
Abstract
New series of 2,4,6-triarylpyridines derivatives 5a–f and 6a–d were designed, synthesized and evaluated for their cytotoxic activity against breast cancer cell lines. Three-component reaction between acetophenones, ammonium acetate and 4-methyl or 2-methylpyridine in the presence of iodine in DMSO lead to title 2,4,6-triarylpyridines. This method allows the simple preparation of the desired products using readily available reagents under mild reaction conditions in good yields. Synthesized compounds were investigated for their cytotoxic activities against MDA-MB-231, MCF-7 and T-47D cell lines. Most of the synthesized compounds showed remarkable cytotoxicity, comparable to standard drug etoposide.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The pyridine substructure is one of the most prevalent heterocycles found in natural products, pharmaceuticals, and functional materials [1]. The 2,4,6-triarylpyridines are significant building blocks in supramolecular chemistry [2] and are also important because of their biological activities [3, 4]. Also, the quaternary 2,4,6-triarylpyridinium derivatives have significant synthetic applications such as in nucleophilic reaction [5], C-alkylation of β-diketones [6], electrophilic amination [7], and azepine synthesis [8]. Therefore, the development of novel and efficient methods for the synthesis of pyridine scaffolds with diverse functionalities and substituents remains an area of current interest, which has been studied in recent year [9,10,11,12,13].
Several series of 2,4,6-triaryl pyridines (Kröhnke pyridines) [14] have been reported that exhibited excellent cytotoxic activities [14,15,16]. These derivatives were synthesized using various methods and procedures. Traditionally, these compounds have been synthesized through the reaction of N-phenacylpyridinium salts with α,β-unsaturated ketones in the presence of NH4OAc [17, 18]. However, the pyridinium salts and the unsaturated ketones have to be synthesized first, and this method is relatively expensive. So, several improved methods have been developed for the synthesis of these pyridines such as the reaction of a ketoketene dithioacetals with methyl ketones in the presence of NH4OAc [19], the reaction of N-phosphinylethanimines with aldehydes [20], the addition of lithiated β-enaminophosphonates to chalcones [21], the solvent-free reaction between acetophenones, benzaldehydes, and NH4OAc in the presence of sodium hydroxide [22], the one-pot reaction of acetophenones, benzaldehydes, and NH4OAc without catalyst under microwave irradiation [23], and the iodine-mediated reaction between alkyl-branched amino acids and acetophenones [24].
Considering the importance of Kröhnke pyridines as anticancer agents, cytotoxic activates of all synthesized compounds were evaluated against three breast cancer cell lines MDA-MB-231, MCF-7, and T-47D [14].
Experimental
All chemicals were purchased from Merck and Fluka companies. All yields refer to isolated products. 1H and 13C NMR spectra were recorded on a Bruker, Rheinstetten, Germany (at 500 and 400 MHz) NMR spectrometer using tetramethylsilane (TMS) as internal standard. Elemental analyses for C, H, and N were performed using a Heraeus CHN-O-Rapid analyzer. Melting points were determined in a capillary tube and are not corrected. The progress of reaction was followed with TLC using silica gel SILG/UV 254 and 365 plates. All products are known compounds, and their structures were deduced by 1H and 13C NMR spectroscopy.
General procedure for the preparation of compounds 5a–f and 6a–d
A solution of 4-methylpyridine 1 or 2-methylpyridine 2 (1 mmol) and iodine (1 mmol) in 3 mL DMSO was heated at 100 °C for 2 h. Then, acetophenones 3 (2 mmol) and ammonium acetate 4 (1 mmol) were added to the reaction mixture and stirring was continued for 3 h. Upon completion of the reaction, monitored by TLC, the reaction mixture was diluted with 10 mL aqueous saturated solution of sodium thiosulphate and extracted with dichloromethane (3 × 4 mL). Combined organic layers were washed with brine solution, dried over anhydrous Na2SO4, solvent was removed under reduced pressure and the resultant crude product was purified by silica gel column chromatography using hexane and ethyl acetate (7:1) as eluents to obtain the desired products 5a–f and 6a–d.
All of synthesized compounds were characterized by their physical constant, comparison with authentic samples, IR, 1H NMR, 13C NMR spectroscopy and elemental analysis.
2,6-diphenyl-4,4′-bipyridine (5a)
1H NMR (500.0 MHz, DMSO-d6): δ 7.39–7.61 (m, 6H, 6CH), 8.07 (d, J = 8.0 Hz, 4H, 4CH), 8.26 (s, 2H, 2CH), 8.55 (d, J = 6.0 Hz, 2H, 2CH), 9.08 (d, J = 6.5 Hz, 2H, 2CH) ppm. 13C NMR (125.7 MHz, DMSO-d6): δ 125.3 (4CH), 127.2 (2C), 128.1 (2CH), 130.0 (2CH), 131.5 (2CH), 131.8 (4CH), 135.2 (C), 144.9 (2C), 145.1 (2C), 159.3 (2C) ppm. Anal Calc. for C22H16N2: C, 85.69; H, 5.23; N, 9.08. Found: C, 85.73; H, 5.21; N, 9.11.
2,6-di-o-tolyl-4,4′-bipyridine (5b)
1H NMR (500.0 MHz, DMSO-d6): δ 2.26 (s, 6H, 2CH3), 7.16–7.33 (m, 4H, 4CH), 7.46 (d, J = 7.5 Hz, 2H, 2CH), 8.01 (d, J = 8.0 Hz, 2H, 2CH), 8.17 (s, 2H, 2CH), 8.23 (d, J = 7.5 Hz, 2H, 2CH), 8.94 (d, J = 6.0 Hz, 2H, 2CH) ppm. 13C NMR (125.7 MHz, DMSO-d6): δ 18.7 (2CH3), 109.3 (2CH), 119.7 (2CH), 120.6 (C), 122.5 (2C), 125.6 (2CH), 126.1 (2CH), 126.6 (2CH), 129.4 (2CH), 130.9 (2CH), 137.0 (C), 144.9 (2C), 158.2 (2C) ppm. Anal Calc. for C24H20N2: C, 85.68; H, 5.99; N, 8.33. Found: C, 85.72; H, 6.02; N, 8.37.
2,6-di-p-tolyl-4,4′-bipyridine (5c)
1H NMR (500.0 MHz, DMSO-d6): δ 2.33 (s, 6H, 2CH3), 7.21 (dd, J = 5.5, 3.0 Hz, 4H, 4CH), 8.06 (d, J = 8.0 Hz, 4H, 4CH), 8.26 (d, J = 5.0 Hz, 2H, 2CH), 8.46 (s, 2H, 2CH), 9.10 (d, J = 6.5 Hz, 2H, 2CH) ppm. 13C NMR (125.7 MHz, DMSO-d6): δ 20.8 (2CH3), 121.7 (2C), 124.4 (2C), 124.5 (C), 127.5 (4CH), 127.6 (4CH), 128.4 (2CH), 128.6 (2CH), 132.1 (2CH), 139.2 (C), 159.3 (2C) ppm. Anal Calc. for C24H20N2: C, 85.68; H, 5.99; N, 8.33. Found: C, 85.71; H, 5.96; N, 8.29.
2,6-bis(4-fluorophenyl)-4,4′-bipyridine(5d)
1H NMR (500.0 MHz, DMSO-d6): δ 7.52–7.60 (m, 4H, 4CH), 8.13 (dd, 3JHH = 4.5, 4JFH = 4.5 Hz, 4H, 4CH), 8.25 (d, J = 4.5 Hz, 2H, 2CH), 8.51 (s, 2H, 2CH), 9.17 (s, 2H, 2CH) ppm. 13C NMR (125.7 MHz, DMSO-d6): δ 122.7 (2CH), 123.5 (4CH), 129.7 (4CH), 132.1 (C), 132.3 (4CH), 133.5 (C), 134.1 (4CH), 143.4 (2C), 151.1 (2C), 163.9 (d, 1JC-F = 253.9 Hz, 2C) ppm. Anal Calc. for C22H14F2N2: C, 76.73; H, 4.10; N, 8.13. Found: C, 76.76; H, 4.07; N, 8.15.
2,6-bis(2-chlorophenyl)-4,4′-bipyridine (5e)
1H NMR (500.0 MHz, DMSO-d6): δ 7.32–7.58 (m, 6H, 6CH), 8.12 (dd, J = 8.2, 6.0 Hz, 2H, 2CH), 8.25 (d, J = 8.0 Hz, 2H, 2CH), 8.55 (d, J = 6.0 Hz, 2H, 2CH), 9.52 (d, J = 6.5 Hz, 2H, 2CH) ppm. 13C NMR (125.7 MHz, DMSO-d6): δ 116.0 (2CH + CH), 116.3 (CH), 120.4 (CH), 120.6 (CH), 124.6 (2CH), 126.1 (CH), 126.9 (CH), 127.4 (2CH), 129.9 (C), 131.8 (CH), 131.9 (CH), 137.0 (2C), 146.5 (C), 146.6 (2C), 149.9 (2C) ppm. Anal Calc. for C22H14Cl2N2: C, 70.04; H, 3.74; N, 7.43. Found: C, 70.07; H, 3.72; N, 7.46.
2,6-bis(4-bromophenyl)-4,4′-bipyridine (5f)
1H NMR (500.0 MHz, DMSO-d6): δ 7.57 (s, 4H, 4CH), 8.14 (s, 2H, 2CH), 8.27 (s, 4H, 4CH), 8.65 (s, 2H, 2CH), 9.15 (s, 2H, 2CH) ppm. 13C NMR (125.7 MHz, DMSO-d6): δ 126.0 (4CH), 127.0 (4CH), 128.7 (C), 129.8 (2C), 130.3 (2CH), 130.9 (2CH), 131.5 (2CH), 133.9 (2C), 136.7 (C), 159.5 (2C) ppm. Anal Calc. for C22H14Br2N2: C, 56.68; H, 3.03; N, 6.01. Found: C, 56.72; H, 3.06; N, 5.97.
2′,6′-diphenyl-2,4′-bipyridine (6a)
1H NMR (500.0 MHz, DMSO-d6): δ 7.06 (t, J = 7.0 Hz, 1H, CH), 7.16 (t, J = 8.0 Hz, 2H, 2CH), 7.20 (d, J = 8.0 Hz, 4H, 4CH), 7.44 (d, J = 9.0 Hz, 1H, CH), 7.91 (t, J = 6.0 Hz, 1H, CH), 8.05 (d, J = 8.0 Hz, 4H, 4CH), 8.46 (s, 2H, 2CH), 9.20 (d, J = 7.5 Hz, 1H, CH) ppm. 13C NMR (125.7 MHz, DMSO-d6): δ 123.9 (C), 124.0 (2C), 127.5 (4CH), 127.6 (4CH), 128.3 (2CH), 128.4 (2CH), 132.5, 136.0, 137.9 and 140.9 (4CH), 143.6 (C), 143.8 (2C) ppm. Anal Calc. for C22H16N2: C, 85.69; H, 5.23; N, 9.08. Found: C, 85.72; H, 5.27; N, 9.11.
2′,6′-di-p-tolyl-2,4′-bipyridine (6b)
1H NMR (500.0 MHz, DMSO-d6): δ 2.33 (s, 6H, 2CH3), 7.11 (d, J = 8.5 Hz, 4H, 4CH), 7.28 (t, J = 7.5 Hz, 1H, CH), 7.45 (d, J = 9.5 Hz, 1H, CH), 7.92 (d, J = 4.5 Hz, 1H, CH), 8.06 (d, J = 8.0 Hz, 4H, 4CH), 8.31 (s, 2H, 2CH), 9.12 (d, J = 4.5 Hz, 1H, CH) ppm. 13C NMR (125.7 MHz, DMSO-d6): δ 123.9 (C), 124.2 (2C), 126.4 (2C), 127.5 (4CH), 127.6 (4CH), 128.4 (2CH), 128.6, 132.1, 136.3 and 138.9 (4CH), 144.1 (C), 159.3 (2C) ppm. Anal Calc. for C24H20N2: C, 85.68; H, 5.99; N, 8.33. Found: C, 85.65; H, 6.02; N, 8.30.
2′,6′-bis(4-fluorophenyl)-2,4′-bipyridine (6c)
1H NMR (500.0 MHz, DMSO-d6): δ 7.26 (dd, 3JHH = 7.0, 3JFH = 7.0 Hz, 4H, 4CH),7.58 (d, J = 9.5 Hz, 1H, CH), 7.74 (t, J = 8.0 Hz, 1H, CH), 8.14 (dd, 3JHH = 7.0, 4JFH = 6.5 Hz, 4H, 4CH), 8.23 (d, J = 7.5 Hz, 1H, CH), 8.47 (s, 2H, 2CH), 9.10 (d, J = 5.0 Hz, 1H, CH) ppm. 13C NMR (125.7 MHz, DMSO-d6): δ 122.5 (2C), 124.2 (C), 127.9 (4CH), 128.2 (2CH), 129.2 (4CH), 131.0, 132.9, 137.1 and 137.4 (4CH), 138.6 (C), 143.0 (2C), 151.5 (2C) ppm. Anal Calc. for C22H14F2N2: C, 76.73; H, 4.10; N, 8.13. Found: C, 76.72; H, 4.07; N, 8.16.
2′,6′-bis(4-bromophenyl)-2,4′-bipyridine (6d)
1H NMR (500.0 MHz, DMSO-d6): δ 7.11 (d, J = 8.0 Hz, 1H, CH), 7.24 (t, J = 7.0 Hz, 1H, CH), 7.52 (d, J = 8.9 Hz, 4H, 4CH), 7.93 (t, J = 8.0 Hz, 1H, CH), 8.12 (d, J = 8.0 Hz, 4H, 4CH), 8.42 (s, 2H, 2CH), 9.12 (d, J = 6.5 Hz, 1H, CH) ppm. 13C NMR (125.7 MHz, DMSO-d6): δ 121.1 (2C), 121.2 (2CH), 123.5 (2CH), 123.6 (2CH), 124.5 (2CH), 124.6 (2CH), 132.5 (CH), 136.0 (2CH), 137.9 (2C) and 140.9 (CH), 143.6 (2C), 143.8 (C), 157.3 (C) ppm. Anal Calc. for C22H14Br2N2: C, 56.68; H, 3.03; N, 6.01. Found: C, 56.68; H, 2.99; Br, 34.31; N, 6.04.
In vitro cytotoxicity assay
Three breast cancer cell lines (MDA-MB-23, MCF-7, and T-47D, 5 × 104 cells/mL) in 96-well culture plates were incubated for 48 h with different concentrations of 2,4,6-triaryl pyridines 5a–f and 6a–d dissolved in DMSO/ethanol as solvent (the final volume of solvent/medium was less than 1% in all experiments). Etoposide and solvent were used as positive and negative controls, respectively. After incubation, the solvent/medium was removed and 200 μL of MTT solution (1 mg/mL, final concentration) was added to all wells. After 4 h of treatment, the culture medium was replaced with DMSO (100 μL) to each well. The absorbance was determined at 492 nm with a multi-well plate reader (Gen5, Power wave xs2, BioTek, America). The IC50 values (all experiments were performed at least three times and) for all compounds were calculated by nonlinear regression analysis [25, 26].
Docking study
The docking study was carried out using AutoDock Tools (version 1.5.6), and the pdb structure of 4FM9 was taken from the protein data bank. Subsequently, water and additional molecules were removed from the enzyme structure. The 3D structure of compound 5e was depicted using MarvineSketch 5.8.3, 2012, and ChemAxon (http://www.chemaxon.com). After AutoDock format of topoisomerase II and compound 5e were prepared. AutoDock format of enzyme was used as an input for the AUTOGRID program. The center of the grid box was placed in coordinates x = 27.42, y = 25.50, and z = 30.68 and the dimensions of the grid box were set at 60 × 60 × The flexible ligand docking system was carried out by 25 runs of the AUTODOCK search by the Lamarckian genetic algorithm. The highest binding energy conformation of the compound 5e was selected by analyzing the interactions between topoisomerase II and this compound, and the results were visualized using Discovery Studio (4.0 Client, Figs. 1 and 2).

Compound 5e in the active site of topoisomerase II

Binding mode of the compound 5e in the active site of topoisomerase II
Results and discussion
Chemistry
In order to achieve a more efficient synthetic process, minimize by-products, decrease the number of separate reaction steps, improving the yields and reaction times and also in extending our research on the synthesis and study of heterocyclic and pharmaceutical compounds, [27,28,29,30,31,32,33] in this work, at first, the reaction of 4-methylpyridine 1 (1 mmol), acetophenone 3a (1 mmol), ammonium acetate 4 in the presence of iodine was investigated in various reaction temperature and solvents (Table 1). Although the 2,4,6-triarylpyridine 5a was not generated in the absence of DMSO (entry 1), the reaction lead to desired product 5 in 75% yield in presence of DMSO (entry 2).

We examined the reaction in various solvents such as DMF, THF, toluene, dichloroethane (DCE), and CH3CN (entries 3–7), and it was found that the best yield was achieved in DMSO. Also, we tested various amount of catalyst iodine for the reaction as 0.5 and 0.2 equiv. per 1 mmol substrate and the 2,4,6-triarylpyridine 5a was produced in 60% and 51% yield, respectively (entries 8, 9). The reaction did not preceded in better yield at 120 °C and 80 °C (entries 10–11).
Encouraged by these results, the reactions for produce desired 2,4,6-triaryl pyridines 5a–f and 6a–d were performed using 4-methylpyridine 1 or 2-methylpyridine 2, ammonium acetate 4 and various acetophenones 3 in the presence of 1 equiv. of iodine at 100 °C in 3 mL DMSO (Scheme 1). The structures of the isolated products 5a–f and 6a–d were assigned on the basis of 1H and 13C NMR spectroscopy.

New method for the synthesis of 2,4,6-triarylpyridines 5a–f and 6a–d
A proposed mechanism for the formation of the target 2,4,6-triarylpyridine derivatives is depicted in Scheme 2. Initially, the methylpyridine (1 or 2) iodinates by iodide to produce iodinated intermediate 7. This intermediate undergoes the Kornblum oxidation [34, 35] at 100 °C in DMSO to give aldehyde 8. Next, acetophenones 3 may condense with the aldehyde 8 to generate α,β-unsaturated compound 9. Alternatively, the condensation of NH4OAc with acetophenones 3 leads to the formation of enamine intermediate 10. The reaction of enaminone 10 and α,β-unsaturated compound 9 affords intermediate 11 which undergoes ring closing condensation to give dihydopyridine 12. Finally, the dihydopyridine 9 oxidizes to corresponding 2,4,6-triarylpyridine 5a–f and 6a–d.

Proposed mechanism for formation 2,4,6-triarylpyridine 5a–f and 6a–d
Cytotoxicity
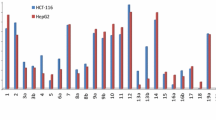
The synthesized 2,4,6-triaryl pyridines 5a–f and 6a–d were evaluated for in vitro cytotoxicity against breast cancer cell lines MDA-MB-231, MCF-7 and T-47D using the MTT assay [36]. The IC50 values of these compounds in comparison with standard drug etoposide are listed in Table 1. Results of the MTT assay demonstrated that compounds 5e, 5b, 5d and 5c with IC50 values 8.74 ± 0.7, 9.45 ± 0.4, 11.59 ± 0.7 and 17.18 ± 0.4 respectively were more potent than etoposide (IC50 = 22.56 ± 0.53 μM) against MDA-MB-231 cells. Moreover, compound 6d was as potent as etoposide against the latter cell line. Compounds 5d, 5e, and 6b (IC50s = 8.12 ± 0.54, 13.1 ± 0.54, and 16.81 ± 0.65 μM, respectively) showed high cytotoxic effects against MCF-7 cell line in comparison etoposide (IC50 = 17.76 ± 0.68 μM). Among the synthesized compounds, cytotoxic activities of the compounds 5b, 5d, 5e, 6a, 6b, and 6d (IC50 values in the range of 8.61 ± 0.32–13.97 ± 0.4 μM) were superior to that of standard drug etoposide (IC50 = 21.18 ± 0.63 μM) against T-47D cells. Compound 5f was also as potent as etoposide against T-47D cells (Table 2).

To obtain an optimized cytotoxic agent, 4-methylpyridine 1 (series 5a–f), 2-methylpyridine 2 (series 6a–d) and substituted acetophenones 3 were used for the synthesis of the target 2,4,6-triaryl pyridines.
In the 4-methylpyridine series 5a–f, 2,6-diphenyl-4-(pyridin-4-yl)pyridine 5a showed moderate cytotoxic effect against all three cell lines studied. Introduction of methyl substituents on 2-positions of 2 and 6-phenyl rings, as in compound 5b, led to a significant increase in cytotoxicity against MDA-MB231 and T-47D. Changing the position of the methyl groups in the mentioned phenyl rings from C-2 to C-4, producing 5c diminished the activity against MDA-MB231 and loss of cytotoxic activity against MCF-7 and T-47D cells. Among the synthesized compounds, 2,6-bis(4-fluorophenyl)-4-(pyridin-4-yl)pyridine 5d and 2,6-bis(2-chlorophenyl)-4-(pyridin-4-yl)pyridine 5e of 4-methylpyridine series showed excellent cytotoxic effects against all the studied cell lines. It is worthy to note that compounds 5d and 5e were the most potent compounds against MCF-7 and MDA-MB231 cells, respectively. The comparison of IC50 value of 4-fluoro compound 5d with their 4-bromo analog 5f revealed that the fluorine atom is more effective than bromine atom in this series.
2-Methylpyridine derivative 6a without substituents on the 2 and 6-phenyl rings showed no activity against MDA-MB231 and MCF-7 cells; however, it showed good activity against T-47D. Compound 6b having 4-methyl group on 2 and 6-phenyl rings did not show activity against MDA-MB231. On the other hand, this compound showed high cytotoxic activity against MCF-7 and T-47D. Among the synthesized compounds, compound 6b was the most potent compound against T-47D. Replacement of methyl groups of compound 6b with fluorine atom, as in compound 6c, led to loss of cytotoxic activity. In contrast, 4-bromo derivative 6d was a good cytotoxic agent against all the three cell lines MDA-MB-231, MCF-7 and T-47D.
The cytotoxicity of the most potent compound 5e, as the most potent compound against MDA-MB231, was also evaluated against normal cell line HDF [34, 35]. The result revealed that compound 5e did not show cytotoxic activity against HDF (IC50 > 100 µM).
Docking studies
Several cytotoxic agents whit 2,4,6-triarylpyridine scaffold were reported that displayed high inhibitory activity against topoisomerase II [16]. Therefore, to find a possible interaction mode of the compound 5e as one of the most potent compound against studied cancer cell lines, molecular modeling was performed in the active site of topoisomerase II (PDB code: 4FM9). As can be seen in Fig. 1, the active site binding pocket of topoisomerase II includes protein residues and DNA bases [37].
Detailed binding mode of compound 5e in the active site of this enzyme revealed that this compound as well fitted in the active site of enzyme and interacted with both protein and DNA parts (Fig. 2). 2-Chlorophenyl moieties of the compound 5e interacted with DNA bases cytosine (DC5) and adenine (DA6) via a hydrophobic interaction and a π-anion interaction. These moieties also formed two hydrophobic interactions with Glu503 and Ala505, and one of them created a π-anion interaction with Arg487. The latter amino acid established a hydrophobic interaction with 4-pyridine group. 4-Pyridine group also interacted with Glu506 through a π-anion interaction. Furthermore, the central pyridine moiety formed a hydrophobic interaction with Ala505.
Conclusions
In conclusion, new derivatives of 2,4,6-triaryl pyridine 5a–f and 6a–d were synthesized and evaluated as cytotoxic agents. For the synthesis of target compounds, an efficient procedure and simple starting materials were used and these compounds. The cytotoxic assay was done against MDA-MB-231, MCF-7, and T-47D using MTT assay. Among the synthesized compounds 5e, 5b, 5d and 5c against MDA-MB-231, compounds 5d, 5e, and 6b against MCF-7, and compounds 5b, 5d, 5e, 6a, 6b, and 6d against T-47D were more potent than etoposide. Compound 5e as one of the most potent compounds against all three studied cell lines was docked into active site of topoisomerase II. Obtained results revealed that this compound interacted as well with both amino acids and DNA bases in the active site of topoisomerase II.
References
G.D. Henry, Tetrahedron 60, 6043 (2004)
M. Borthakur, M. Dutta, S. Gogoi, R.C. Boruah, Synlett 2008, 3125 (2008)
I.J. Enyedy, S. Sakamuri, W.A. Zaman, K.M. Johnson, S. Wang, Bioorg. Med. Chem. Lett. 13, 513 (2003)
A. Shrestha, S. Park, S. Shin, T.M. Kadayat, G. Bist, P. Katila, Y. Kwon, E.S. Lee, Bioorg. Chem. 79, 1 (2018)
Y. Asanuma, H. Eguchi, H. Nishiyama, I. Tomita, S. Inagi, Org. Lett. 19, 1824 (2017)
J. Marquet, M. Moreno-Manas, P. Pacheco, M. Prat, A.R. Katritzky, B. Brycki, Tetrahedron 46, 5333 (1990)
R.A. Abramovitch, J.M. Beckert, P. Chinnasamy, H. Xiaohua, W. Pennington, A.R.V. Sanjivamurthy, Heterocycles 28, 623 (1989)
A.R. Katritzky, J.M. Aurrecoechea, K.K. Quian, E. Anna, G.J. Palenik, Heterocycles 25, 387 (1987)
R.M. Martin, R.G. Bergman, J.A. Ellman, J. Org. Chem. 77, 2501 (2012)
J.M. Neely, T. Rovis, J. Am. Chem. Soc. 135, 66 (2013)
J. Wu, W. Xu, Z.X. Yu, J. Wang, J. Am. Chem. Soc. 137, 9489 (2015)
X. Zhang, Z. Wang, K. Xu, Y. Feng, W. Zhao, X. Xu, Y. Yan, W. Yi, Green Chem. 18, 2313 (2016)
R.S. Rohokale, B. Koenig, D.D. Dhavale, J. Org. Chem. 81, 7121 (2016)
R. Karkia, R.K.Y. Junb, T.M. Kadayat, S. Shin, T.B.T. Magar, G. Bist, A. Shrestha, Y. Na, Y. Kwon, E.S. Lee, Eur. J. Med. Chem. 113, 228 (2016)
R. Karkia, C. Song, T.M. Kadayat, T.B.T. Magar, G. Bist, A. Shrestha, Y. Na, Y. Kwon, E.S. Lee, Bioorg. Med. Chem. 23, 3638 (2015)
R. Karkia, C. Park, K.Y. Jun, J.G. Jee, J.H. Lee, P. Thapa, T.M. Kadayat, Y. Kwon, E.S. Lee, Eur. J. Med. Chem. 84, 555 (2014)
F. Kröhnke, W. Zecher, J. Curtze, D. Drechsler, K. Pfleghar, K.E. Schnalke, W. Weis, Angew. Chem. Int. Ed. 1, 626 (1962)
F. Kröhnke, Synthesis 1, 1 (1976)
K.T. Potts, M.J. Cipullo, P. Ralli, G. Theodoridis, J. Am. Chem. Soc. 103, 3584 (1981)
T. Kobayashi, H. Kakiuchi, H. Kato, Bull. Chem. Soc. Jpn 64, 392 (1991)
F. Palacios, A.M.O. de Retana, J. Oyarzabal, Tetrahedron Lett. 37, 4577 (1996)
G.W.V. Cave, C.L. Raston, Chem. Commun. 22, 2199 (2000)
S. Tu, T. Li, F. Shi, F. Fang, S. Zhu, X. Wei, Z. Zong, Chem. Lett. 34, 732 (2005)
J.C. Xiang, Y. Cheng, Z.X. Wang, J.T. Ma, M. Wang, B.C. Tang, Y.D. Wu, A.X. Wu, Org. Lett. 19, 2997 (2017)
Z. Tashrifi, M. Mohammadi-khanaposhtani, M. Shafiee Ardestani, M. Safavi, K. Rad-Moghadam, M. Mehrdad, B. Larijani, M. Mahdavi, Lett. Drug Des. Discov. 6, 213 (2019)
F. Khalili, S. Akrami, M. Safavi, M. Mohammadi-Khanaposhtani, M. Saeedi, S.K. Ardestani, B. Larijani, A. Zonouzi, M.B. Tehrani, M. Mahdavi, Anti-Cancer Agents Med. Chem. 19, 265 (2019)
M. Nikpassand, L. Zare Fekri, M. Nabatzadeh, Comb. Chem. High Throughput Screen. 20, 533 (2017)
M. Nikpassand, L. Zare Fekri, S. Sanagou, Dye. Pigment. 136, 140 (2017)
H. Taherkhorsand, M. Nikpassand, Comb. Chem. High Throughput Screen. 21, 65 (2018)
M. Nikpassand, D. Pirdelzendeh, Dye. Pigment. 130, 314 (2016)
M. Nikpassand, L. Zare Fekri, P. Farokhian, Ultrason. Sonochem. 28, 341 (2016)
M. Nikpassand, L. Zare Fekri, L. Karimian, M. Rassa, Curr. Org. Synth. 12, 358 (2015)
L. Zare Fekri, M. Nikpassand, S. Pourmirzajani, B. Aghazadeh, RSC Adv. 8, 22313 (2018)
M. Mohammadi-Khanaposhtani, S. Rezaei, R. Khalifeh, S. Imanparast, M.A. Faramarzi, S. Bahadorikhalili, M. Safavi, F. Bandarian, E.N. Esfahani, M. Mahdavi, B. Larijani, Bioorg. Chem. 80, 288 (2018)
M. Mohammadi-Khanaposhtani, K. Fahimi, E. Karimpour-Razkenari, M. Safavi, M. Mahdavi, M. Saeedi, T. Akbarzadeh, Lett. Drug Des. Discov. 16, 818 (2019)
M. Mohammadi-Khanaposhtani, M. Safavi, R. Sabourian, M. Mahdavi, M. Pordeli, M. Saeedi, S.K. Ardestani, A. Foroumadi, A. Shafiee, T. Akbarzadeh, Mol. Divers. 19, 787 (2015)
R. Karkia, C. Park, K.Y. Jun, J.G. Jee, J.H. Lee, P. Thapa, T.M. Kadayat, Y. Kwon, E.S. Lee, Euro. J. Med. Chem. 84, 555 (2014)
Acknowledgements
Financial support from the Research Council of Islamic Azad University of Rasht branch is sincerely acknowledged.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Baloutaki, B.A., Sayahi, M.H., Nikpassand, M. et al. An efficient method for the synthesis of new derivatives of 2,4,6-triarylpyridines as cytotoxic agents. Res Chem Intermed 46, 1153–1163 (2020). https://doi.org/10.1007/s11164-019-04025-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-019-04025-6