 as the link length between the donor and the spacer π, and L2 as the link length between the acceptor and the π-spacer (Fig. 1). Also, the dihedral angle between Donor and π-spacer (Φ) (Fig. 1) has been introduced in order to study the molecular structure effect. The values of the selected structural parameters (L1, L2 and Φ) calculated in neutral, cationic and anionic states are listed in Table 1.
In neutral state, the calculated L2 bond length presents low values compared to L1 bond length. This result implies a strong conjugation between the π-bridge to the acceptor moiety than between the phenyl ring of donor TPA and the π-bridge [18]. When we compare the bond lengths (L1 and L2) for studied dyes, we note that D2 and D5 gave relatively the low values. This can be due to steric reasons [18].
In addition, it is essential to study how the doped dye molecule becomes responsible for charge transport. Noting that to obtain the optimized doped structure, we started from the optimized structure of the neutral form. We can conclude from Table 1 that the positive doping effect favors the quinoid forms of the studied molecules which was confirmed by many research works [26,27,28,29,30].
On the other hand, the dihedral angles Φ in neutral state for Di = 1–6 rank in the order of D1 > D4 > D3 > D6 > D2 > D5. The large dihedral angle between the donor and π-bridge may hamper the conjugation action of the whole molecule and lead to low intermolecular charge transfer. The dihedral angles between furan, oxazole and TPA donor moiety in D2 and D5 were 1.66° and 1.26°, respectively, which indicates a quasi-planar configuration of D2 and D5. Thus, the linked furan and oxazole units are favorable to transfer the electron from the electron donor to the electron acceptor leading to the largest ICT absorption wavelength and indicating a strong conjugation effect. However, the trigonal geometry of TPA donor and linked furan and oxazole units can induce dye aggregations because of π–π interactions among dye molecules [31]. This suggests that the oxygen atom of the furan and oxazole rings induces a coplanar structure by hydrogen bonding interaction between oxygen and hydrogen of the ring.
Thus, the introduction of nitrogen in thiophene, furan and pyrrole connectors, affording a thiazole, oxazole and imidazole units, clearly limits the steric hindrance between the neighboring heterocycles and the donor unit (TPA) resulting in a slight reduction in the dihedral angles [32]. In addition, the dihedral angles calculated for doped structures are decreased compared to their corresponding in neutral states. This indicates that the doping of studied molecules improves their planarity and forces the attractive interactions between atoms [33].
Electronic properties
In order to check if the method used to calculate the electronic properties of the studied dyes is appropriate, it is necessary to carry out a comparison between the energy difference obtained by the theoretical approach and the experimental method. It was necessary to make the comparison between the energy gap obtained theoretically and experimentally [13]. The energies of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) were deduced from the optimized ground-state geometries, for the studied dyes obtained at a B3LYP/6-31G(d, p) level. The energy gap (Egap) has been evaluated as the difference between LUMO and HOMO levels (Egap = ELUMO – EHOMO). Their values are listed in Table 2.
As shown in Table 2, the value of the D1 (2.46 eV) obtained at B3LYP/6-31G (d, p) level of the band gap is close to that obtained experimentally for same dye [17]. The calculated band gap of the studied dyes increases in the following order D4 < D5 < D1 < D6 < D2 < D3. The weaker Egap of D4 and D5 indicates a significant effect of thiazole and oxazole units as π-spacers inserted between the donor moiety and acceptor group, which can lead to band gap narrowing in the studied dyes. On the other hand, concerning the comparison between D1/D4, D2/D5 and D3/D6, it suggests that the replacement of carbon atoms in D1, D2 and D3 by nitrogen atoms in D4, D5 and D6 leads to increase in the energy gap.
The LUMO energy values of all studied dyes are less negative than the conduction band of the semiconductor ECB(TiO2) which means that excited electrons of the studied compounds are efficiently injected into the TiO2 conduction band in the following order: D3 > D2 > D6 > D1 > D5 > D4. However, the introduction of thiophene, thiazole and oxazole bridged groups in D1, D4 and D5, respectively, stabilizes their LUMO levels compared to the LUMO of other dyes.
On the other hand, the HOMO energies of all studied dyes show lower energy levels than the E(I−/I3−) redox potential. Therefore, the oxidized dyes could be regenerated electron from the reduced species in the electrolyte for an efficient charge separation. The energy barrier between the E(I−/I3−) redox potential and the EHOMO of the dyes was increased in the following order: D3 < D2 < D6 < D5 < D1 < D4. This regeneration was affected by the nature of the π-bridge group, which was decreased in D3 and D2 dyes having pyrrole and furan bridge groups, while it was increased in D1 and D4 dyes having thiophene and thiazole π-spacers. Thus, when we compare D1/D4, D2/D5 and D3/D6, we observe a slight increase and decrease in LUMO and HOMO energy values, respectively, when adding a C-N bond in bridge groups of D1, D2 and D3 dyes. This result indicates that the dyes with more C-N bonds in bridged group provides a high driving force for regeneration of the dye by the electrolyte (I−/I3−) and more sufficient driving forces for electron injection into the conducting bond of TiO2.
Intramolecular charge transfer (ICT) is strongly related to the electron distribution of the frontier molecular orbitals (FMOs). The FMOs for the six compounds are shown in Figs. 3, 4.


Frontier molecular diagram of the studied dyes
The electronic distribution of the HOMO of D1 and D4 is mainly distributed over the donor and π-spacer, while that of D2, D3, D5 and D6 is almost delocalized over the whole molecule. On the other hand, the LUMO orbitals are distributed mainly on the electron acceptor units and partly on its adjacent π-spacer ring. These electron density distributions are very interesting for effective charge separation and electron injection, which indicate that the transfer of electrons goes from the donor to the acceptor via π-spacer. The acceptor group of all dyes has considerable contribution to the LUMOs, which could lead to a strong electronic coupling with TiO2 surface and thus improve the electron injection efficiency.
Moreover, the HOMO of all dyes presents a bonding character, which facilitates the mobility of electrons of the donor unit to the LUMO of the acceptor unit. Meanwhile, the LUMO of all the compounds presents an anti-bonding character. Absorption is achieved in this system by the transition of a π-electron from the HOMO to LUMO level, and this transition can be classified as a π–π* ICT. Therefore, we can deduce that electrons on the donor/π-spacer units are responsible for the absorption in the ultraviolet, while the acceptor/π-spacer units are mainly responsible for the absorption phenomenon in the visible and near-UV.
NBO analysis
To understand the origins of the photoexcitation mechanism, the natural bond orbital (NBO) analysis was carried out based on the optimized structure of the ground state using B3LYP/6-31G(d,p) level, and the detailed results are tabulated in Table 3.
Based on the principle of D-π-A architecture, the positive charges of the donor group and π- spacer of all dyes indicated them being an effective electron-pushing unit. On the other hand, the negative charges in the acceptor group show that they are effective electron-withdrawing unit. Therefore, during the photoexcitation, the electrons could be successively transferred from the donor group to the acceptor group, passing through the π-spacer, and finally injected into the semiconductor TiO2 conduction band. The NBO charges of all sensitizers indicate that pyrrole as a π-spacer could greatly favor charge transfer.
Ionization potentials, electron affinities, reorganization energy
Ionization potentials (IPs), electronic affinities (EAs), electron extraction potentials (EEPs) and hole extraction potentials (HEPs) are essential parameters for assessing the ability of the molecule to give and accept electrons to generate excitons. They can provide a qualitative indication of the charge injection and transport ability. In addition, for p-type materials, the improvement of hole and electron injection/transport is based on the lower IP, which signifies a more favored release of electrons and creation of the hole, and the small EA value which can favor more efficient and faster transfer of electrons from the excited dye to the semiconductor [36, 37].
The adiabatic ionization potential (IPa), the vertical ionization potential (IPv), the adiabatic (EAa) and the vertical (EAv) electronic affinity have been calculated as shown below [36]:
where E(M), E+(M+) and E−(M−) represent, respectively, the energies of the neutral, cation and anion species in their ground-state geometries, while E+(M) and E−(M) represent, respectively, the energies of the cation and anion species with the geometries of the neutral species. The electron extraction potential (EEP) and hole extraction potential (HEP) have been calculated as follows [36]:
where E(M+) and E(M−) represent the energies of the neutral species with the geometries of the cation and anion species, respectively.
The IPs, EAs, EEPs and HEPs are calculated at B3LYP/6-31G(d,p) level of theory for each studied molecule and listed in Table 4.
As shown in Table 4, for the studied dyes, the IP values increase in the following order: D3 < D2 < D6 < D1 < D5 < D4, showing that D3 attained the lowest IP value, followed by the D2 dye, implying that the bridge group of D3 promotes a faster rate of the release of electrons and formation of holes [38]. Nevertheless, the values of EA decrease in the following order: D3 < D2 < D6 < D1 < D5 < D4, suggesting that D3 dye undertakes a faster rate of injecting an electron directly to the TiO2 semiconductor [38]. On the other hand, the hole and electron extraction potentials HEP and EEP of the studied compounds exhibit almost, respectively, the same trend as described above for IP and EA.
In addition, the Marcus theory (Eq) is used to shed light on the charge transfer rate [26].
In Eq. (7), kinject is the electron injection rate constant (in S−1) from the photoexcited dye to the conduction band of the semiconductor TiO2, kBT is the thermal energy of Boltzmann, (h) is the Planck’s constant, ΔGinject is the free energy of injection, (λ) is the system reorganization energy, and the coupling constant between the reagent and the product potential curves are given by |VRP|. It is clear that the reorganization energy (λ) is among the key parameters that have a dominant impact on the charge transfer rate. Indeed, the greater mobility of carriers for any organic compound corresponds to its small reorganization energy [39]; it is calculated using the following formulas [40]:
where λ+ and λ−are the reorganization energies of holes and electrons, respectively.
According to the values of the hole and electron reorganization energies listed in Table 4, the values of λ+are less than those of λ−for all dyes; it indicates that these organic molecules are more appropriate for hole-transport materials. Furthermore, the small total reorganization energy value mitigates the parasitic charge recombination process and shows a more effective hole-charge separation [40]. For the studied dyes, D3 has the lowest total reorganization energy, which reflects its most favorable charge-transport properties and the least degree of charge recombination. These results indicate that the introduction of pyrrole as π-spacer in D3 is appropriate for generating and transporting free holes and electrons as the p-type materials in organic solar cell devices. This can be explained by the Coulomb interaction between nitrogen and hydrogen, which could greatly favor charge transfer [41].
Electron injection investigations and photovoltaic properties
In order to better study the electrochemical properties of the dyes in their excited state, we introduce electron injection driving force (ΔGinject) which could also influence the electron injection rate (Eq. 7). It is related to the driving force of the electron injection from the photoinduced excited states of organic dyes to TiO2 cluster surface. The calculation detail for the determination of ΔGinject is expressed in Eq. (10), according to Koopman's theorem [42]:
where \(E_{{\text{dye*}}}^{{{\text{OX}}}}\) is the oxidation potential of the dye in the excited state and \(E_{{{\text{TiO}}_{{2}} }}^{{{\text{CB}}}}\) is the reduction potential of the semiconductor conduction band (TiO2). In this work, we consider \(E_{{{\text{TiO}}_{{2}} }}^{{{\text{CB}}}}\) = − 4.0 eV for TiO2 [43], and \(E_{{\text{dye*}}}^{{{\text{OX}}}}\) is calculated on the basis of Eq. (11) [43].
where \(E_{{{\text{dye}}}}^{{{\text{OX}}}}\) presents the ground-state oxidation potential of the dye and E0 0 is the vertical electron transition energy corresponding to the wavelength of the maximum absorption (λmax). Based on Koopman’s theorem, ground-state oxidation potential energy is related to ionization potential energy. Thus, \(E_{{{\text{dye}}}}^{{{\text{OX}}}}\) can be estimated as negative of EHOMO.
Furthermore, the dye must be regenerated by electron transfer from the redox electrolyte, which is then reduced to the counter electrode. The driving force of regeneration ΔGreg can be expressed as follows [44]:
where \(E_{{I^{ - } /I_{3}^{ - } }}^{{{\text{redox}}}}\) = 4.8 eV is the redox potential of the iodide/triiodide pair. The obtained \(E_{{\text{dye*}}}^{{{\text{OX}}}}\), \(E_{{{\text{dye}}}}^{{{\text{OX}}}}\), E0 0, ΔGinject and ΔGreg values are listed in Table 5.
The results listed in Table 5 show that the energy differences of the driving forces ΔGreg and ΔGinject are negative, which indicate that the use of dyes in DSSCs is possible. It should be noted, however, that D3 dye has a higher regenerative motive power (ΔGreg) energy difference than the other dyes. The regeneration of D3 therefore seems more favorable. In addition, the dyes with the nitrogen atom (D3 and D6) have an electron injection energy force difference (ΔGinject) greater than that of the other dyes. The injection of the electron could therefore be favored in this case. On the other hand, previous research has demonstrated that the driving force of 0.20 eV was necessary for efficient dye regeneration [45].
The power conversion efficiency (PCE) of solar cells is generally the most commonly used parameter to compare the performance of various solar cells; it can be calculated from Eq. (13).
where JSC is the short-circuit current density, VOC is the maximum open-circuit voltage, FF is the fill factor, and Pinc is the incident photon to current efficiency. VOC is theoretically calculated as the energy difference between the LUMO level of dye and the conduction band of semiconductor (TiO2) [46].
The obtained VOC values of the studied dyes calculated according to Eq. (14) range from 1.295 to 1.701 eV. These values are sufficient for a possible efficient electron injection. However, we can note from Eq. (14) that the higher the ELUMO value, the larger the VOC value, which explains why the highest value of VOC is those of D3.
Absorption spectra
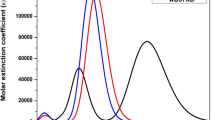
The choice of the exchange–correlation functional is very important to describe precisely the excitation energy and the visible spectra of study dyes. Then, the hybrid functional (BHandHLYP) and the corrected functional (B3LYP) were tested to study the functional influence on the maximum absorption wavelengths of D1 calculated in a tetrahydrofuran (THF) solution which it was used in the experimental study. So, the electron absorption spectrum of the dye in a THF solution was simulated to study the solvation effect. The absorption spectra in THF are shown in Fig. 5. Table 6 shows the wavelength of the maximum absorption of D1 calculated using BHandHLYP/6-31G + (d) and B3LYP/ 6-31G + (d) methods against the available experiment value.

Absorption spectra of all the studied dyes calculated at the TD-BHandHLYP/6–31G + (d,p) level in THF solvent
The value of the absorption wavelength calculated by the hybrid functional BHandHLYP shows that it is in a good agreement with the experimental value and better than these determined by B3LYP functional. Therefore, after choosing the adequate method, we proceed to calculate the maximum wavelength of absorption (λmax), corresponding oscillator strength (ƒ), the light-harvesting efficiencies (LHE) and the main configuration of each excitation. The obtained results are summarized in Table 7.
Accepted manuscript
The calculated λmax in the gas phase is in the following order: D4 > D5 > D1 > D2 > D6 > D3; upon incorporation of the sulfur atom into the thiophene and thiazole π-spacers, the maximum absorption of D1 and D4 was shifted to red, relative to D2 and D5 having bridges with oxygen atom and to D3 and D6 having bridges with nitrogen atom, and this is due to the more effective π-electron delocalization of the spacers with sulfur atom than oxygen and nitrogen atoms. In addition, D3 and D6 showed hypsochromatic shifted absorption peaks, while D4 and D5 showed bathochromic displacement. These maximum absorptions correspond to the electron transition from HOMO to LUMO. The λmax is a function of the electron availability [47]. This indicates an intramolecular charge transfer (ICT) from the donor (TPA) through the π-spacer to the cyanoacrylic acid acceptor, which would be beneficial for electron injection into the conduction band of TiO2.
The solvent effect was calculated by applying the conductive-type polarizable continuum model (C-PCM) in a tetrahydrofuran (THF) solvent. A notable redshift is observed for compounds in a solution of tetrahydrofuran (THF) compared to that in the gas phase. This may be ascribed to the H-aggregation of the dyes or the deprotonation of cyanoacrylic acid upon adsorption onto the TiO2 surface [48].
Besides VOC, λ and ΔGinj, another factor related to efficiency of DSSC is the performance of the dyes responsible of the incident light. Based on the light-harvesting efficiency (LHE) of the dyes, the value has to be as high as possible to maximize the photocurrent response. The LHE can be expressed as follows [43]:
where ƒ is the oscillator force of the dye.
As shown in Table 6, the LHE values for gas phase dyes are within a narrow range (0.869–0.928). This means that all sensitizers give a photocurrent quasi-similar. However, the dye with thiophene spacer is more efficient in absorbing photons and injecting photoexcited electrons to the conduction band of TiO2. We note that the transition to the solvated phase allows a remarkable increase at the level of the LHE for all the dyes.
Because of the above data, we could draw a conclusion that the large LHE, ΔGinject, VOC as well as small total could have a high efficiency. Thus, the performance of DSSCs sensitized by dye 3 might be superior to the other dyes, due to its favorable performances of the above factors based on our computed results.
Conclusions
The DFT and TD-BHandHLYP methods were employed to investigate the structural, optoelectronic, photophysical and charge transfer properties of six organic triphenylamine-based dyes with difference π-bridge groups. The results indicate that the experimental maximum absorption wavelength is well reproduced by BHandHLYP hybrid functional for the studied D1 dye.
The results obtained show that the absorption maxima of the studied systems vary between 403.55 and 445.14 nm, which is very suitable for an efficient harvest of light. For the molecules bridged by thiazole and oxazole (D4 and D5), we noted a significant redshift values of absorption maxima and bathochromic ICT absorption peak, which can explicated by the low gap energy observed for D4 and D5 compared to the other compounds. The absorption spectra of the six dyes in tetrahydrofuran are all redshifted significantly, as compared to that in vacuo which indicate that the solvent had a great effect on optical properties. On the other hand, the pyrrole linkage improves VOC leading to higher power conversion efficiency (PCE). Moreover, the results revealed better charge-transport properties for the dye D3 compared to the other dyes, such as a: lower rate of charge recombination, faster charge injection and faster dye regeneration processes.
As a result, dye D3 delivers the best performance among the dyes studied, exhibiting electron injection driving force of 2.238, with LHE of 0.928 and VOC of 1.869. However, although D3 has the largest VOC over the other two dyes, it still exhibits the lowest power conversion efficiency.
The overall results reveal that the molecules Di, i = 1–6 can therefore be considered as promising materials in organic photovoltaic cell applications, and thus, the change of the π-conjugation spacer in organic dyes is an effective way to improve the photovoltaic performance.
Furthermore, the theoretical results obtained are in agreement with the experiment results, which indicate that the computational methods used would be efficient to predict the optoelectronic and photovoltaic properties of other sensitizers for photovoltaic applications.
References
B. O’Regan, M. Grätzel, Nature 353, 737 (1991)
J.-X. Cheng, Z.-S. Huang, L. Wang, D. Cao, Dyes Pigm. 131, 134 (2016)
Y. Wu, W. Zhu, Chem. Soc. Rev. 42, 2039 (2013)
X. Liu, Y. Luo, H. Li, Y. Fan, Z. Yu, Y. Lin, L. Chen, Q. Meng, Chem. Commun 0, 2847 (2007)
J. Wu, Z. Lan, J. Lin, M. Huang, Y. Huang, L. Fan, G. Luo, Chem. Rev. 115, 2136 (2015)
J. Wu, Z. Lan, J. Lin, M. Huang, Y. Huang, L. Fan, G. Luo, Y. Lin, Y. Xie, Y. Wei, Chem. Soc. Rev. 46, 5975 (2017)
A. Mahmood, Sol. Energy 123, 127 (2016)
A. Carella, F. Borbone, R. Centore, Front. Chem. 6, 481 (2018)
M. Liang, W. Xu, F. Cai, P. Chen, B. Peng, J. Chen, Z. Li, J. Phys. Chem. C 111, 4465 (2007)
R. Li, X. Lv, D. Shi, D. Zhou, Y. Cheng, G. Zhang, P. Wang, J. Phys. Chem. C 113, 7469 (2009)
N.P. Liyanage, A. Yella, M. Nazeeruddin, M. Grätzel, J.H. Delcamp, A.C.S. Appl, Mater. Interfaces 8, 5376 (2016)
S. Ennehary, H. Toufik, S. M. Bouzzine, and F. Lamchouri, J. Comput. Electron. (2020).
Y.K. Eom, J.Y. Hong, J. Kim, H.K. Kim, Dyes Pigm. 136, 496 (2017)
A. Irfan, A.G. Al-Sehemi, S. Muhammad, M.S. Al-Assiri, A.R. Chaudhry, A. Kalam, M. Shkir, J. King Saud Univ. Sci. 27, 361 (2015)
L.L. Estrella, S.H. Lee, D.H. Kim, Dyes Pigm. 165, 1 (2019)
A.D. Becke, J. Chem. Phys. 98, 5648 (1993)
R. Krishnan, J.S. Binkley, R. Seeger, J.A. Pople, J. Chem. Phys. 72, 650 (1980)
Z.M.E. Fahim, S.M. Bouzzine, A.A. Youssef, M. Bouachrine, M. Hamidi, Comput. Theoret. Chem. 1125, 39 (2018)
V. Barone, M. Cossi, J. Phys. Chem. A 102, 1995 (1998)
T. Yanai, D.P. Tew, N.C. Handy, Chem. Phys. Lett. 393, 51 (2004)
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996)
A. Üngördü, Chem. Phys. Lett. 733, 136696 (2019)
M.P. Balanay, D.H. Kim, J. Mol. Struct. (Thoechem) 910, 20 (2009)
W.-L. Ding, D.-M. Wang, Z.-Y. Geng, X.-L. Zhao, W.-B. Xu, Dyes Pigm. 98, 125 (2013)
Gaussian 09, R.A.: 1, mj frisch, gw trucks, hb schlegel, ge scuseria, ma robb, jr cheeseman, g. Scalmani, v. Barone, b. Mennucci, ga petersson et al., gaussian. Inc Wallingford CT. 121, 150 (2009).
A. Aicha Youssef, S. Mohamed Bouzzine, Z. Mohyi Eddine Fahim, İ. Sıdır, M. Hamidi, M. Bouachrine, Phys. B Condensed Matter 560, 111 (2019)
H. Toufik, S. M. Bouzzine, O. Ninis, M. Aberkane, F. Lamchouri, M. Hamidi, and M. Bouachrine, Жypнaл Фiзичниx Дocлiджeнь 1702 (2012).
H. Toufik, S.M. Bouzzine, O. Ninis, F. Lamchouri, M. Aberkane, M. Hamidi, M. Bouachrine, Res. Chem. Intermed. 38, 1375 (2012)
M. Lazrak, H. Toufik, S.M. Bouzzine, H. Bih, F. Lamchouri, IOP Conf. Ser. Earth Environ. Sci. 161, 012021 (2018)
M. Lazrak, H. Toufik, S. M. Bouzzine, H. Bih, and F. Lamchouri, 10 (n.d.).
J.H. Bae, S.J. Lim, J. Choi, S.B. Yuk, J.W. Namgoong, J.H. Ko, W. Lee, J.P. Kim, Dyes Pigm. 162, 905 (2019)
C. Figueira, P. Lopes, C. Gomes, L. F. Veiros, P. Gomes, Exploring the influence of steric hindrance and electronic nature of substituents in the supramolecular arrangements of 5-(Substituted Phenyl)-2-Formylpyrroles (2015).
Thin Film Physics Division, Department of Physics, Chemistry, and Biology (IFM), Linköping University, Sweden and S. Khromov, Doping Effects on the Structural and Optical Properties of GaN (Linköping University Electronic Press, Linköping, 2013).
J.B. Asbury, Y.-Q. Wang, E. Hao, H.N. Ghosh, T. Lian, Res Chem Intermediat 27, 393 (2001)
A. Hagfeldt, M. Graetzel, Chem. Rev. 95, 49 (1995)
L.L. Estrella, M.P. Balanay, D.H. Kim, J. Phys. Chem. A 120, 5917 (2016)
J.C. Delgado, Y. Ishikawa, R.G. Selsby, Photochem. Photobiol. 85, 1286 (2009)
W.R. Duncan, O.V. Prezhdo, Annu. Rev. Phys. Chem. 58, 143 (2007)
B.C. Lin, C.P. Cheng, Z.P.M. Lao, J. Phys. Chem. A 107, 5241 (2003)
G.R. Hutchison, M.A. Ratner, T.J. Marks, J. Am. Chem. Soc. 127, 2339 (2005)
H. Li, L. Yang, R. Tang, Y. Hou, Y. Yang, H. Wang, H. Han, J. Qin, Q. Li, Z. Li, Dyes Pigm. 99, 863 (2013)
R. Manne, T. Åberg, Chem. Phys. Lett. 7, 282 (1970)
J. Preat, C. Michaux, D. Jacquemin, E.A. Perpète, J. Phys. Chem. C 113, 16821 (2009)
Z.M.E. Fahim, S.M. Bouzzine, Y. Ait Aicha, M. Bouachrine, M. Hamidi, Res. Chem. Intermed. 44, 2009 (2018)
K. Hara, T. Sato, R. Katoh, A. Furube, Y. Ohga, A. Shinpo, S. Suga, K. Sayama, H. Sugihara, H. Arakawa, J. Phys. Chem. B 107, 597 (2003)
W. Sang-aroon, S. Laopha, P. Chaiamornnugool, S. Tontapha, S. Saekow, V. Amornkitbamrung, J Mol Model 19, 1407 (2013)
A. SalimiBeni, M. Zarandi, B. Hosseinzadeh, A. NajafiChermahini, J. Mol. Struct. 1164, 155 (2018)
L.-Y. Lin, C.-H. Tsai, K.-T. Wong, T.-W. Huang, L. Hsieh, S.-H. Liu, H.-W. Lin, C.-C. Wu, S.-H. Chou, S.-H. Chen, A.-I. Tsai, J. Org. Chem. 75, 4778 (2010)
Acknowledgements
This work was realized with the support of the National Center for Scientific and Technical Research (CNRST—Morocco) as part of the Research Excellence Awards Program (No. 28USMBA2017).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lazrak, M., Toufik, H., Bouzzine, S.M. et al. Bridge effect on the charge transfer and optoelectronic properties of triphenylamine-based organic dye sensitized solar cells: theoretical approach. Res Chem Intermed 46, 3961–3978 (2020). https://doi.org/10.1007/s11164-020-04184-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-020-04184-x