
Abstract
This study focuses on the search of new applications of polyoxometalates known as decaniobate ions [Nb10O28]6−. The decaniobates can be used as basic solid catalysts in multicomponent reactions to obtain good yields to 4H-pyrans (> 95%) under microwave radiation and solvent-free conditions, using several aldehydes with different electron-withdrawing or electron donor substituents. The synthesis of (TMA)6 [Nb10O28]6H2O was performed following a simple protocol (HPNb), which was modified to decrease the number of hexaniobate species that are formed as impurities (HPNb-HF).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyoxometalates (POMs) are anionic metal-oxide clusters based on octahedral MO6 units sharing their edges or corners where M represents the metal atom. The diverse applications of polyoxometalates in chemistry and the pharmaceutical industry have led to the study of less common polyoxometalates; an example is the decaniobate ion ([Nb10O28]6−), where the more representative compound is TMA6[Nb10O28] (TMA+: tetramethyl ammonium ion) that is stable at pH 5–8 and is insoluble in nonaqueous solvents [1]. The possibilities of the applications of this class of polyoxometalates have been explored in electrocatalysts [2] and photocatalysts [3, 4].
The decaniobates possess more negative charge than other POMs of group IV (Mo, W) due to the difference in their maximum oxidation states, thus this property should be more exploited [5]. Recently, Tsukuda and colleagues demonstrated that decaniobate clusters [Nb10O28]6− can act efficiently as a new base catalyst for aldol-type condensation reactions, including Knoevenagel and Claisen–Schmidt reactions [6], and in CO2 fixation [7]. The basic catalysis was ascribed to the negative charge on the surface oxygens, which was demonstrated by density functional theory calculations. However, other types of basic reactions should be explored for new applications in the field of catalysis using this type of POMs.
One interesting reaction requiring the use of base catalysts is the synthesis of 4H-pyrans, where a reaction between an aldehyde, malononitrile and a β-dicarbonyl compound is involved (Fig. 1). The reaction mechanism initially proceeds by Knoevenagel condensation where the strong basic sites of the catalyst promote the reaction by abstracting a proton from the active methylene compound, which reacts with the aldehyde forming an alkene intermediate followed by a Michael-type addition affording the 4H-pyran compound [8, 9]. Generally, this multicomponent reaction (MCR) is carried out with piperidine, dibutylamine, morpholine or metal alkoxides, using volatile solvents leading to long reaction times and tedious catalyst recovery [8,9,10,11,12]. Different types of basic solids or hybrids such as hydrotalcites, mesoporous Ca-MCM or oxides have been explored in this reaction [13, 14]. Although decaniobates ([Nb10O28]6−) can also act as basic solids, to date the reports on this solid are scarce in the organic synthesis field [6].

Synthesis of 6-amino 4H-pyran derivative via a three-component coupling of aldehyde, malononitrile and β-dicarbonyl compound
Although MCRs are a valuable synthetic tool in the synthesis of heterocyclic compounds and have advantages such a high atom economy, alternative sources of heating are being considered in order to reduce reaction times [15, 16]. One of the most widely used in chemistry is microwave radiation, which has shown that it can reduce the reaction times by generating a direct energy transfer to the reactants, provoking instantaneous superheating [17,18,19]. The aim of this work was to study a new application of decaniobates in MCR, to establish a clean and efficient method for the synthesis of 4H-pyran derivatives with excellent yields under microwave radiation at short reaction times.
Materials and methods
Preparation of decaniobate salts
A decaniobate salt [N(CH3)4]6[Nb10O28].6H2O (HPNb) was synthesized according to the procedure reported by Casey et al. [1]. In a typical procedure, Nb2O5 (2.1 mmol) with an alcoholic solution of 3.3 mmol of tetramethylammonium hydroxide pentahydrate [N(CH3)4]OH.5H2O was heated to 120 °C in a Teflon-lined Parr vessel for 18 h, after which it was allowed to cool to room temperature and subsequently filtered, and the obtained solid was washed with acetone. To avoid the formation of hexaniobate phases, Nb2O5 was treated with HF [20] and then the decaniobate salt was prepared. The final solid was denoted as HPNb-HF.
Characterization
Raman spectroscopy experiments were carried out at room temperature using a Confocal Raman Microscope (Witec, alpha 300) with a 50× objective lens, a Nd: YAG green laser with 532 nm wavelength and 800 scans. Spectra for each solid were taken over the range of 20 and 3000 cm−1, scanning at a step size of 1.0 cm−1 with an integration time constant of 10 s.
Thermogravimetric analysis was performed using a Setaram thermobalance. The decaniobate powder (40.0 mg) was transferred to an alumina crucible and heated using a 5 °C min−1 heating rate up to 800 °C, under an N2 atmosphere flowing at 20 cm3 min−1.
The X-ray diffraction measurements were carried out with a Rigaku Miniflex II using Cu Kα radiation (λ = 1.54056 Å).
The XRD patterns were studied in the 2θ range of 10°–60°, using a count time of 1 s and a step size of 0.05°.
IR spectra were obtained with a Nicolet iS50 spectrometer by the ATR method. IR spectra were processed with a resolution of 4 cm−1 and a spectral range of 4000–400 cm−1.
Temperature-programmed desorption (TPD) of CO2 was performed to study the materials basicity. The equipment used was a Micromeritics AutoChem 2920. In addition, volumetric titration with benzoic acid was carried out to measure the basicity of the solid catalysts using the method reported by Tanabe et al. [21]. In this method, for 0.25 g of catalyst, a benzoic acid solution 0.1 N and 1 mL of bromothymol blue as indicator were used.
General procedure for the synthesis of 4H-pyrans
The 1,3-Diketo compound, malononitrile and aromatic aldehydes were used in the synthesis of 4H-pyrans. All chemicals were purchased from Aldrich and used without further purification. A mixture of ethyl acetoacetate or methyl acetoacetate (1 mmol), aldehyde (1 mmol), and malononitrile (1 mmol), and the catalysts (100 mg) were placed in a microwave tube containing a magnetic stirrer under solvent-free conditions. This was heated under microwave irradiation at 80 °C. The equipment used was an Anton Paar Monowave 400.
The reaction progress was monitored by TLC (1:2 EtOAc: petroleum ether as mobile phase). The catalyst was recovered by filtration after precipitation of decaniobate in acetone (2 × 1 mL). The crude product was recrystallized using hot ethanol and no further recrystallization was required. The yield was expressed as the ratio of moles of products/product to moles of initial aldehyde (Table 1).
All the products were identified by comparison of melting point (mp) and nuclear magnetic resonance (NMR) data. 1H and 13C NMR spectra were performed on a Bruker (400 MHz) spectrometer using TMS as internal reference. 13C NMR spectra were recorded at 100 MHz.
Reuse and stability tests of the catalyst were carried out by running five consecutive experiments, under the same reaction conditions. After each test, the catalyst was separated from the reactions mixture by filtration, washed with acetone (2 × 1 mL), dried under vacuum, and thn reused.
Results and discussion
To confirm the presence of decaniobate phase ([Nb10O28]6−), different techniques such as Raman spectra, XRD, TGA and FTIR were used. In the synthesis of (TMA)6 [Nb10O28]6H2O (HPNb) HF was also incorporated with the aim of avoiding the presence of hexaniobate phase that is generated as impurity by excess of basic medium in the synthesis process.
Figure 2 shows the Raman spectra of HPNb and HPNb-HF in the region of 200–1200 cm−1. For comparison purposes, the Raman spectrum of amorphous Nb2O5 used for the synthesis of HPNb is shown. Figure 2a displays the band at 694 cm−1 related to the Nb2O5 T-phase consisting of 4 × 4 blocks forming the corner-shared octahedral NbO6; each block is connected, sharing the edges of the octahedron. Additionally, the band observed around 170 cm−1 is associated with O–Nb–O stretch vibrations [22]. The spectra of HPNb (Fig. 2b) evidenced the appearance of a new signal around 935 cm−1 indicating the formation of decaniobate species [23]. A small band near 200 cm−1 indicates the presence of the hexaniobate phase [23]. However, when the solid is synthesized in the presence of HF, the band at 200 cm−1 disappears but the band at 935 cm−1 decreases significantly due to that at lower pH, as can be seen in Fig. 2c. In the presence of HF, the decaniobate phase dissociates rapidly, leaving niobic acid [23]. Other bands near 420 and 700 cm−1 are related to TMA+ ions [24].

Raman spectrum of a amorphous Nb2O5; b HPNb; c HPNb-HF
The X-ray diffraction patterns of HPNb and HPNb-HF are shown in Fig. 3. The powder XRD pattern of HPNb agrees well with that of (TMA)6[Nb10O28].6H2O reported previously [1]. The X-ray diffraction pattern reported by Ohlin et al. [1] was initially considered (code CCDC 722216). Furthermore, the signals around 2θ = 10° and 20° are related to the reflection angles of the decaniobate phase, which is composed of six NbO6 octahedral systems in a 2 × 3 arrangement attached to two NbO6 octahedra above and two more sharing the edges below [25]. However, the signals at 2θ = 10.1° and 10.99° decrease when the decaniobate is prepared in the presence of HF. This confirms that in an acid medium, the decaniobate structure is unstable, which both promotes the dissociation towards niobic acid and loss of the decaniobate crystalline phase.

X-ray diffraction patterns. a HPNb (Theoretical); b HPNb-HF, c HPNb
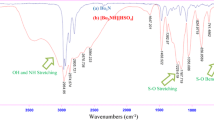
Figure 4 shows the FTIR spectra of HPNb and HPNb-HF. The FTIR bands at 1000 and 1400 cm−1 are characteristic of CH2 and CH3 stretching bending of TMA+, while the bands at 1000 and 911 cm−1 correspond to Nb=O vibrations, partly overlapped with the band of TMA ion [26]. The bands at 826 and 750 cm−1 were assigned to Nb–O asymmetric stretching. Two symmetric stretching bands of Nb–O bond appear at 587 and 546 cm−1 [26], which are indistinguishable in Fig. 4. However, a shift to higher frequencies is evidenced, which is related to the large cluster size, suggesting an increase in the number of Nb–O coordination species [27].

FTIR spectra of a HPNb; b HPNb-HF
In our spectrum a signal appears at 1177 cm−1, which was not detected by Ohlin et al. [26]. In order to clarify this point, various experiments were done. At the beginning, the catalyst was rigorously washed with water (more than eight times) in order to remove traces of TMAOH. The absence of TMAOH was evaluated by FTIR (Fig. S2). Although there is a slight decrease in the intensity of the bands between 1000 and 1400 cm−1, the band at 1177 cm− 1 is retained, which confirms the absence of TMAOH in the structure. Moreover, in comparison with the spectrum of TMAOH (source of TMA+), it was found that those bands belong to the TMA+ ion. However, the intensity of these bands in the spectrum of HPNb is much greater than in TMA-OH (Fig. S3), given that the TMA+ ions are present as stabilizing agents of the solid structure, thus discarding the presence of TMA-OH species, which can have an important catalytic effect.
Figure 5 displays the TGA curves of HPNb and HPNb-HF. In the decaniobate salt, the first signal in the range 80–150 °C can be attributed to loss of physically adsorbed water, while the second signal between 210 and 380 °C can be associated with loss of TMA+ ligands. However, given the low concentration of the decaniobate phase, this weight loss is less evident for the HPNb-HF solid. Finally, the third mass loss is associated with decomposition, which occurs as a consequence of the collapse of decaniobate structure and its transformation to oxide Nb2O5 at a temperature above 560 °C [28].

Thermogram of a HPNb, b HPNb-HF
TPD-CO2 studies were used to compare the basicity of decaniobate solids. Figure 6 depicts the temperature-programmed desorption spectra of two catalysts (HPNb and HPNb-HF). In the case of HPNb (Fig. 6a), the peak observed at around 300 °C could be assigned to TMA+ cations originating from the structure of decaniobate, in agreement with thermogravimetric analysis results. Indeed, the quantification of basicity strength using this technique is hard since at this temperature the loss of TMA+ also occurs. A new peak in the desorption patterns of HPNb appears at 380 °C, indicating a distinct behavior in comparison with HPNb due to the desorbed CO2.

TPD-CO2 profiles of a HPNb; b HPNb-HF
As a comparative method, the basicity of the solids was determined by the volumetric titration method with benzoic acid. It was found that the basicity of the decaniobate HPNb (0.12 mmol of benzoic acid/g) is three times greater than that of the HPNb-HF (0.04 mmol of benzoic acid/g), which confirms the low concentration of decaniobate phase.
To optimize the reaction temperature in the microwave equipment, three temperatures (60, 80, 120 °C) were tested in the reaction model between malononitrile, ethyl acetoacetate benzaldehyde and HPNb. At lower temperature (60 °C), the reaction does not yield 4H-pyran but only the Knoevenagel intermediate, while at 120 °C the reaction yields other unidentified products. Thus, the chosen temperature was 80 °C. Table 1 summarizes the catalytic activity results of the basic solid catalysts to obtain 4H-pyrans using a solvent-free reaction medium and microwave radiation heating at 80 °C. In all cases, the desired products were obtained with a selectivity of 100%, without side products and recovering, almost quantitatively, the unchanged starting materials. The selectivity was 100%. No side products were detected (Table 1).
No remarkable differences were attained using HPNb or HPNb-HF in the synthesis of ethyl-6-amino-5-cyano-2-methyl-4-phenyl-4H-pyran-3-carboxylate (yield: 96%, selectivity: 100%), indicating that the reaction occurs regardless of the amount of decaniobate phase, since a low basicity is enough to promote the reaction. In the absence of catalyst at 80 °C, only the product of condensation between benzaldehyde and malononitrile (Knoevenagel intermediate) was obtained (Table 2).
Several aldehydes with different substituents were tested with HPNb, and the results (entries 2–14) show that aldehydes exhibiting electron-withdrawing substituents favor shorter reaction times (entries 2, 3, 7 and 10) compared to the aldehydes having electron donor groups [29] (entries 4, 5, 6, and 9). Although the reaction systems show very similar yields, it is well known that the reduction of the reaction time is a parameter of great importance in organic synthesis. This protocol offers several advantages, such as using a reusable and efficient heterogeneous basic catalyst.
The reuse of the catalyst was evaluated in four cycles of reaction using HPNb. For this purpose, after completion of the reaction, the resulting mixture was stirred with acetone and centrifuged for recovering the insoluble catalyst, which was subsequently dried under vacuum and reused under the same procedure as described before. The catalyst was easily isolated from the reaction mixture due to the insolubility of the decaniobate salt in acetone and dichloromethane. This solid was characterized by FTIR (Fig. S1). It can be seen that after the reuse experiments the most characteristic signals of the decaniobate are preserved but in minor intensity, which is possibly due to the adsorption of the substrate on the catalyst surface.
Conclusion
A new application of decaniobates in the synthesis of 4H-pyran derivatives was studied. Although the synthesis of decaniobate modified with HF decreases the formation of hexaniobate phases, it is not convenient since the decaniobate phase dissociates rapidly to niobic acid. However, the reaction to afford 4H-pyrans was successfully promoted regardless of the type of solid employed and it was favored under microwave irradiation with short reaction times (1 h) and under solvent-free conditions. After completion of the reaction, the recovered catalyst was reused, and it retained its initial catalytic activity after four cycles.
References
C.A. Ohlin, E.M. Villa, W.H. Casey, Inorg. Chim. Acta. 362, 1391 (2009)
Y. Ye, C. Chen, H. Feng, J. Zhou, J. Ma, J. Chen, Open J. Inorg. Chem. 3, 59 (2013)
L. Shen, Y.-Q. Xu, Y.-Z. Gao, F.-Y. Cui, C.-W. Hu, J. Mol. Struct. 934, 37 (2009)
P. Huang, C. Qin, Z.-M. Su, Y. Xing, X.-L. Wang, K.-Z. Shao, J. Am. Chem. Soc. 134, 14004 (2012)
M. Nyman, Dalton Trans. 40, 8049 (2011)
S. Hayashi, S. Yamazoe, K. Koyasu, T. Tsukuda, RSC Adv. 6, 16239 (2016)
S. Hayashi, S. Yamazoe, K. Koyasu, T. Tsukuda, Chem. An Asian J. 12, 1635 (2017)
R. Pagadala, S. Maddila, S. Jonnalagadda, J. Heterocycl. Chem. 52, 1226 (2015)
H. Valizadeh, A. Azimi, J. Iran. Chem. Soc. 8, 123 (2011)
R.M.N. Kalla, M.R. Kim, I. Kim, Tetrahedron Lett. 56, 717 (2015)
G.P. Lu, C. Cai, J. Heterocycl. Chem. 48, 124 (2011)
S. Balalaie, S. Ramezanpour, M. Bararjanian, J.H. Gross, Catal. Commun. 38, 1078 (2008)
S. Sadjadi, M.M. Heravi, V. Zadsirjan, V. Farzaneh, Appl. Surf. Sci. 426, 881 (2017)
E. Nope, J.J. Martínez, H.A. Rojas, Á.G. Sathicq, G.P. Romanelli, Res. Chem. Intermed. 43, 2103 (2017)
E. Ruijter, R. Scheffelaar, R.V. Orru, Angew Chem. Int. Ed. 50, 6234 (2011)
L.M. Sanchez, H.J. Thomas, G.P. Romanelli, Mini Rev. Org. Chem. 12, 115 (2015)
N.R. Guha, D. Bhattacherjee, P. Das, Tetrahedron Lett. 55, 2912 (2014)
E.D.J.M. Prieto, B. Rivas, J. Sánchez, Cienc en Desarro. 4, 219 (2013)
K. Badamali, R. Luque, J.H. Clark, S.W. Breeden, Catal. Commun. 10, 1010 (2009)
R. Das, J. Ray, P. Pramanik, J. Mater. Res. 15, 2273 (2000)
K. Tanabe, T. Yamaguchi, J. Res. Inst. Catal. Hokkaido Univ. 11, 179 (1964)
P. Chagas, H.S. Oliveira, R. Mambrini, M. Le Hyaric, M.V. de Almeida, L.C. Oliveira, Appl. Catal. A Gen. 454, 88 (2013)
M. Aureliano, C.A. Ohlin, M.O. Vieira, M.P. Marques, W.H. Casey, L.A. de Carvalho, Dalton Trans. 45, 7391 (2016)
A.L. Shiguihara, M.A. Bizeto, V.R. Constantino, J. Braz. Chem Soc. 21, 1366 (2010)
E.J. Graeber, B. Morosin, Acta Crystallogr. Sect B. 33, 2137 (1977)
J.-H. Son, C.A. Ohlin, W.H. Casey, Dalton Trans. 42, 7529 (2013)
A. Fielicke, G. Meijer, G. Helden, J. Am. Chem. Soc. 125, 3659 (2003)
J. Niu, X. Fu, J. Zhao, S. Li, P. Ma, J. Wang, Cryst. Growth Des. 10, 3110 (2010)
S.W. Kshirsagar, N.R. Patil, S.D. Samant, Synth. Commun. 41, 1320 (2011)
Acknowledgements
We thank COLCIENCIAS for the financial support under Project No. 110965843004. EN, GPR and AGS thank to UNLP, CONICET, MYNCIT and ERANET-1.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gutierrez, L.F., Nope, E., Rojas, H.A. et al. New application of decaniobate salt as basic solid in the synthesis of 4H-pyrans by microwave assisted multicomponent reactions. Res Chem Intermed 44, 5559–5568 (2018). https://doi.org/10.1007/s11164-018-3440-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-018-3440-y