
Abstract
Super bases are extremely important compounds with high proton affinities (PAs) and many applications in organic, inorganic, polymer, and photochemistry. Here, we have compared and contrasted the basicity of 2,4,6-cycloheptatrienesilylene (1), with its benzo-substituted derivatives including: 4,5-benzo-2,4,6-cycloheptatrienesilylene (2), 3,4-benzo-2,4,6-cycloheptatrienesilylene (3), 2,3-benzo-2,4,6-cycloheptatrienesilylene (4), dibenzo[a,c]-2,4,6-cycloheptatrienesilylene (5), dibenzo[a,d]-2,4,6-cycloheptatrienesilylene (6), dibenzo[a,e]-2,4,6-cycloheptatrienesilylene (7), dibenzo[a,b]-2,4,6-cycloheptatrienesilylene (8), and tribenzo[a,c,e]-2,4,6-cycloheptatrienesilylene (9), at B3LYP/6-311++G** level. All scrutinized silylenes (1–9) and their corresponding protonated forms (1H–9H) appear as minima on their energy surfaces. The conductor-like polarizable continuum model is applied to predict the pka values for nucleophilic silylenes (1–9) in dimethyl sulfoxide, using thermodynamic cycles of Gibbs free energies. In most cases, the scrutinized 1–9 show relatively high basicity, which qualify them for being categorized as super bases or proton sponges. The overall trend of basicity (7 > 6 > 3 > 1 > 2 > 4 > 5 > 8 > 9) appears consistent with both proton affinity in solution phase (PA2) and nucleophilicity (N). Among our scrutinized silylenes, 7 shows the highest basicity, Mulliken electronegativity (\({\mathcal{X}}\)), pka, N, PA, the lowest singlet–triplet energy gap (ΔEs–t), absolute chemical hardness (ηabs), band gap (ΔEH–L), divalent angle (C–Si–C, \({\hat{\text{A}}}\)), and Si–C bond length (\({\dot{\text{A}}}\)). The least basic silylene turns out to be 9, which is the most non-planar structure. It shows the lowest dipole moment (D), nucleus independent chemical shift value (NICS (1)), N, PA, the widest dihedral angle (C–Si–C–C, \({\hat{\text{D}}}\)), and the highest ΔEH–L. Our investigation introduces novel silylenic super bases with possible applications in organic chemistry.
Graphical abstract
Following up on our previous report (Kassaee et al. [41]), we investigate the basicity of novel one-, two-, three-, and four-cyclic conjugated silylenes (1–9). Most of these silylenes turn out as a super base for showing high pKa and proton affinity.

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The acid–base concept of organic compounds have been considered from theoretical and experimental aspects [1,2,3,4,5]. The basicity of carbenes [6,7,8,9], silylenes [10,11,12], and germylenes [13,14,15,16] have been studied at theoretical levels. Silylenes mostly prefer to exist as singlets and can interact as a Lewis acid or base. The basicity of silylenes may be triggered by their nucleophilicity or proton affinity. Theoretical studies have been reported on the geometrical and thermodynamic properties of two-, three-, and poly-cyclic hydrocarbons including biphenyl, indene, azulene, naphthalene, anthracene, phenanthrene and pyrene, at various theoretical levels [17,18,19,20,21]. Furthermore, stability, electron and proton affinities, basicity and nucleophilicity have been assessed for heterocyclic [22,23,24,25] and polycyclic hydrocarbons [26, 27] and silylenes [28]. The stability of a conjugated base has a significant effect on the acidity of the corresponding acid. Conjugation, electronegativity, hybridization, and solvation appear as the most prominent contributing factors to the stability of the conjugated base and consequently the higher strength of its acid [29,30,31]. Also, the hard and soft acids and bases (HSAB) are widely used to determine the stability of many compounds [32]. Effects of aromatic rings on stabilization of carbenes are probed, at B3LYP/6-311++G** level. As the extent of conjugation increases, the stability of carbene improves [33,34,35]. In some cases, higher aromaticity may lead to more kinetic stability. For instance, di(triptycyl)carbene appears as a persistent triplet dialkyl carbene [36]. The stability of NHX (X = C, Si, and Ge) type polycyclic structures are also scrutinized, at theoretical levels [37]. Theoretical studies are reported on some cyclohepta-2,4,6-trienylidenes fused with benzene rings, which may be considered analogs of our novel one-, two-, three-, and four-cyclic conjugated silylenes (1–9, Scheme 1). The singlet carbene analogs of 1, 2, and 4 were found not to be minima for showing two imaginary frequencies [38]. In another survey, during the rearrangement of benzofulvene to naphthalene, the six-membered carbenic analogs of 2–4 were characterized as intermediates, through flash vacuum pyrolysis (FVP) [39]. Thermodynamic parameters of the carbenic analog of 5 has been scrutinized [40]. The most similar report includes the MNDO study on the carbenic analogs of 1–5 that we reported earlier [41]. Several methods are used to calculate the acidity of organic and inorganic compounds in either gas or solution phase [42,43,44,45,46,47,48,49,50]. The gas-phase acidity is mainly influenced by ion-pairing, aggregation effects, and entropic terms. Theoretical pKa values are usually obtained considering thermodynamic cycles which combine gas-phase and liquid-phase calculations, using different solvents and solvation methods, including CPCM [51,52,53,54,55,56]. Following up on our quest for stable group 14, here we have scrutinized thermodynamic and structural parameters of novel one-, two-, three-, and four-cyclic conjugated silylenes (1–9, Scheme 1). To this end, basicity of 1–9 are estimated by considering their corresponding protonated structures (1H–9H, Scheme 2).

2,4,6-Cycloheptatrienesilylene (1) with one, two, and three fused benzene rings (2–9), scrutinized at B3LYP/6-311++G** level

The protonated structures of 2,4,6-cycloheptatrienesilylene (1H), with one, two, and three fused benzene rings (2H–9H), scrutinized at B3LYP/6-311++G** level
Computational methods
All structures are optimized using Gaussian 98 via B3LYP/6-311++G** Pople’s basis set. Harmonic vibrational frequencies (\({\nu_{\min}}\)), ΔEH–L, ΔEs–t, the changes of enthalpies (ΔHs–t), and Gibbs free energies (ΔGs–t) are also calculated at 298.15 K and 1.0 atm. In order to determinate the acidity (pKa) of 1–9, ΔGs–t are calculated in gas and solution phases using DMSO as solvent. To predict the Gibbs free energy of solvation, the CPCM [57,58,59,60], which is based on the polarized continuum model (PCM), is applied. The nucleophilicity index [61] (N = EHOMO(Nu) − EHOMO(TCNE)) and the electrophilicity index [62] (ω = μ2/2η) are provided where μ is the chemical potential [63] (μ = (EHOMO − ELUMO)/2), and η is the chemical hardness [64] (η = ELUMO − EHOMO). In addition, the gas-phase proton affinities (PA = − ΔHProtonation) [65, 66], ionization potential (IP) [67,68,69], electron affinity (EA) [69,70,71], Mulliken electronegativity (\({\mathcal{X}}\) = (IP + EA)/2), and absolute hardness (ηabs = (IP − EA)/2) are calculated [72,73,74,75]. Furthermore, dipole moments (D) and structural parameters such as bond lengths (Å), divalent and dihedral angles (\({\hat{\text{A}}}\) and \({\hat{\text{D}}}\), respectively), and symmetries are determined. The NICS [76,77,78,79,80] values and the natural bond orbital (NBO) [81,82,83,84,85] charges are computed to give clear evidences of stabilization and basicity.
Results and discussion
To scrutinize the effects of fused benzene ring(s) on 2,4,6-cycloheptatrienesilylene (1), we characterized singlet (s) and triplet (t) states of 1 fused with zero-, one-, two-, and three-conjugated phenyl rings (1, 2–4, 5–8, and 9, respectively, Scheme 1 and Table 1). The NBO charges, structural features, and thermodynamic parameters including ΔEs–t, ΔHf, ΔGf, and ΔEH–L, N, ω, μ, and also, NICS (1) data are obtained (Tables 1, 2).
Silylenes 2–6 are planar while 1 and 7–9 appear non-planar (Table 1). The highest \({\hat{\text{D}}}\) is displayed by 9 (42.22°) that exhibits the lowest dipole moment (D, 1.82 Debye). In addition, 9 carries nearly the highest NBO positive charge on its Si atom (1.003). This appears consistent with its NICS (1) value (− 0.65 ppm, Table 2) and indicates the lower ring current of 9 compared to 1–8. Also, 7 shows nearly the lowest NBO positive charge on its Si atom (0.774) which is consistent with its highest N (4.01 eV).
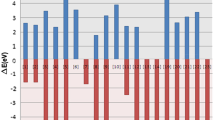
The more negative values of ΔEs–t for 1–9 indicate the higher stability of singlet states compared to their corresponding triplets. The lower absolute value of ΔEs–t reflects the lower stability, and higher chemical activity. The results show that all our silylenes are rather nucleophilic. For instance, 1 and 3 show 3.80 and 3.85 eV for N and 2.71 and 3.62 eV for ω, respectively (Table 2). Structure 7 appears as our most nucleophilic (N = 4.01 eV) silylene. Also, 7 has the lowest ΔEs–t (− 15.71 eV), ΔHs–t (− 15.71 eV), ΔGs–t (− 14.75 eV), and ΔEH–L (− 2.54 eV). In contrast, 9 shows the lowest N (3.55 eV), and the highest ΔEH–L (− 3.32 eV). The overall order of silylenes ΔEs–t, ΔHf, and ΔGf is: 5 > 2 > 9 > 4 > 8 > 6 > 3 > 1 > 7. The overall trend for N shows 7 > 6 > 3 > 1 > 2 > 4 > 5 > 8 > 9. For all structures, we diagnosed the aromaticity by NICS at 1 Å above the center of aromatic system (NICS (1)). The number of benzene rings make a difference in the diatropic current at the center of the aromatic system. All scrutinized structures obey Hückel rule (4n + 2; 1: 6πe−, 2–4: 10πe−, 5–8: 14πe−, and 9: 18πe−). Evidently, planarity plays a dominant role in aromaticity of 1–9. Two-cyclic species (2–4) show higher aromaticity with respect to the remaining structures for being more planar. On the other extreme, 9 is the most non-planar silylene with the least NICS (1) value (− 0.65 ppm). Silylenes with one benzene ring (2–4) appear more planar and aromatic than those with two benzene rings (5–8), which turn out more planar and aromatic than the silylene with three benzene rings (9). Hence, the overall trend of NICS (1) values is: 3 > 4 > 2 > 7 > 6 > 5 > 1 > 8 > 9. Evidently, non-planar 1 that has no stabilizing benzo-substituent falls between 5 and 8. The N, PA2 and NICS (1) values show apparent correlation with the corresponding electrostatic potential (ESP) maps (Fig. 1). For instance, silylenes 2–4 illustrate the higher total electron densities at the centre of the aromatic systems that are consistent with NICS (1) values.

Correlation between N, PA1, NICS (1), and the corresponding ESP map for silylenes (1–9), at B3LYP/6-311++G** level
To determine the ηabs and \({\mathcal{X}}\), the EA and IP are computed (Table 3). The accuracy of \({\mathcal{X}}\) is dominated by IP which has higher values than EA. The positive and negative electron affinities suggest that the addition of an electron is an endothermic and exothermic processes, respectively. Structures 3 and 7 show the lowest and the highest \({\mathcal{X}}\) values (2.90 and 4.34 eV, respectively). The latter data on 7 appears consistent with its highest value of N (4.01 eV), the lowest ηabs (0.10 eV) and IP (6.76 eV). Therefore, 7 can easily donate its valence electrons and turn out as the most basic silylene. The proton affinities in gas (PA1) and solution (PA2) phases show different values for 1–9. The PA2 values indicate that silylenes have less tendency to react with proton because the solvation effects [dielectric constant (ε) of DMSO = 47 F/m] [86] that stabilize the initial reactants (H+ and M, M = 1–9) with respect to that in gas phase. For instance, 5 shows 243.89 and 178.72 kcal/mol for PA1 and PA2, respectively. The overall trend of PA2 appears consistent with N: 7 > 6 > 3 > 1 > 2 > 4 > 5 > 8 > 9.
A thermodynamic cycle is employed to account for deprotonation of HA+ (HA+ = 1H–9H) in gas and solution phases (Scheme 3).

Thermodynamic cycles in gas and solution phases for determining the pKa of protonated silylenes (HA+ = 1H–9H) and basicity of A = 1–9, at B3LYP/6-311++G** level
Our silylenes 1–9 are represented by A. Their changes of Gibbs free energy for deprotonation in gas, solution, and solvation in DMSO are shown by \(\Delta G^{\circ}_{\text{gas}} , \Delta G^{\circ}_{\text{sol}} ,\;{\text{and}}\;\Delta G^{\circ}_{\text{s}}\), respectively.
The criteria for acidity is given by
where
Thermodynamic parameters such as PA and N correlate with pKa values. Relative basicity of 1–9 can indicate the relative stability of divalent structures with respect to their corresponding protonated forms. In other words, a stronger acid generates a weaker conjugated base [87]. The results show high basicity, PA, and pKa for 1–9 that are similar to those reported in literature for super bases or proton sponges [88,89,90,91]. The proton sponge definition that was first introduced by Alder et al. has been attributed to the compounds with intramolecular hydrogen bonding after protonation such as 1,8-bis(dimethylamino)naphthalene [92]. Therefore, most of our scrutinized 1–9 may be considered as super bases or proton sponges for showing PA, intrinsic gas phase basicity (GB), and pKa approximately above 245.3 kcal/mol, 239 kcal/mol, and 7.5 (in DMSO), respectively [93, 94]. To determine the pka values, we calculate the absolute Gibbs free energies in gas and solution phases (\(G^{\circ}_{\text{Gas}}\), \(G^{\circ}_{\text{DMSO}}\), respectively) and \(\Delta G^{\circ}_{\text{S}}\), for 1–9 and their protonated structures (1H–9H, Schemes 1, 2, 3, Tables 4 and 5).
The \(\Delta G^{\circ}_{\text{S}}\) for 1H–9H show higher values than their corresponding 1–9. This may be attributed to the solvent stabilization of the positive charge on the former. Using Eqs. 1 and 2, we may get rather accurate pka values in DMSO. A higher pka represents lower acidity and indicates higher basicity of 1–9. Structures 1H–9H benefit from aromatization that disperse the positive charge on Si atom. This aromatization and solvent effects, makes 1H–9H more stable than 1–9. The non-planarity caused by the three fused benzene rings in 9 diminishes the possibility of their electron delocalization. The overall trend of pka values with respect to N, and PA2 is: 7 > 6 > 3 > 1 > 2 > 4 > 5 > 8 > 9.
A higher pka may imply a higher tendency for a silylene to react as a nucleophile and/or a base. Structure 7 is the most basic structure among our scrutinized silylenes (pka = 12.19). Evidently, PA2 shows a direct relationship with pKa [95]. An excellent correlation between pKa and PA2 (R2 = 0.9916, Fig. 2) indicates that the basicity of a silylene may enhance as its PA2 increases. Almost all probed silylenes (except 8 and 9) represent super basicity for showing relatively high PA and pKa (approximately 7–12).

Correlation between pKa and PA2 for silylenes 1–9, at B3LYP/6-311++G** level
Conclusions
In this study, effects of fused benzene rings on 2,4,6-cycloheptatrienesilylene (1) basicity is probed. To this end, we have scrutinized the novel one-, two-, three-, and four-cyclic conjugated silylenes (1–9) and determined their basicity using the corresponding protonated structures (1H–9H), at B3LYP/6-311++G** level. Most of the silylenes show relatively high basicity that can be fall in the category of super bases and proton sponge compounds. All of the scrutinized structures appear as minima on their energy surfaces for showing no imaginary frequencies. The structural and thermodynamic parameters for 1–9 are also calculated. The overall order of silylenes ΔEs–t, ΔHf, and ΔGf is: 5 > 2 > 9 > 4 > 8 > 6 > 3 > 1 > 7. To suggest the basicity trend of 1–9, the CPCM is applied in DMSO as a solvent. The thermodynamic cycles of Gibbs free energies give the relative pka values for protonated silylenes (1H–9H). Aromaticity, planarity, N, PA, steric and solvation effects appear as significant factors in our study of basicity. The overall trend of basicity, PA2, and N is: 7 > 6 > 3 > 1 > 2 > 4 > 5 > 8 > 9. Among our scrutinized silylenes, 7 shows the highest basicity, N, PA, \({\mathcal{X}}\); and the lowest ΔEH–L, ΔEs–t, \({\hat{\text{A}}}\), Si–C bond length, and ηabs, while 9 shows the lowest N, PA, NICS (1), dipole moment, pKa and the highest ΔEH–L, and \({\hat{\text{D}}}\). The results suggest reaching at excellent basicity values for 1–9 silylenes with high pka and PAs values in gas phase and DMSO.
References
M.A. Montes-Morán, D. Suárez, J.A. Menéndez, E. Fuente, Carbon N. Y. 42, 1219 (2004)
M. Alcami, O. Mo, M. Yanez, J. Phys. Org. Chem. 15, 174 (2002)
A.L. Llamas-Saiz, C. Foces-Foces, J. Elguero, J. Mol. Struct. 328, 297 (1994)
E.D. Raczyńska, M. Decouzon, J. Gal, P. Maria, G. Gelbard, F. Vielfaure-Joly, J. Phys. Org. Chem. 14, 25 (2001)
Y. Nonoguchi, S. Sudo, A. Tani, T. Murayama, Y. Nishiyama, R.M. Uda, T. Kawai, Chem. Commun. 53, 10259 (2017)
Y.-J. Kim, A. Streitwieser, J. Am. Chem. Soc. 124, 5757 (2002)
J.A. Platts, Phys. Chem. Chem. Phys. 2, 3115 (2000)
A. Beste, O. Krämer, A. Gerhard, G. Frenking, Eur. J. Inorg. Chem. 1999, 2037 (1999)
D. Martin, O. Illa, A. Baceiredo, G. Bertrand, R.M. Ortuno, V. Branchadell, J. Org. Chem. 70, 5671 (2005)
A. K. Biswas, M. K. Si and B. Ganguly, New J. Chem. 42, 11153 (2018)
P.V. Bharatam, R. Moudgil, D. Kaur, Organometallics 21, 3683 (2002)
M. Driess, S. Yao, M. Brym, C. van Wüllen, D. Lentz, J. Am. Chem. Soc. 128, 9628 (2006)
A.K. Biswas, B. Ganguly, Chem. Eur. J. 23, 2700 (2017)
A. Sojoudi, F.A. Shakib, M.R. Momeni, M. Imani, S. Shojaee, Comput. Theor. Chem. 1009, 81 (2013)
S.S. Kostina, T. Singh, W.J. Leigh, Organometallics 31, 3755 (2012)
M. Haeberlein, J.S. Murray, T. Brinck, P. Politzer, Can. J. Chem. 70, 2209 (1992)
A. Comandini, K. Brezinsky, J. Phys. Chem. A 115, 5547 (2011)
S. Ketrat, S. Müller, M. Dolg, J. Phys. Chem. A 111, 6094 (2007)
D. Wang, A. Violi, D.H. Kim, J.A. Mullholland, J. Phys. Chem. A 110, 4719 (2006)
R.W. Alder, S.P. East, J.N. Harvey, M.T. Oakley, J. Am. Chem. Soc. 125, 5375 (2003)
C.E.H. Dessent, Chem. Phys. Lett. 330, 180 (2000)
R.-E. Li, J.-H. Sheu, M.-D. Su, Inorg. Chem. 46, 9245 (2007)
T. Kosai, S. Ishida, T. Iwamoto, Angew. Chem. Int. Ed. 55, 15554 (2016)
N. Peran, Z.B. Maksić, Chem. Commun. 47, 1327 (2011)
I. Despotović, Z.B. Maksić, R. Vianello, Eur. J. Org. Chem. 2007, 3402 (2007)
M. Meot-Ner, J. Phys. Chem. 84, 2716 (1980)
V.J. Vandiver, C.S. Leasure, G.A. Eiceman, Int. J. Mass Spectrom. Ion Process. 66, 223 (1985)
E. Ohta, T. Ogaki, T. Aoki, Y. Oda, Y. Matsui, H. Ikeda, in AIP Conference Proceedings, vol. 1702 (AIP Publishing, 2015), p. 90060
A. Streitwieser Jr., J.H. Hammons, Prog. Phys. Org. Chem. 3, 41 (1965)
R.G. Pearson, J. Songstad, J. Am. Chem. Soc. 89, 1827 (1967)
A. Bagno, G. Scorrano, R.A.M. O’Ferrall, Rev. Chem. Intermed. 7, 313 (1987)
T.-L. Ho, Chem. Rev. 75, 1 (1975)
H.L. Woodcock, D. Moran, B.R. Brooks, P.R. Schleyer, H.F. Schaefer, J. Am. Chem. Soc. 129, 3763 (2007)
J.K. Kendall, H. Shechter, J. Org. Chem. 66, 6643 (2001)
C. Trindle, J. Org. Chem. 68, 9669 (2003)
E. Iiba, K. Hirai, H. Tomioka, Y. Yoshioka, J. Am. Chem. Soc. 124, 14308 (2002)
L. Pause, M. Robert, J. Heinicke, O. Kühl, J. Chem. Soc. Perkin Trans. 2, 1383 (2001)
A. Kuhn, M. Vosswinkel, C. Wentrup, J. Org. Chem. 67, 9023 (2002)
L.T. Scott, M.M. Hashemi, T.H. Schultz, M.B. Wallace, J. Am. Chem. Soc. 113, 9692 (1991)
K. Hirai, T. Itoh, H. Tomioka, Chem. Rev. 109, 3275 (2009)
M.Z. Kassaee, M.R. Nimlos, K.E. Downie, E.E. Waali, Tetrahedron 41, 1579 (1985)
T. Noda, K. Suzuki, N. Katada, M. Niwa, J. Catal. 259, 203 (2008)
F. Chevallier, Y.S. Halauko, C. Pecceu, I.F. Nassar, T.U. Dam, T. Roisnel, V.E. Matulis, O.A. Ivashkevich, F. Mongin, Org. Biomol. Chem. 9, 4671 (2011)
V.E. Matulis, Y.S. Halauko, O.A. Ivashkevich, P.N. Gaponik, J. Mol. Struct. THEOCHEM 909, 19 (2009)
I.E. Charif, S.M. Mekelleche, D. Villemin, N. Mora-Diez, J. Mol. Struct. THEOCHEM 818, 1 (2007)
H.A. De Abreu, W.B. De Almeida, H.A. Duarte, Chem. Phys. Lett. 383, 47 (2004)
R. Casasnovas, J. Frau, J. Ortega-Castro, A. Salva, J. Donoso, F. Muñoz, J. Mol. Struct. THEOCHEM 912, 5 (2009)
V.S. Bryantsev, Chem. Phys. Lett. 558, 42 (2013)
R.W. Taft, Prog. Phys. Org. Chem. 14, 247–350 (1983)
I.A. Topol, G.J. Tawa, R.A. Caldwell, M.A. Eissenstat, S.K. Burt, J. Phys. Chem. A 104, 9619 (2000)
M.D. Liptak, G.C. Shields, J. Am. Chem. Soc. 123, 7314 (2001)
A.M. Toth, M.D. Liptak, D.L. Phillips, G.C. Shields, J. Chem. Phys. 114, 4595 (2001)
J.T. Muckerman, J.H. Skone, M. Ning, Y. Wasada-Tsutsui, Biochim. Biophys. Acta (BBA) Bioenerg. 1827, 882 (2013)
J.C. Kromann, F. Larsen, H. Moustafa, J.H. Jensen, PeerJ 4, e2335 (2016)
M.D. Liptak, K.C. Gross, P.G. Seybold, S. Feldgus, G.C. Shields, J. Am. Chem. Soc. 124, 6421 (2002)
M.D. Liptak, G.C. Shields, Int. J. Quantum Chem. 85, 727 (2001)
J. Tomasi, M. Persico, Chem. Rev. 94, 2027 (1994)
M. Cossi, V. Barone, B. Mennucci, J. Tomasi, Chem. Phys. Lett. 286, 253 (1998)
V. Barone, M. Cossi, J. Tomasi, J. Comput. Chem. 19, 404 (1998)
M. Cossi, V. Barone, J. Chem. Phys. 109, 6246 (1998)
L.R. Domingo, P. Pérez, Org. Biomol. Chem. 9, 7168 (2011)
P.K. Chattaraj, S. Giri, S. Duley, J. Phys. Chem. A 116, 790 (2011)
B. Smit, D. Frenkel, Mol. Phys. 68, 951 (1989)
R.G. Pearson, Inorg. Chem. 27, 734 (1988)
E.P.L. Hunter, S.G. Lias, J. Phys. Chem. Ref. Data 27, 413 (1998)
L.L. Lohr, H.B. Schlegel, K. Morokuma, J. Phys. Chem. 88, 1981 (1984)
P.K. Nayak, N. Periasamy, Org. Electron. 10, 1396 (2009)
K. Fukui, H. Kato, T. Yonezawa, Bull. Chem. Soc. Jpn. 33, 1197 (1960)
S. Janietz, D.D.C. Bradley, M. Grell, C. Giebeler, M. Inbasekaran, E.P. Woo, Appl. Phys. Lett. 73, 2453 (1998)
R.A. Kendall, T.H. Dunning Jr., R.J. Harrison, J. Chem. Phys. 96, 6796 (1992)
J.-L. Bredas, Mater. Horizons 1, 17 (2014)
R.G. Pearson, J. Org. Chem. 54, 1423 (1989)
R.G. Parr, R.G. Pearson, J. Am. Chem. Soc. 105, 7512 (1983)
R.G. Pearson, Proc. Natl. Acad. Sci. 83, 8440 (1986)
M.V. Putz, N. Russo, E. Sicilia, Theor. Chem. Acc. 114, 38 (2005)
Z. Chen, C.S. Wannere, C. Corminboeuf, R. Puchta, P.R. Schleyer, Chem. Rev. 105, 3842 (2005)
P.R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao, N.J.E. Hommes, J. Am. Chem. Soc. 118, 6317 (1996)
A. Stanger, J. Org. Chem. 71, 883 (2006)
P.R. Schleyer, M. Manoharan, Z.-X. Wang, B. Kiran, H. Jiao, R. Puchta, N.J.R.E. Hommes, Org. Lett. 3, 2465 (2001)
M. Randić, Chem. Rev. 103, 3449 (2003)
E.D. Glendening, C.R. Landis, F. Weinhold, Wiley Interdiscip. Rev. Comput. Mol. Sci. 2, 1 (2012)
E.D. Glendening, F. Weinhold, J. Comput. Chem. 19, 610 (1998)
F. Weinhold, C.R. Landis, Valency and Bonding: A Natural Bond Orbital Donor–Acceptor Perspective (Cambridge University Press, Cambridge, 2005)
A.E. Reed, L.A. Curtiss, F. Weinhold, Chem. Rev. 88, 899 (1988)
F. Weinhold, J. Comput. Chem. 33, 2363 (2012)
A. Luzar, J. Stefan, J. Mol. Liq. 46, 221 (1990)
I.A. Koppel, R.W. Taft, F. Anvia, S.-Z. Zhu, L.-Q. Hu, K.-S. Sung, D.D. DesMarteau, L.M. Yagupolskii, Y.L. Yagupolskii, J. Am. Chem. Soc. 116, 3047 (1994)
H. Chen, D.R. Justes, R.G. Cooks, Org. Lett. 7, 3949 (2005)
E.D. Raczynska, M. Decouzon, J. Gal, P. Maria, K. Wozniak, R. Kurg, S. N. Carins, ChemInform. 31, 6202 (2000)
M.P. Vlasenko, V.A. Ozeryanskii, J. Phys. Org. Chem. 30, e3609 (2017)
B. Kovačević, Z.B. Maksić, Chem. Eur. J. 8, 1694 (2002)
R.L. Benoit, D. Lefebvre, M. Fréchette, Can. J. Chem. 65, 996 (1987)
V.A. Ozeryanskii, M.P. Vlasenko, A.F. Pozharskii, Tetrahedron 69, 1919 (2013)
Q. Jie, J. Guo-Zhu, J. Phys. Chem. A 48, 12983 (2013)
B. Kovačević, Z.B. Maksić, Org. Lett. 3, 1523 (2001)
Acknowledgements
Moral support of Mr. Karim Ayoubi-Chianeh is appreciated. Financial support of Tarbiat Modares University (TMU) is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ayoubi-Chianeh, M., Kassaee, M.Z. Novel silicon super bases at DFT level of theory: effects of fused benzene rings on the basicity of 2,4,6-cycloheptatrienesilylene. Res Chem Intermed 45, 4677–4691 (2019). https://doi.org/10.1007/s11164-019-03856-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-019-03856-7