
Abstract
The synthesis of polymer supported zinc–salen complex (PS-Zn–salen) is described. The mononuclear zinc(II)–salen complex was characterized by Fourier-transform NMR spectroscopy, energy-dispersive X-ray spectroscopy, Fourier transform infrared spectrophotometry (FT-IR), thermogravimetric analysis, scanning electron microscopy, surface area and pore size distribution by Brunauer–Emmett–Teller. The synthesized PS-Zn–salen complex used as a recyclable heterogeneous catalyst for the efficient synthesis of hydantoins, thiohydantoins and Schiff bases in an aqueous medium. The isolated yields of hydantoins, thiohydantoins and Schiff bases achieved up to 89, 95 and 94%, respectively. In spite of conventional heterogeneous catalysts, current PS-Zn–salen complex shows thermal stability up to 280 °C. Moreover, the catalyst could be recovered easily by simple filtration and reused for next run with slightly declining its activity up to six successive runs. The FT-IR spectrum of recycle catalyst after 6th run confirmed that the catalyst was stable during the course of a reaction. The leaching of metal from the PS-Zn–salen is negligible, which was confirmed by AAS and hot filtration test.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The compound hydantoin, or imidazolidine-2,4-dione, was first separated by Nobel laureate Adolph von Baeyer in 1861 from the hydrogenolysis of allantoin and several acyclic structures [1]. Hydantoins have several uses in current organic synthesis and much important analogue in a number of therapeutic agents, which have an extensive array of biological activities [2]. The most conversant medicinal application of a hydantoin is as the drug phenytoin, which has a controlling effect on the central nervous system (CNS), and has been used by people with epilepsy as an anticonvulsant to prevent psychomotor epilepsy for more than 60 years. Merrit and Putnam first recognized that phenytoin was active against induced seizures in cats, subsequently becoming accessible for medical use and has been functional as the foremost drug for the treatment of tonic–clonic seizures [2, 3]. Additionally, hydantoin derivatives have synthetic value, e.g. as precursors to α-amino acid and pyruvic acid derivatives [4,5,6]. Also, it has been observed primarily that these are undesired by-products in the production of peptides [7, 8]. The fosphenytoin is a sodium channel antagonist and nilutamide, an androgen antagonist, used in the treatment of epilepsy and anticancer therapy, respectively [9].
Thiohydantoins are closely associated analogues of hydantoins containing sulphur atom and have a large number of medicinal and industrial applications [10, 11]. There are various routes of synthesis for hydantoin and its derivatives, depending on the choice of starting materials [12, 13]. Usually, phenytoin and its derivatives are synthesized by the well-known Bucherer–Bergs reaction [14].
In the preceding decades, many homogeneous catalytic methodologies were described to obtain hydantoin and their derivatives. Ghandi [15] has demonstrated a facile and suitable synthetic route to obtain some asymmetrical 1,3-diarylsubstituted 4,5-dihydroxy-2-imidazolidinones by the reaction of aqueous glyoxal with N-heteroaryl-N′-phenylureas using formic acid. Here in the same year, a process for the synthesis of enantiomerically pure hydantoins from optically pure α-amino amides [16]. Another novel synthetic approach for the synthesis of 2-hydantoins has been developed by using unprotected amino acids under acylation conditions with potential applications towards the preparation of different heterocyclic compounds from peptides [17]. Under ambient condition, Chandrashekar et al. [18] attempted to synthesize the prodrug fosphenytoin sodium via imidate ester. Subsequently, several homogeneous approaches were established to synthesize hydantoin and its derivatives using aldehyde extracted from almond kernels [19], α1-adrenoceptor antagonists using 2-methoxyphenylpiperazine [20], treating methyl phenyl acetate with di-tert-butyldiaziridinone [21], eco-friendly dialkylphosphate-mediated solvent-free condition [22], various amine-alkyl terminal fragments at N1-position [23] and 2-thiohydantoin-asparagine from N-acetyl-2-thiohydantoin-asparagine [24]. Likewise, hybrids sandwiched between phenytoin and thiosemicarbazide, 1,3,4-oxadiazole, 1,3,4-thiadiazole or 1,2,4-triazole, have shown anticonvulsant activity [25]. Moreover, the different products of N-(2,4-dioxo 4,4-diphenyl imidazolidin-4-yl) benzamides capable of anti-HIV and antiviral agents were described [26]. Recently, 5-substituted-3-(alkoxycarbonyl)alkyl-hydantoin products were synthesized by mechanochemistry from amino esters or dipeptides, through one-pot or two-step cyclization reaction containing aminoacid unsymmetrical urea and carboxy-imidazolyl-dipeptide ester intermediates [27].
Furthermore, syntheses of hydantoin and thiohydantoin derivatives using microwave activation [28,29,30,31] and ultrasound-activation [32] have also been successfully reported. Conversely, the homogenous reactions employed for the preparation of hydantoins and thiohydantoins confines their practical application in product refining from the reaction mixture. In order to develop more environmentally benign methods for the synthesis of hydantoins, heterogeneous catalytic systems have become an active area of research in recent years. The synthesis of phenytoin (5,5-diphenylhydantoin) was carried out using MgAl calcined hydrotalcites under eco-friendly conditions with appreciable conversion (80–95%) and selectivity (90–95%) [33]. Safari et al. [34] incorporated magnetic Fe3O4 nanoparticles as a heterogeneous catalyst for the synthesis of 5,5-disubstituted hydantoins. Consecutively, the researchers have developed a simple magnetic Fe3O4–chitosan nanoparticles for the large scale preparation of 5,5-diphenylhydantoins and 5,5-diphenyl-2-thiohydantoins, with low catalyst loading, reusability, and ease of operation [35]. With the hope of meeting a few principles of green chemistry, the same group designed renewable hybrid Fe3O4-chitosan nanocatalysts to synthesize 5-substituted hydantoins with high yields in short duration [36].
Among different heterogeneous catalysts, Zn–salen complex has received a remarkable attentions due to their prospective use in various fields, such as enantioselective alkynylation of ketones [37], CO2 fixation with epoxides at atmospheric pressure [38], detection of H2O2 in cells [39], photo catalyst for the activation of molecular oxygen to degrade organic pollutants in aqueous solution under visible light irradiation (λ ≥ 420 nm) [40]. However, Zn–salen complexes were used in quantitative analysis of applications, viz. fluorometry, as solvent extraction reagents and as solid phase extraction sorbents [41].
From the literature survey, Co-salen complexes immobilized onto the HEMA-resin [42], polystyrene supported salen type bis-thiopseudourea Pd(II)-complex [43] and reusable salen Mn(III) complexes are reported [44] for the various applications, but there is no report on polymer supported zinc–salen complex (PS-Zn–salen) for the preparation of phenytoins. In our previous work, we exploited some polymer-supported benzimidazolium based ionic liquid for efficient adsorption of hexavalent chromium [45] and the PS-Zn–salen complexes for one pot oxidative esterification of aldehyde to carboxylic ester [46]. In the continuation of our work, we have revealed the facile access to polymer supported zinc–salen complex: Highly efficient heterogeneous catalyst for hydantoins, thiohydantoins and Schiff bases synthesis in aqueous medium.
Experimental
Materials
All chemicals were used reagent grade with the highest purity available. ZnCl2, 4-vinylbenzyl chloride, DMSO-d6, CDCl3 were purchased from Sigma Aldrich (India). 4-Hydroxybenzaldehyde, formaldehyde and other aldehydes, Azobisisobutyronitrile (AIBN) were obtained from Avra synthesis Pvt. Ltd. and ethanol from Changshu Yangyuan chemical China. Chloroform, acetonitrile, urea, thiourea, and potassium thiocyanate from SDFCL and THF procured from Spectrochem Pvt. Ltd. and used as received unless otherwise mentioned.
Characterization techniques
NMR spectra were recorded in DMSO-d6 and CDCl3 on a Bruker spectrometer operating at 400 MHz and chemical shifts are given in ppm downfield from TMS (δ = 0.00). Surface morphology and the elemental composition of the complex were investigated by scanning electron microscope (SEM) along with energy dispersive X-ray (EDX) spectroscopy (Carl Zeiss EVO/18SH, UK). An accelerating voltage of 10 kV was applied to obtain SEM images. The sample was coated with carbon tape, and gold particles were sputtered over the sample using a gold sputter coater, after which the polymeric material was directly analyzed using the scanning electron microscope. FT-IR spectra were obtained from IR Affinity Shimadzu using the KBr pellet method. Thermal stability of PS-Zn–salen was confirmed using TGA thermo analyser SII, 7200 (Seiko, Japan). Atomic absorption spectrometer (AA240, Varian, Victoria, Australia) was used to determine leaching Zn in reaction. Brunauer–Emmett–Teller (BET) surface area was measured at the temperature of 77 K liquid N2 using a Quantachrome NOVA-1000 instrument analyser. Specific surface area, average pore diameter and pore volume are measured using N2 adsorption–desorption isotherms. Before analysis, samples were degassed at 150 °C for 5 h under vacuum for the removal of moisture and unwanted adsorbents from the sample.
Synthesis and characterization of PS-Zn–salen complex
Synthesis of polyvinyl benzyl chloride (PVBC) 1a, poly 1-(4-sec-butyl)benzyl)-1H-benzo[d]imidazole, 1b and 5-(chloromethyl)-2-hydroxybenzaldehyde 1c
The synthesis of compounds 1a, 1b were carried out according to Khiratkar et al [47] and 1c according to Naik et al. [48], and the characterization data of the corresponding compounds match with the literature reports.
1a:
1H NMR (400 MHz, CDCl3) δ = 7.08 (Ar–H, br), 6.5 (Ar–H, br), 4.56 (CH2–Cl, br), 1.71 (br), 1.60 (br), 1.42 (br), 0.95 (br). 13C NMR (100 MHz, CDCl3) 145.56, 134.97, 128.57, 46.37, 40.36, 31.60, 25.28. FT-IR (KBr) in cm−1: 3024, 2922, 2848, 1610, 1510, 1442, 1221, 1265, 1109, 1018, 912, 823, 707, 669, 559.
1b:
1H NMR (400 MHz, CDCl3) δ = 7.99 (NCHN, br), 7.7 (Bim–H, br), 7.19 (Bim–H, br), 6.6 (Ar–H, br), 6.17 (Ar–H, br), 4.95 (Bim–CH2–Ar, br), 1.84 (br), 1.48 (br), 1.25 (br), 0.84 (br). 13C NMR (100 MHz, CDCl3) 143.85, 143.09, 133.95, 128.03, 127.27, 122.97, 122.35, 120.37, 110.06, 67.99, 48.33, 29.71, 25.62. FT-IR (KBr) in cm−1: 2918, 1492, 1363, 1330, 1259, 1197, 1178, 844, 817, 738, 634, 574.
1c:
1H NMR (400 MHz, CDCl3) δ = 11.08 (s, 1H, Ph-CHO), 9.90 (s, 1H, Ph–OH), 7.6–7.55 (m, 2H, arom.), 7.02–7.00 (d, J = 8.89 Hz, 1H, CH arom), 4.6 (s, 2H, –CH2–).13C NMR (100 MHz, CDCl3) 196.19, 161.64, 137.64, 133.64, 118.33, 45.24. FT-IR (KBr) in cm−1: 3203, 3243, 2875, 1647, 1579, 1481, 1379, 1259, 1186, 1145, 1080, 991, 876, 766, 717, 667, 572, 497.
Synthesis of 1-(4-(sec-butyl)benzyl)-3-(3-formyl-4-hydroxybenzyl)-1H-benzo[d]imidazol-3-ium chloride (IL) 1d
A mixture of poly 1-(4-sec-butyl)benzyl)-1H-benzo[d]imidazole (20 mmol, 5.28 gm) and 5-(chloromethyl)-2-hydroxybenzaldehyde (20 mmol, 3.4 gm) was put into a 100-mL round bottom flask in acetonitrile medium. The mixture was stirred at room temperature for 24 h. The precipitated compound was then washed with diethyl ether to obtain white powder of 1d. Yield: 68%.
1H NMR (400 MHz, DMSO-d6) δ = 10.25 (CH=O, s), 8.4 (C–NH–C, s), 8.1–7.5 (Bim H, br), 7.2–6.2 (Ar, br), 5.8 (CH2, br), 5.5 (CH2, br), 2, 1.23, 1.09, 0.85 (aliphatic proton). FT-IR (KBr) in cm−1: 3396, 2926, 1614, 1556, 1487, 1442, 1371, 1282, 1249, 1188, 744, 632.
Synthesis of 3,3′-((((1E,1′E)-(ethane-1,2-dylbis(azanylylidene))bis(methanylylidene))bis(4-hydroxy-3,1-phenylene))bis (methylene))bis(1-(4-(sec-butyl)benzyl)-1H-benzo[d]imidazol-3-ium) chlorideie [salen ligand] 1e
The salen ligand was prepared by slightly changing the procedure from Morris et al. [49]. A solution of 1, 2-diethyl amine (7.5 mmol, 0.45 gm) and IL (1d) (15 mmol, 6.51 gm) in ethanol was stirred for 30 min at room temperature and refluxed for 2 h. The ligand was isolated by filtration and purified by repeated washing with ethanol to remove unreacted amine. The product was then dried under vacuum to acquire the yellow powder of salen ligand 1e.
1H NMR (400 MHz, DMSO-d6) δ = 13.18 (OH, br), 10.26 (CH=O, s), 8.5 (C–NH–C, s), 8.4–7.5 (Bim H, br), 7.2–6.8 (Ar, br), 5.6 (CH2, br), 5.5 (CH2, br), 3.5, 2, 1.23, 1.09, 0.85 (aliphatic proton). FT-IR (KBr) in cm−1: 3354, 3053, 3024, 2968, 2920, 2850, 1631, 1558, 1494, 1423, 1369, 1334, 1284, 1188, 742, 634, 574.
Synthesis of Zn–salen complex 1f
The Zn–salen complex was prepared by slight modification of the method of Morris et al. [49]. Initially salen ligand 1e (1 mmol, 0.892 g) in 10 mL ethanol was put into a 100-mL flask followed by drop-wise charging of ZnCl2 (1 mmol, 0.136 g) carried out at room temperature for 1 h. The mixture was then refluxed at 75–80 °C for 2 h. The product was easily separated after the reaction. The product was repeatedly washed with ethanol and dried in a hot air oven to get a yellow fine powder of Zn–salen complex 1f (Fig. 1).

Preparation of polymer supported Zinc-centered salen complex (1f)
General procedure for synthesis of Aryl thiourea derivatives 2a–2f
The library of aryl thiourea derivatives 2a–2f was prepared according to the literature [50], and the products were confirmed by FT-IR analysis (Supplementary data).
General procedure for the synthesis of hydantoin and thiohydantoin derivatives (3a–j) catalyzed by (1f)
Slightly altering the process from the literature for preparation of hydantoin and thiohydantoin derivatives was achieved by refluxing a mixture of benzil (1 mmol, 210 mg), urea or thiourea derivative (1.5 mmol), in aqueous ethanol (H2O/EtOH, 7:3) in the presence of 10 mg catalyst at 65 °C for 4 h. The reaction progress was monitored by TLC (petroleum ether:ethyl acetate, 7:3 v/v). After the completion of the reaction, the heterogeneous catalyst was separated by filtration and the filtrate was acidified with concentrated HCl to precipitate the desired product. The resulting solids were recrystallized from aqueous ethanol to give pure products [35].
(3a) 5,5-Diphenylimidazolidine-2,4-dione
(Yield = 65%); 1H NMR (400 MHz, DMSO-d6) δ = 11.06 (s, 1H), 9.27 (s, 1H), 7.41–7.31(m, 10H). 13C NMR (100 MHz, DMSO-d6) 174.70, 155.86, 139.84, 128.39, 127.91, 126.48, 70.12. FT-IR (KBr) in cm−1: NH (3265, 3194), C=O (1770, 1710). GC–MS m/z: calcd. for C15H12N2O2: 252.0899, found 252.1824.
(3b) 1-Methyl-5,5-diphenylimidazolidine-2,4-dione
(Yield = 51%); 1H NMR (400 MHz, DMSO-d6) δ = 9.64 (s, 1H), 7.40–7.35 (m, 10H), 2.94 (s, 3H). 13C NMR (100 MHz, DMSO-d6) 173.28, 155.49, 139.62, 128.50, 128.12, 126.64, 69.18, 24.49. FT-IR (KBr) in cm−1=NH (3280), C=O (1770, 1695).
(3c) 1,3-Dimethyl-5,5-diphenylimidazolidine-2,4-dione
(Yield = 89%); 1H NMR (400 MHz, DMSO-d6) δ = 7.02–6.8 (m, 10H), 6.5 (s, 6H). 13C NMR (100 MHz, DMSO-d6) 169.28, 138.02, 127.23, 127.20, 126.91, 92.28, 25.65,FT-IR (KBr) in cm−1=C=O (1739), C=S (1178).
(3d) 5,5-Diphenyl-2-thioxoimidazolidin-4-one
(Yield = 95%); 1H NMR (400 MHz, DMSO-d6) δ = 12.13 (s, 1H), 11.30 (s, 1H), 7.42–7.31 (m, 10H). 13C NMR (100 MHz, DMSO-d6) 181.32, 175.15, 138.35, 128.72, 128.39, 126.55, 72.95. FT-IR (KBr) in cm−1: NH (3251, 3155), C=O (1745, 1722), C=S (1155). GC–MS m/z: calcd. for C15H12N2OS: 268.0670 and found 268.1799.
(3e) 1,5,5-Triphenyl-2-thioxoimidazolidin-4-one
(Yield = 83%); 1H NMR (400 MHz, DMSO-d6) δ = 11.92 (s, 1H), 7.52–7.42 (m, 13H), 7.34 (d, J = 9.72 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) 181.01, 173.13, 138.13, 134.94, 128.98, 128.91, 128.85, 128.66, 126.76, 71.86. FT-IR (KBr) in cm−1=NH (3294), C=O (1737), C=S (1172).
(3f) 1-(4-Chlorophenyl)-5,5-diphenyl-2-thioxoimidazolidin-4-one
(Yield = 76%); 1H NMR (400 MHz, DMSO-d6) δ = 12 (s, 1H), 7.59 (d, J = 8.63 Hz, 2H), 7.50–7.40 (m, 12H). 13C NMR (100 MHz, DMSO-d6) 180.61, 172.94, 138.01, 133.94, 131.74, 130.78, 128.90, 128.70, 129.79, 71.96. FT-IR (KBr) in cm−1=NH (3319), C=O (1735), C=S (1176).
(3g) 5,5-Diphenyl-2-thioxo-1-(p-tolyl)imidazolidin-4-one
(Yield = 39%); 1H NMR (400 MHz, DMSO-d6) δ = 11.89 (s, 1H), 7.50–7.41 (m, 10H), 7.32 (d, J = 8.51 Hz, 2H), 7.20 (d, J = 8.51 Hz, 2H), 2.36 (s, 3H). 13C NMR (100 MHz, DMSO-d6) 181.19, 173.18, 138.64, 138.17, 138, 129.48, 128.90, 126.75, 71, 20. FT-IR (KBr) in cm−1=NH (3153), C=O (1747), C=S (1172).
(3h) 1-(4-Fluorophenyl)-5,5-diphenyl-2-thioxoimidazolidin-4-one
(Yield = 88%); 1H NMR (400 MHz, DMSO-d6) δ = 11.95 (s, 1H), 7.50–7.40 (m, 12H), 7.37 (t, J = 8.8 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) 180.91, 173.09, 163.17, 160.72, 138.07, 131.31, 128.89, 126.70, 116.02, 71.88. FT-IR (KBr) in cm−1=NH (3221), C=O (1734), C=S (1153).
(3i) 1-(4-bromophenyl)-5,5-diphenyl-2-thioxoimidazolidin-4-one
(Yield = 74%); 1H NMR (400 MHz, DMSO-d6) δ = 11.97 (s, 1H), 7.73 (d, J = 8.74 Hz, 2H), 7.50–7.40 (m, 10H), 7.35 (d, J = 8.74 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) 180.53, 172.87, 137.99, 132.18, 131.99, 131.07, 128.90, 128.69, 126.77, 122.27, 71.96. FT-IR (KBr) in cm−1=NH (3315), C=O (1735), C=S (1174).
(3j) 1-(naphthalen-1-yl)-5,5-diphenyl-2-thioxoimidazolidin-4-one
(Yield = 54%) 1H NMR (400 MHz, DMSO-d6) δ = 12.10 (s, 1H), 8.12–8.06 (m, 2H), 7.68 (d, J = 8.07 Hz, 1H), 7.58–7.44 (m, 13H), 7.29 (d, J = 8.51 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) 181.38, 173.45, 138.33, 138.09, 133.77, 129.96, 129.78, 129.36, 129.11, 128.73, 128.34, 127.36, 126.91, 126.56, 125.67, 121.26, 72.44. FT-IR (KBr) in cm−1=NH (3151), C=O (1762), C=S (1178).
General procedure for synthesis of Schiff bases 4a–4i catalyzed by (1f)
Varying the procedure from the literature, 10 mg catalyst was added to a solution of ethylenediamine (1 mmol, 60 mg) and 2 mmol of benzaldehyde derivative in 10 mL distilled water and refluxed at 80 °C for 2 h. The reaction mixture was poured onto ice, and the resulting precipitate was dissolved in ethyl acetate and the catalyst was separated by filtration. Evaporate off EtOAc to get Schiff bases, which can be further recrystallized using ethanol [51].
(4a) 2,2′-((1E,1′E)-(Ethane-1,2-diylbis(azanylylidene))bis (methanylylidene))diphenol
(Yield = 79%); 1H NMR (400 MHz, CDCl3) δ = 13.19 (s, 2H), 8.35 (s, 2H), 7.30–7.21 (m, 4H), 6.94 (d, J = 8.35 Hz, 2H), 6.8 (t, J = 7.75 Hz, 2H), 3.93 (S, 4H). 13C NMR (100 MHz, CDCl3) 166.52, 161.02, 132.41, 131.50, 118.69, 118.65, 116.97, 59.77. GC–MS m/z: calcd. for C16H16N2O2: 268.1212 and found 268.2494.
(4b) 3,3′-((1E,1′E)-(Ethane-1,2-diylbis(azanylylidene))bis (methanylylidene))diphenol
(Yield = 86%); 1H NMR (400 MHz, DMSO-d6) δ = 9.4 (s, 2H), 8.23 (s, 2H), 7.22 (t, J = 7.6 Hz, 2H), 7.14 (s, 2H), 7.10 (d, J = 7.34 Hz, 2H), 6.8 (d, J = 8.09 Hz, 2H), 3.82 (s, 4H). 13C NMR (100 MHz, DMSO-d6) 161.89, 157.54, 137.44, 129.59, 119.28, 117.79, 113.64, 60.87.
(4c) (1E,1′E)-N,N′-(Ethane-1,2-diyl)bis(1-p-tolylmethanimine)
(Yield = 70%);1H NMR (400 MHz, CDCl3) δ = 8.24 (s, 2H), 7.59 (d, J = 7.31 Hz, 4H), 7.19 (d, J = 7.63 Hz, 4H), 3.94 (s, 4H), 2.36 (s, 6H). 13C NMR (100 MHz, CDCl3) 162.84, 141.12, 133.92, 129.56, 128.36, 62, 21.76.
(4d) (1E,1′E)-N,N′-(Ethane-1,2-diyl)bis(1-(4-methoxyphenyl) methanimine)
(Yield = 70%); 1H NMR (400 MHz, CDCl3) δ = 8.20 (s, 2H), 7.64 (d, J = 8.56 Hz, 4H), 6.90 (d, J = 8.53 Hz, 4H), 3.91 (t, 4H), 3.82 (s, 4H). 13C NMR (100 MHz, CDCl3) 162.03, 161.71, 129.76, 129.35, 114.07, 61.86, 55.47.
(4e) (1E,1′E)-N,N′-(Ethane-1,2-diyl)bis(1-(3,4,5-trimethoxy-phenyl)methanimine)
(Yield = 88%); 1H NMR (400 MHz, CDCl3) δ = 8.29 (s, 2H), 7.07 (s, 4H), 4.07 (s, 4H), 4.00 (s, 12H), 3.9 (s, 6H). 13C NMR (100 MHz, CDCl3) 162.21, 153.34, 140.30, 131.61, 105.08, 61.44, 60.82, 56.12.
(4f) 5,5′-((1E,1′E)-(Ethane-1,2-diylbis(azanylylidene))bis(methanylylidene))bis(2 ethoxyphenol)
(Yield = 78%); 1H NMR (400 MHz, DMSO-d6) δ = 9.14 (s, 2H), 8.16 (s, 2H), 7.22 (s, 2H), 7.07 (d, J = 8.30 Hz, 2H), 6.95 (d, J = 7.92 Hz, 2H), 3.79 (s, 6H), 3.33 (t, 4H). 13C NMR (100 MHz, DMSO-d6) 172.55, 184.28, 134.55, 134.35, 133.22, 26.33.
(4g) (1E,1′E)-N,N′-(Ethane-1,2-diyl)bis(1-(4-bromophenyl) methanimine)
(Yield = 92%); 1H NMR (400 MHz, CDCl3) δ = 8.20 (s, 2H), 7.56 (d, J = 8.35 Hz, 4H), 7.52 (d, J = 8.35 Hz, 4H), 3.9 (s, 4H). 13C NMR (100 MHz, CDCl3) 161.56, 135.11, 131.95, 129.62, 125.21, 61.55.
(4h) (1E,1′E)-N,N′-(Ethane-1,2-diyl)bis(1-(4-chlorophenyl) methanimine)
(Yield = 76%); H NMR (400 MHz, CDCl3) δ = 8.22 (s, 2H), 7.62 (d, J = 7.70 Hz, 4H), 7.36 (d, 7.70 Hz, 4H), 3.95 (s, 4H). 13C NMR (100 MHz, CDCl3) 161.62, 136.95, 134.90, 129.18, 61.75.
(4i) (1E,1′E)-N,N′-(Ethane-1,2-diyl)bis(1-(4-nitrophenyl) methanimine)
(Yield = 75%) 1H NMR (400 MHz, CDCl3) δ = 8.37 (s, 2H), 8.25 (d, J = 7.62 Hz, 4H), 7.87 (d, J = 7.64 Hz, 4H), 4.06 (s, 4H). 13C NMR (100 MHz, CDCl3) 160.66, 149.40, 141.72, 129.09, 124.20, 61.78, 30.04.
Results and discussion
Catalyst characterization
Fourier transform infrared (FT-IR) spectroscopy
The FT-IR spectra of the IL (1d), salen ligand (1e) are compared with that of Zn–salen complex (1f) (Fig. 2) to define the coordination sites that may be involved in chelation. The most significant IR spectral bands in (1d) and (1e) for (O–H) appeared at 3396 cm−1 and 3354 cm−1, respectively. Similarly, stretching bands observed at 1656 cm−1 is due to (aldehyde C=O) in IL (1d) and 1631 cm−1 for (C=N) in the ligand (1e). The formation of Schiff base Zn(II)-complex is evidenced by the disappearance of (O–H) stretching band in (1f). Conclusive evidence of the bonding is also shown by the appearance of the new band in the region (532 and 416 cm−1) in the spectrum of the complex due to (Zn–O) and (Zn–N) stretching vibrations. For the Schiff base, the (C=N–) stretching of salen ligand (1e) is observed as a strong band at 1631 cm−1. This band undergoes a small shift on complexation (~ 9); such a small shift has been observed at 1622 cm−1 (1f), which revealed that azomethine nitrogen is involved in coordination to the metal [52,53,54].

FT-IR spectrum of 1d, 1e and 1f
Scanning electron microscopy (SEM) images and energy dispersive X-ray analysis (EDX)
The surface morphology and elemental analysis of polymer supported Zn–salen complex was characterized by SEM/EDX as shown in Fig. 3. The scanning electron micrograph (SEM) images were reported for polymer supported salen ligand (A) and polymer supported Zn–salen complex (C). The polymer supported Zn–salen complex showed morphological changes (A and C) that indicate the formation of the desired complex. The surface of polymer supported Zn–salen complex reveal micron sized particles deposited over the surface (Fig. 3c). The comparative spectrum of energy-dispersive X-ray spectroscopy, ligand (B) and a metal complex (D) confirmed the presence of Zn in the metal complex (D).

SEM/EDX of polymer supported salen ligand (a/b) and polymer supported Zn–salen complex (1f) (c/d)
Thermal analysis
Thermal stability of PS-salen (1e) and PS-Zn–salen (1f) were studied using TGA under a nitrogen atmosphere with a heating rate of 10 °C min−1 over a temperature range of 30–800 °C (Fig. 4). There is negligible weight loss in 1f (4.250%) compared to 1e (8.5%) below 120 °C indicating the presence of physically adsorbed solvent. Weight loss of 62.56% 1e, 36.48% 1f in the region of 235–470 and 280–550, respectively, may be due to a salen group of the ligand and the complex begins to crumble. The observations clearly indicate the stability of PS-Zn–salen is more compared to the PS-salen ligand. Further weight loss in both 1e and 1f perceived to decomposition of poly 1-(4-sec-butyl)benzyl)-1H-benzo[d] imidazole. From TGA analysis it is observed that the weight loss is negligible up to 280 °C in 1f and is found to be thermally stable; therefore, it is concluded that the polymer supported Zn–salen complex (1f) catalyst is suitable for high-temperature reaction.

Thermogravimetric analysis (TGA) thermogram of PS-salen (1e) and PS-Zn–salen (1f)
BET analysis
Specific surface area and pore size distribution of the Zn–salen complex was measured using adsorption and desorption isotherm of nitrogen molecules. The BET specific surface area of synthesized Zn–salen complex was found to be 4.783 m2/g (Fig. 5). The total pore volume and average pore diameter of Zn–salen complex was 0.015 cc/g and 1.562 nm, respectively (Fig. 6).

BET isotherm of PS-Zn–salen

Pore diameter and pore volume of PS-Zn–salen
Catalytic activity of polymer supported Zn–salen complex
Owing to our curiosity in the progress of more ecological catalytic reaction conditions and our interest in the green chemistry of ILs [55, 56], we report here new results under heterogeneous catalysis to support the formation of hydantoin/thiohydantoin/Schiff bases in an aqueous medium. To begin our study, we examined the synthesis of hydantoins and thiohydantoins using benzil and urea/thiouera derivatives (Scheme 1) and Schiff bases (Scheme 2) by reacting ethylenediamine with aromatic aldehydes in the presence of polymer supported Zn–salen complex under a variety of reaction conditions. The results are summarized in Tables 1 and 2 showing the optimization of the amount of catalyst, temperature, time on the synthesis of 5,5-diphenyl-2-hydantoin (Scheme 3) and Schiff bases, respectively (Scheme 4).

Synthesis of 5,5-diphenylhydantoins and 5,5-diphenyl-2-thiohyd-antoins

Synthesis of Schiff bases

Synthesis of 5,5-diphenyl-2-hydantoin

Synthesis of Schiff base
Initially, in order to optimize the reaction parameters such as amount of catalyst, temperature and time, we carried out the model reaction for the synthesis of 5,5-diphenyl-2-hydantoin by reacting benzil with urea under different conditions shown in Table 1. A control reaction was attempted to test the necessity of a catalyst by stirring the reagents in the absence of the catalyst. It is worth emphasizing that reactions accomplished 65% yield in the presence of catalyst, but in the absence of catalyst, gave poor results (yield 20%; Table 1, entry 1). Likewise, the reaction carried out with ligand (1e) does not show much progress in the product (Table 1, entry 2). The amount of catalyst loading was tested on the model reaction (Table 1, entries 3–6). The optimal amount of the catalyst was 10 mg (Table 1, entry 4). However, it is exciting to note that there is no change in yields irrespective of loading more amount of the catalyst (Table 1, entries 5–6). Thus, it was evident that the amount of catalyst plays an important role in the formation of products.
With the optimized amount of catalyst (10 mg) in hand, we examined reaction (Scheme 3) using benzil and urea at different temperatures and time in aqueous ethanol (30%). We also investigated the reaction using the ratio of benzil to urea 1.0:1.5 between the reagents toward a more efficient and greener process to yield the corresponding 5,5-diphenyl-2-hydantoin (65% yield) at 65 °C Table 3. The yield improved as the reaction temperature was raised from room temperature to 65 °C. At 65 °C, moderate yield (65%) of product was obtained in moderate yield of 65% in 240 min (Table 1, entries 7–10). A further increase in temperature did not improve the product yield, but somewhat decreased it (Table 1, entries 11 and 12). It is worth mentioning that the catalytic activity enhances with increasing duration of the reaction from 60 min to 240 min (Table 1, entries 13–16) but remained almost the same with a variation of time from 240 to 300 min (Table 1, entry 17). Thus the formation of hydantion proceeded smoothly in the presence of aqueous ethanol, 10 mg cat., and 65 °C in 240 min. and gave the desired product in admirable yield. Similarly optimization of all the parameters like amount of catalyst, temperature and time for the synthesis of Schiff bases catalyzed by 1f, were carried out and summarized in Table 2. Further, to examine the substrate choice of this reaction, urea with different substituents were used under the above-optimized conditions. From the results shown in Table 4, it is apparent that the reactions progressed effectively and gave the desired products in good to excellent yields in 240 min.
A variety of substituted urea and thiourea, with either electron-donating or electron-withdrawing groups, provided encouraging results in this reaction. For example, methyl and dimethyl urea afforded the corresponding hydantoin products in appreciable yields (Table 4, entries 2 and 3). Thiourea (Table 4, entry 4), phenyl (Table 4, entry 5) and halo phenyl sub-stituted thiourea were equally responsive to these conditions providing thiohydantoins in 74 to 88% yield (Table 4, entries 6, 8 and 9). P-tolylthiourea 1-naphthyl thiourea could also be achieved using this methodology, albeit in significantly lower yields indicating the admirable catalytic activity of catalyst (Table 4, entry 7 and 10). The effect of various electron-donating or electron-withdrawing groups on synthesis of Schiff base derivatives summarized in Table 5.
A plausible mechanism for the hydantoin, thiohydantoin catalyzed by PS-Zn–salen complex is shown in Scheme 5. Initially, (1f) as a Lewis acid, and OH group of the base through hydrogen bonding activated the carbonyl group of benzil for the nucleophilic substitution of urea that leads to the formation of corresponding intermediate (I). The ring closure of this intermediate, via the electrophilic nitrogen atom of urea offered an intermediate (II). The stability of the intermediate (II) controls the selectivity of the main product (hydantoin/thiohydantoin) in the presence of (1f). Furthermore, the catalyst prevents the reaction pathway (b) and follows the reaction pathway (a). In the presence of KOH as a catalyst, the pathway (b) might be possible [33]. Therefore, the selectivity of the major product depends on the competitive paths (a) and (b).

Possible reaction pathway for the synthesis of hydantoin/thiohydantoin
Stability and recyclability
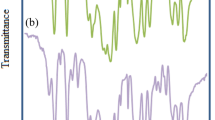
In continuation of our efforts to develop an efficient and environmentally benign protocol for the organic synthesis, our aim was to provide, improved, water-tolerant and recoverable catalyst. The recyclability of the catalyst was studied using the above model reaction. After separation of the product, by filtration, the catalyst was washed with ethanol and reused in the next run without further purification. A mixture of benzil and thiourea in (1:1.5) ratio was added to the aqueous ethanol (30%) and stirred under the same conditions. The data listed in (Fig. 7) indicated that the Zn–salen catalyst was successfully used in six successive runs and presented steady activity to afford an average yield of 88.5% with no significant loss of performance. Compared with the traditional solvents and catalysts, the easy reusability is also a prominent property of the complex for environmental protection and commercial reasons. High stability of complex 1f was further confirmed by using FT-IR and showed that the structure of the catalyst stayed intact during the catalysis even after six runs (Fig. 8). Therefore, the present PS-Zn–salen complex is very attractive for the synthesis of hydantoins, thiohydantoins and Schiff bases. Moreover yield obtained in an aqueous medium using 1f catalyst, towards condensation reaction is better than or analogous to the literature reports with respect to catalyst loading and duration of reaction (Table 6).

Recyclability of catalyst (1f) recovers up to 6th run

FT-IR spectrum of catalyst (1f) recovers up to 6th run
Leaching studies
To determine the degree of leaching of Zn from the heterogeneous catalyst, the catalyst was removed by filtration and the Zn content of the filtrate was determined by AAS. During the course of reactions, only 0.184 ppm of Zn was lost into solution after the first run. After the 6th recycle loss of 1.155 ppm of Zn was observed, which shows the average amount of leaching of Zn per cycle is about 0.192 ppm.
To define whether the catalyst is truly functioning as heterogeneous or if it is just a reservoir for more active soluble Zinc species, we have performed a hot filtration test [57]. This test includes filtering off the solid catalyst during some point of the reaction at the optimized temperature. The substrates (urea and benzil) allowed reacting at reaction temperature with Ps-Zn–salen for 1 h, filtering the catalyst and continued the reaction in the filtrate. We found that after hot filtration of the catalyst, there is no acceptable change in conversion of product. On the basis of these outcomes, we may conclude that the catalysis reaction is indeed heterogeneous in nature and the leached zinc species did not catalyzed the reaction.
Conclusions
In conclusion, we have successfully synthesized a new polymer supported Zn–salen (PS-Zn–salen) complex. The existence of organic functionalities in the complex was clearly quantified by Fourier transform infrared spectrophotometry (FT-IR), energy-dispersive X-ray spectroscopy (EDX), scanning electron microscopy (SEM) and Fourier transform NMR spectroscopy (FT-NMR). The heterogeneous catalyst shows high thermal stability up to 280 °C and has displayed admirable activity in the synthesis of hydantoins, thiohydantoins and Schiff bases in an aqueous medium. It could be reused six times with slightly decline the activity, representing a robust solid catalyst. The stability of the catalyst was further confirmed by FT-IR analysis, which revealed that the Zn–salen complex catalyzed the condensation reactions without deterioration of the ordered structure. The high yield of condensation compounds (up to 95%), the use of a relatively low catalyst, the easy work up, an environmentally benign protocol, mild reaction conditions, high atom economy, shorter reaction times are the advantages of this protocol over the other procedures reported. Other studies of its applicability in synthetic conversions are currently on-going in our laboratory.
References
M. Meusel, M. Gutschow, Org. Prep. Proced. Int. 36, 391 (2004)
E. Ware, Chem. Rev. 46, 403 (1950)
C.A. Lopez, G.G. Trigo, Adv. Heterocycl. Chem. 38, 177 (1985)
W.C. Groutas, M.A. Stanga, J.C. Castrisos, E.J. Schatz, J. Enzyme Inhib. 3, 237 (1990)
N.A. Meanwell, H.R. Roth, E.C.R. Smith, D.L. Wedding, J.J. Wright, J. Org. Chem. 56, 6897 (1991)
R. Sarges, P.J. Oates, Prog. Drug Res. 40, 99 (1993)
F. Albericio, G. Barany, Int. J. Peptide Protein Res. 30, 177 (1978)
N.O. Mahmoodi, Z. Khodaee, ARKIVOC iii, 29 (2007)
M. Lamothe, M. Lannuzel, M. Perez, J. Comb. Chem. 4, 73 (2002)
H.R. Henze, P.E. Smith, J. Am. Chem. Soc. 65, 1090 (1943)
H.C. Carrington, W.S. Waring, J. Chem. Soc. 1950, 354 (1950)
W.J. Brouillette, M.L. Brown, T.M. Delorey, G.J. Ling, J. Pharm. Sci. 79, 871 (1990)
A. Kiasat, M. Nikmanesh, IJHC 2, 20 (2012)
C. Montagne, M. Shipman, Synlett 14, 2203 (2006)
M. Ghandi, A. Olyaei, F. Salimi, Molecules 11, 768 (2006)
D. Zhang, X. Xing, G.D. Cuny, J. Org. Chem. 71, 1750 (2006)
S. Reyes, K. Burgess, J. Org. Chem. 71, 2507 (2006)
C.R. Elati, S. Gangula, A. Naredla, S. Ashok, A. Bhattacharya, R. Bandichhor, Synth. Commun. 38, 2950 (2008)
A. Ashnagar, N.G. Naseri, M. Amini, Int. J. Chem. Tech. Res. 1, 47 (2009)
J. Handzlik, D. Maciąg, M. Kubacka, S. Mogilski, B. Filipek, K. Stadnicka, K. Kiec-Kononowicz, Bioorg. Med. Chem. 16, 5982 (2008)
B. Zhao, H. Du, Y. Shi, J. Am. Chem. Soc. 130, 7220 (2008)
V. Kumar, H. Rana, R. Sankolli, M.P. Kaushik, Tetrahedron Lett. 52, 6148 (2011)
J. Handzlik, E. Szymanska, J. Chevalier, E. Otrebska, K. Kiec-Kononowicz, J.M. Pages, S. Alibert, Eur. J. Med. Chem. 46, 5807 (2011)
G.E. Delgado, K.N. Varela, R.V. Araque, J.A. Rodriguez, A.J. Mora, L.E. Seijas, Av. Quim. 9, 3 (2014)
S. Botros, N.A. Khalil, B.H. Naguib, Y. EI-Dash, Eur. J. Med. Chem. 60, 57 (2013)
B. Vinod, D. Selvakumar, BioMedRx 1, 363 (2013)
L. Konnert, L. Gonnet, I. Halasz, J.-S. Suppo, R. Marcia de Figueiredo, J.-M. Campagne, J.S. Suppo, R. Marcia de Figueiredo, J.M. Campagne, J. Org. Chem. 81, 9802 (2016)
S. Paul, M. Gupta, R. Gupta, A. Loupy, Thieme Stuttgart 1, 75 (2002)
G.G. Muccioli, J.H. Poupaert, J. Wouters, B. Norberg, W. Poppitz, G.K. Scriba, D.M. Lambert, Tetrahedron 59, 1301 (2003)
E. Gallienne, G.G. Muccioli, D.M. Lambert, M. Shipman, Tetrahedron Lett. 49, 6495 (2008)
A. Gbaguidi, S.S. Kpoviessi, C.N. Kap, G.G. Muccioli, D.M. Lambert, G.C. Accrombessi, M. Mansourou, J.H. Poupaert, AJPAC 5, 168 (2011)
N.M. Arani, J. Safari, Ultrason. Sonochem. 18, 640 (2011)
D. Sachdev, A. Dubey, Catal. Commun. 11, 1063 (2010)
J. Safari, L. Javadian, C. R. Chimie 16, 1165 (2013)
J. Safari, L. Javadian, RSC Adv. 4, 48973 (2014)
J. Safari, L. Javadian, IJC 6, 57 (2016)
P.G. Cozzi, Angew. Chem. 115, 3001 (2003)
X.D. Lang, Y.C. Yu, L.N. He, J. Mol. Catal. A Chem. 420, 208 (2016)
J. Jing, J.L. Zhang, Chem. Sci. 4, 2947 (2013)
T. Araya, S. Quan, J. Man-Ke, M. Wan-Hong, H. Johnson, H. Ying-Ping, Water Air Soil Pollut. 227, 284 (2016)
N.R. Bader, Rasayan J. Chem. 3, 660 (2010)
A. Bukowska, W. Bukowski, React. Funct. Polym. 68, 657 (2008)
S. Keesara, M.R. Mandapati, S. Parvathaneni, Appl. Catal. A 496, 58 (2015)
Y. Chen, R. Tan, Y. Zhang, G. Zhao, W. Zheng, R. Luo, D. Yin, Appl. Catal. A 491, 106 (2015)
A.G. Khiratkar, S.S. Kumear, P.R. Bhagat, RSC Adv. 6, 37757 (2016)
K.R. Balinge, A.G. Khiratkar, P.R. Bhagat, J. Mol. Liq. 242, 1085 (2017)
A.G. Khiratkar, P.N. Muskawar, P.R. Bhagat, RSC Adv. 6, 105087 (2016)
P.U. Naik, G.J. McManus, M.J. Zaworotko, R.D. Singer, Dalton Trans. 36, 4834 (2008)
G.A. Morris, H. Zhou, C.L. Stern, S.B.T. Nguyen, Inorg. Chem. 40, 3222 (2001)
S.R. Mathapathi, J.F. Sakhare, M.B. Swami, J.K. Dawle, Der Pharma Chemica 4, 2248 (2012)
A. Dhakshinamoorthy, K. Kanagaraj, K. Pitchumani, Tetrahedron Lett. 52, 69 (2011)
M. Ferguson, N. Giri, X. Huang, D. Apperley, S.L. James, Green Chem. 16, 1374 (2014)
D.N. Kumar, B.S. Garg, Spectrochim. Acta. A. 64, 141 (2006)
R.I. Kureshy, N.H. Khan, S.H.R. Abdi, S.T. Patel, P. Iyer, J. Mol. Catal. A Chem. 150, 163 (1999)
P.N. Muskawar, K. Thenmozhi, J.M. Gajbhiye, P.R. Bhagat, Appl. Catal. A 482, 214 (2014)
P.N. Muskawar, S.S. Kumar, P.R. Bhagat, Mol. Catal. A Chem. 380, 112 (2013)
D. Nakatake, R. Yazaki, Y. Matsushima, T. Ohshima, Adv. Synth. Catal. 358, 2569 (2016)
Acknowledgements
The authors thank VIT Vellore for providing “VIT SEED GRANT” for carrying out this research work and SIF DST-VIT-FIST for NMR, GC–MS, FT-IR and SEM/EDX VIT University, Vellore for providing necessary facility to bring this article to this level. We are also thankful to the Dean, School of Advanced Sciences, and all the Scholars of our Research Laboratory, “Smart Materials Laboratory for Bio-Sensing and Catalysis” VIT University Vellore, who have encouraged the authors during this period.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Balinge, K.R., Khiratkar, A.G., Muskawar, P.N. et al. Facile access to polymer supported zinc–salen complex: highly efficient heterogeneous catalyst for synthesizing hydantoins, thiohydantoins and Schiff bases in aqueous medium. Res Chem Intermed 44, 2075–2097 (2018). https://doi.org/10.1007/s11164-017-3215-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-017-3215-x