
Abstract
We prepared a number of heterogeneous catalysts by exchanging the structural iron of magnetite with molybdenum ions. To obtain the optimum value, Mo at various concentrations was coprecipitated with iron species (Fe3−x Mo x O4, x = 0.028, 0.069, 0.13, and 0.21). Characterization revealed that all the samples had inverse spinel structure with excellent stability and magnetic properties. Higher Mo contents (x = 0.13 and 0.21) significantly improved the specific surface area of magnetite, leading to higher capacity for methylene blue (MB) adsorption. The catalytic performance of the samples for degradation of MB solution through Fenton reaction was then assessed. The Fe2.62Mo0.21O4 sample showed substantial activity, removing MB completely within 150 min. This enhanced activity is discussed based on the enlarged surface area, the role of surface Mo4+/Mo6+ redox pairs, and oxygen vacancies. Kinetic studies revealed that MB degradation by Fe3−x Mo x O4 nanoparticles in presence of H2O2 was well fit by a zeroth-order kinetics model. These results support use of such Fe3−x Mo x O4 materials as active magnetically separable heterogeneous catalysts, capable of degrading various contaminants through Fenton reaction.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The textile industry is one of the most water-consuming industries and produces large quantities of wastewater at the end of the cycle, carrying a wide variety of chemicals, mostly dyes, that are considered recalcitrant due to their stable structure [1]. As a result, they are recognized as common environmental pollutants that must be properly treated before disposal [2]. Various treatment methods are available globally to minimize the impact of dyes on human health and the environment. Advanced oxidation processes (AOPs) are among the most effective treatment techniques for decomposition of organic pollutants by in situ-generated active species. The Fenton reaction is a well-known and frequently studied oxidation process, due to its simple equipment, ease of operation, and higher efficiency. In this reaction, highly active and nonselective hydroxyl radicals are produced from the breakdown of H2O2 by the catalytic action of iron (Eq. 1).
The Fenton process has been widely applied for treatment of various dye wastewaters at laboratory or pilot-plant scale [1]. This is especially reported for treatment of methylene blue (MB) solution, due to its wide applications and recalcitrant nature [3]. The degradation rate is high in the conventional Fenton reaction, using iron salts at acidic condition of pH 3.0, because of the availability of the reactants in the reaction solution. Furthermore, use of external energy sources such as UV light and/or ultrasound energy helps achieve higher efficiencies [4, 5]. However, the soluble iron is precipitated as ferric hydroxide at higher pH condition, resulting in catalyst withdrawal from the reaction medium [6], making catalyst recovery impracticable [7, 8]. In light of this, recent studies in this field have mainly focused on production of heterogeneous catalysts that can achieve higher efficacy in the Fenton reaction with stable structure, easy recovery, and reusability [9, 10]. Alteration of the properties of common iron minerals has also been examined using various routes to achieve the goals mentioned above. Increasing the specific surface area, improving the surface characteristics by introducing more active sites and species, and increasing the electron mobility in the catalyst structure are some of the various changes applied [7, 11].
Over the last decade, a tremendous number of publications have reported on synthesis and versatile applications of engineered magnetite nanoparticles (NPs). This has given rise to compounds with pronounced properties that are not observed in pure magnetite. Such engineered magnetite NPs have attracted extensive interest due to their applications in various fields of environmental analytical chemistry and catalysis as sensors, enzyme carriers, oxidants, activators for degradation of organic contaminants to purify water and wastewater, and for polymerization of organic compounds [12,13,14]. Currently, several methods such as coprecipitation, hydrothermal, thermal, emulsion, microbial, and green synthesis procedures are used to produce magnetite NPs with desired properties [15,16,17,18]. These materials can be easily separated from the reaction vessel by a magnet and used in several applications, in turn greatly reducing the cost of such processes. Due to the Fe2+ cations at octahedral sites in its structure, magnetite has been reported to be a more appropriate candidate for use in Fenton treatment systems compared with other iron oxides (Eq. 2) [19]. Recently, transition-metal-substituted magnetites have been developed as a class of heterogeneous catalysts with significantly enhanced activity for Fenton oxidation of organic pollutants. In such materials, exchange of structural iron of magnetite with other transition metal(s) results in more active sites, with higher electron mobility and H2O2 decomposition rate, and thereby improved oxidation of pollutants [7]. To date, transition metals from the fourth series of the Periodic Table have been widely studied, while studies on transition metals from other periods remain scarce. Hence, in our previous study [20], niobium-substituted magnetite was prepared and characterized and its Fenton activity comprehensively evaluated. In this study, molybdenum was chosen from the fifth period as well. From the isomorphic substitution viewpoint, Mo and iron species have similar ionic radius [Mo4+ (65 pm) and Mo6+ (59 pm) versus Fe2+ (61 pm) and Fe3+ (55 pm)]. Therefore, the structural iron in magnetite could, theoretically, be replaced by Mo. In this work, a series of Fe3−x Mo x O4 samples were synthesized by coprecipitation method and their main physicochemical properties determined. Subsequently, the adsorption capacity and Fenton catalytic activity of the samples for MB solution were evaluated under various operational conditions. Furthermore, the kinetics of MB degradation via Fenton reaction was identified. Finally, the stability of the samples was assessed at different pH conditions through a number of leaching experiments.
Experimental
Materials
Chemicals were of reagent grade, and solutions were prepared using deionized water. Hydrazine, iron(II) chloride tetrahydrate (FeCl2·4H2O), and molybdenum(V) chloride (MoCl5) were obtained from Sigma Aldrich. Hydrogen peroxide (H2O2, 30% w/w), methylene blue (MB), sodium hydroxide (NaOH), sodium chloride (NaCl), sulfuric acid (H2SO4), and hydrochloric acid (HCl) were purchased from Merck.
Synthesis of Fe3−x Mo x O4 nanoparticles
A modified method was used for synthesis of the samples [21]. First, oxygen was removed from the utilized water and solutions via nitrogen sparging. To synthesize magnetite, 0.90 mol L−1 FeCl2·4H2O was prepared in HCl solution, followed by addition of a small amount of hydrazine into the solution and setting the pH below 1.0, to avoid oxidation of Fe2+ cations. The solution was then heated up to 100 °C. A solution of the same volume comprising NaNO3 (0.90 mol L−1) and NaOH (4.0 mol L−1) was added dropwise into the heated iron mixture under mechanical stirring (500 rpm), and the procedure was continued for 2 h at 90 °C. Then, the solution was kept aside to return to room temperature. N2 gas flow was maintained during all procedures to inhibit oxidation of Fe2+ cations. Finally, the resulting black particles were centrifuged at 3500 rpm for about 5 min, then washed with boiling deionized (DI) water and again centrifuged for several times. The particles were collected after washing and dried in a vacuum oven at 100 °C for 24 h. The same procedure was followed for synthesis of Fe3−x Mo x O4 samples by dissolving a preset quantity of MoCl5 in HCl solution. Finally, the samples were ground and sieved using a 200-mesh screen.
Characterization
A PANalytical Empyrean (DY 1032, Westborough, MA) diffractometer with Cu Kα radiation (λ = 0.15418 nm) was employed to record XRD patterns from 2θ of 20° to 80° and determine the crystallite structure of the samples. The BET method (Micromeritics, TriStar II 3020) was used to measure the specific surface area, pore size, and pore volume of the samples. A PHI Quantera (II) with monochromatic Al Kα (1486.6 eV) source and spherical capacitor analyzer (SCA) was employed to explore the surface characteristics using X-ray photoelectron spectroscopy (XPS) of the samples. To calibrate the binding energies for the charge shift, the C 1s line of carbon located at binding energy of 284.8 eV was set as a reference. The morphology and particle size of the samples were determined from TEM images (FEI Tecnai G2). The magnetic characteristics of the samples were studied by vibrating-sample magnetometry (Lake Shore VSM, 7400 Series). The solid addition method was applied to detect the point of zero charge pH (pHPZC) of the synthesized samples. For this, a number of 250-mL conical flasks were filled with 45 mL NaCl solution (0.1 mol L−1) and their initial pH (pH0) was set from 4.0 to 11.0 using addition of either NaOH (0.1 mol L−1) or HCl (0.1 mol L−1). Then, a total volume of precisely 50 mL NaCl solution was obtained in each flask by adding NaCl solution. Thereafter, 0.05 g of each sample was mixed in each flask and shaken at 170 rpm. After 24 h, the final pH (pHf) of the solutions was obtained, subtracted from the initial pH value (*pH = pHi − pHf), and the resulting values plotted against pHi. pHPZC was taken as the point of intersection of the resulting curve with the abscissa at *pH = 0 [22].
Adsorption experiments
A batch system was used to study the adsorption of MB on the synthesized catalysts at 25 °C. A predetermined quantity of each catalyst (1 g L−1) was dispersed in MB solutions of diverse concentrations (25, 50, 100, and 200 mg L−1) with pHi of 7 and shaken constantly using a mechanical mixer (170 rpm) for 2 h to achieve adsorption equilibrium. Then, the adsorbent was separated from the solution by centrifugation at 3500 rpm for 5 min. A UV–Vis spectrophotometer (Spectroquant® Pharo 300) was used to determine the equilibrium concentration of MB at λ max of 664 nm. Previously, a series of standard MB solutions with known concentrations were used to plot a calibration curve. Equation 3 was then used to estimate the total MB adsorbed per unit mass of sample:
where C 0 is the initial and C e the equilibrium MB concentration (mg L−1) in the liquid part, V is the solution volume, and m is the mass of dry catalyst (g). The adsorption isotherms of the samples were also studied using the obtained results.
Fenton experiments
Fenton experiments were carried out for methylene blue solution (100 mL, 50 ppm) using 250-mL Pyrex beakers in batch mode at room temperature. First, the pH of the solution was adjusted to 7 using sodium hydroxide (NaOH, 0.1 mol L−1) with a pHmeter (Eutech, CyberScan pH 300). Next, a predetermined dose of each catalyst was poured into the MB solution and agitated mechanically for 120 min at 170 rpm to achieve sorption equilibrium. Addition of H2O2 at t = 120 min initiated the reaction, which was carried out for more than 180 min. The sample was then separated by centrifugation at 3500 rpm for 5 min, and a 0.20-μm filter (Nylon, Whatman) was used to filter the supernatant to remove the catalyst particles. The concentration of untreated dye was determined immediately by UV–Vis spectrophotometer at λ max = 664 nm.
A Shimadzu TOC-L (CPH CN 200, Japan) analyzer with autosampler was used to measure the total organic carbon (TOC) of the treated MB solutions. All experiments were run in absence of light and at room temperature. Assessments were carried out at least twice, and the results presented as the mean with experimental error <5%. The color and TOC removal (%) were calculated using the following equation (Eq. 4):
where X 0 and X i are the initial and final values for the MB or TOC concentration of the solution.
The kinetics of Fenton oxidation was studied in batch mode by adding the synthesized sample (1 g L−1) into MB solution with concentration of 25, 50, 100, and 200 mg L−1 and pH of 7 and shaking for 10–200 min at 25 °C. The amount of MB left in the supernatant in each time interval was calculated using Eq. (4).
Durability and reusability experiments
The durability of the synthesized samples was examined using a modified procedure [22]. For this, the concentrations of structural iron and molybdenum washed away from the synthesized catalysts were measured under different pH values. The samples (0.02 g) were added into aqueous solutions (100 mL, pH 4, 7, and 10) and stirred in a shaker at controlled temperature for 3 h. The amount of Fe and Mo in the supernatant was determined by inductively coupled plasma–optical emission spectrometry (ICP-OES; PerkinElmer Optima 7000 DV).
Results and discussion
Characteristics of Fe3−x Mo x O4 nanoparticles
A number of molybdenum ferrite NP samples were fabricated by facile coprecipitation method using precursors at various Fe:Mo ratios, and inductively coupled plasma (ICP) analysis was used to measure the amounts of Fe and Mo left over in the synthesis vessel. Subsequently, the values obtained from ICP analysis were applied to determine the chemical formula of each sample. The main chemical formula obtained for x values of 0.05, 0.1, and 0.2 was Fe2.94Mo0.028O4, Fe2.89Mo0.069O4, and Fe2.79Mo0.13O4, respectively. With respect to the amounts used, negligible Fe ions remained unreacted, while the amount of Mo incorporated into the samples was about 56% (x = 0.05), 69% (x = 0.1), and 65% (x = 0.2). In comparison with Nb-substituted magnetite, these results indicate lower incorporation maxima for Mo compared with Nb [20]. Although x = 0.2 was chosen as it gave the highest molar fraction of incorporated Mo, a sample with x = 0.3 was also prepared due to its lower incorporation maximum of Mo. In this sample (Fe2.62Mo0.21O4), the amount of structural Mo in the magnetite was about 70% of the initially utilized value. Moreover, the values obtained from ICP analysis were also used to obtain the approximate quantity of oxygen vacancies in the synthesized Fe3−x Mo x O4 samples. According to these results, the amount of oxygen vacancies increased in parallel with the increasing amount of Mo utilized in the prepared samples, mainly due to the structural cationic deficiency (lower structural Mo4+ than stoichiometric value), and adjustments for structural dislocations derived from the substitutions with unequal charges (Fe2+ by Mo4+). In the latter case, replacement of Fe2+ by Mo4+ could be responsible for the reduction of the same amount of Fe3+ to Fe2+ on the basis of electrovalence equilibrium, or structural dislocations could be adjusted by induction of oxygen vacancies, which are believed to act as active sites for generation of hydroxyl radicals in the Fenton reaction [7].
The XRD spectra of the Fe3−x Mo x O4 catalysts (x ≈ 0.028–0.21) are shown in Fig. 1. Nine Bragg diffraction peaks at 2θ ≈ 18.3°, 30.2°, 35.6°, 37°, 43.3°, 53.7°, 57.2°, 62.8°, and 74.4° are seen in these patterns, corresponding to (111), (220), (311), (222), (400), (422), (511), (440), and (533) crystal planes of the inverse spinel structure of magnetite, respectively. These data fit well with the standard Joint Committee on Powder Diffraction Standards (JCPDS) cards no. 98-009-8087, 98-015-8745, 98-007-7589, and 98-015-7651 for magnetite, confirming the conservation of the magnetite structure after incorporation of Mo. In this group of samples, replacement of Fe2+ (61 pm) by Mo4+ (65 pm) and Mo6+ (59 pm) altered the lattice parameters from 8.394 Å for the unmodified magnetite to 8.378, 8.358, and 8.344 Å. The difference in the intensity of the diffractograms mainly results from the variation in the particle size of the samples. Hence, Scherrer’s equation (Eq. 5) was used to determine the average size of the crystallites using the results obtained from XRD analysis:
where D hkl is the crystallite size, hkl are the Miller indices of the plane being analyzed (in this study, [311]), K is the Scherrer constant (0.94), λ is the wavelength of the X-rays (0.154 nm for Cu Kα radiation), B hkl is the width at half-peak intensity (FWHM) in radians, and θ is the Bragg angle in degrees. The results obtained from this equation were consistent with the data acquired from TEM images and presented in Table 1.

XRD spectra of Fe3−x Mo x O4 samples
Table 1 also presents the results obtained from BET analysis. The effective surface area of the samples increased with greater Mo content due to reduced particle size accompanied by the increased structural Mo in the samples. On the other hand, increase in the Mo content in the samples reduced the pore size, which could be another reason for the enhanced surface area.
Transmission electron microscopy (TEM) revealed the morphology, shape, and size of the Fe3−x Mo x O4 NPs. The micrographs showed the characteristic morphology of crystallized magnetite, with all the samples growing well in octahedral form (Fig. 2). The slight agglomeration observed in the samples is due to their magnetic properties and attractive dipole–dipole forces. However, the extensive particle agglomeration in the Fe2.62Mo0.21O4 sample could result from smaller particle size [23] and/or higher crystallization degree [24]. On the other hand, the size of the samples decreased as the Mo content was increased. The changes in particle size and surface area were especially obvious for the Fe2.62Mo0.21O4 sample, showing a fivefold decrease in particle size from 90 nm to 20 nm and more than threefold increase in specific surface area compared with the unmodified magnetite sample.

TEM micrographs of samples a A, b B, c C, and d D
X-ray photoelectron spectroscopy (XPS) was applied to determine the surface elemental composition of the Fe3−x Mo x O4 samples. This was mainly used to verify the presence of coprecipitated Mo in the modified magnetite and determine its chemical state. On this basis, a wide-ranging XPS survey spectrum helped to determine the elements present on the samples (Fig. 3). Subsequently, the chemical states of elements of interest in the samples were detected by narrow scans of each element and a peak deconvolution process. The binding energy (BE) spectral regions of 222–242 eV, 520–550 eV, and 700–740 eV for Mo 3d, O 1s, and Fe 2p, respectively, were subsequently used for narrow scans.

XPS spectra of Fe3−x Mo x O4 samples
Although the XRD pattern showed the inverse spinel structure of the prepared samples, it could also be related to maghemite with the same ferrite structure as magnetite. To prove the presence of Fe2+ cations in the structure, XPS is a valuable tool to differentiate between Fe2+ and Fe3+ ions. Figure 4a shows the narrow scan Fe 2p spectrum of the Fe2.62Mo0.21O4 sample. The peaks between binding energy of 709.9 eV and 724.5 eV related to Fe 2p 1/2 and Fe 2p 3/2 were used to determine the chemical state of the surface Fe [25]. The obtained results matched well with the reference data for Fe2+ and Fe3+ cations [26]. Furthermore, the presence of satellite peaks (indicated by arrows) adjacent to the main peaks are signatures of Fe2+ and Fe3+ cations. The configuration interaction of valence electron relaxation is the main reason for generation of the satellite peaks [27]. The weight data (%) for Fe species on the surface of the samples are given in Table S1. The amount of surface Fe2+ cations in the samples decreased as the Mo content was increased, as is evident from the Fe2+/Fe3+ ratio values. This indicates that the incorporated Mo4+ cations substituted Fe2+ cations at octahedral sites of magnetite. Similarly, the slight shift towards higher binding energy of the Fe 2p peaks for the modified samples can be attributed to replacement of Fe2+ by Mo and alteration in the Fe2+/Fe3+ ratio (Fig. S1). The data for satellite Fe2+ and satellite Fe3+ are also presented in Table S1, being important for precise quantification of the surface iron species. Details regarding the peak position and corresponding chemical states of the Fe species in the samples were acquired by deconvolution of the Fe 2p spectrum of the Fe2.62Mo0.21O4 sample (Fig. 4a).

a Fe 2p and b Mo 3d 3/2 deconvolutions of the Fe2.62Mo0.21O4 sample
The presence of Mo on the samples was also confirmed by the XPS analysis. Figure 4b shows the XPS spectrum of Mo 3d for the Fe2.62Mo0.21O4 sample. The Mo 3d data agree perfectly with the peaks for Mo (IV) and (VI) at BE of 232.2 and 235.6 eV, which can be attributed to Mo 3d 5/2 and Mo 3d 3/2, respectively.
The magnetic characteristics of the synthesized catalysts were studied by vibrating-sample magnetometry (VSM). The coercivity (Hci), remanence (M r), saturation magnetization (M s), and squareness ratio (SQR = M r/M s) were the main parameters extracted from the hysteresis loops (Fig. 5). The magnetic characteristics of the samples were assessed based on these parameters, as presented in Table 2.

Hysteresis loops for a Fe3O4 and b Fe2.62Mo0.21O4 samples
A slim hysteresis loop (low SQR value) is characteristic of ferromagnetic compounds due to their high magnetic saturation (M s) and low residual magnetization (M r). Based on these results, the magnetic property of magnetite was retained after the alteration, aiding recovery of the samples from the treated solution. Interestingly, the Fe2.62Mo0.21O4 NPs showed improved magnetization with smaller SQR value, which can be ascribed to the very small particle dimensions. Its good magnetic properties are comparable to results obtained for Fe2.79Nb0.19O4 (SQR = 0.157) [20] and Fe2.79Nb0.171Mo0.023O4 (SQR = 0.151) [28], representing the optimized samples among the studied group of catalysts.
Adsorption of methylene blue by Fe3−x Mo x O4 nanoparticles
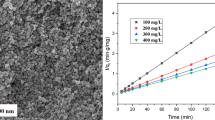
Figure 6a displays the quantity of MB adsorbed on the Fe3−x Mo x O4 samples in the equilibrium state after 120 min of stirring. The amount of MB adsorbed was significantly higher for the Mo-substituted magnetite samples compared with pure magnetite. The rise in q e paralleled the increasing Mo content of the samples. In contrast to unmodified magnetite, the q e was enhanced from 4.9 to 9.3, 11.8, 13.8, and 19.7 mg/g for the Fe3−x Mo x O4 (x = 0.028, 0.069, 0.13, and 0.21) samples at neutral condition, respectively. The substantial amount of MB (about 40%) adsorbed on the Fe2.62Mo0.21O4 sample can be attributed to its large specific surface area [29]. As explained above, the surface area of the Fe3−x Mo x O4 samples increased with the amount of Mo introduced into the magnetite structure. Therefore, the available sites for MB adsorption increased. This indicates that Mo substitution significantly improved the surface characteristics of magnetite for enhanced adsorption capacity.

MB elimination via a adsorption, b degradation, and mineralization by Fe3−x Mo x O4 samples at pH 7 and 10
On the other hand, the surface charge of the samples also plays a key role in the adsorption of MB. The point of zero charge (PZC) is the pH at which the charge on the surface of the catalyst is zero. Accordingly, the surface of magnetite is positively charged at pH values below the PZC, and vice versa. Based on the fact that the pHPZC values for the Fe3−x Mo x O4 samples were below 7 (Fig. S2), the charge on the surface of the samples was negative at neutral condition. Moreover, as a cationic dye, MB is adsorbed on the negatively charged surface of the catalyst. Therefore, MB adsorption onto the studied samples took place under both neutral and basic conditions. However, experiments at acidic condition (pH 3) showed negligible adsorption of MB on the samples due to charge repulsion. The adsorption experiments were repeated at pH 10 to evaluate the effects of pH on the adsorption capacity of the samples (Fig. 6a). The MB adsorbed at pH 10 was greatly improved within 120 min of agitation. This can be ascribed to a rise in surface hydroxyl groups in basic condition [30]. Another potential driving force for the enhanced adsorption properties of the Fe3−x Mo x O4 samples could be surface oxygen vacancies that act as active sites for enhanced adsorption capacity. These results indicate that both the surface properties of the heterogeneous catalyst and the characteristics of the probe molecule had crucial effects on the adsorption efficacy of the samples.
Degradation and mineralization of MB by Fe3−x Mo x O4 samples
The activity of the synthesized catalysts was examined based on their efficacy for oxidation of MB via Fenton reaction. Once adsorption equilibrium was reached, the MB degradation process was initiated by addition of H2O2 to the system. The removal of MB (C t /C 0) through adsorption and Fenton reaction is depicted in Fig. S3. All the Mo-substituted magnetite NPs showed remarkable activity for decolorization of MB solution in presence of H2O2. The MB removal efficiency using the Fe2.94Mo0.028O4, Fe2.89Mo0.069O4, Fe2.79Mo0.13O4, and Fe2.62Mo0.21O4 samples was about 48, 52, 56, and 63% more than that of pure magnetite within the reaction duration of 150 min. However, the MB decolorization rate was significantly improved to about 10–25% when the pH of the solution was increased to 10 (Fig. 6b). This enhancement of the degradation efficiency at pH 10 mainly occurs because of the large number of surface OH-groups and corresponding higher adsorption capacity of the samples.
However, there are reports of low adsorption of some anionic dyes on the surface of structurally modified magnetite samples. It is reported that degradation of these anionic dyes through Fenton reaction took place via some other oxidation mechanisms [29].
The MB mineralization effectiveness of the Fe3−x Mo x O4 samples is also shown in Fig. 6b. The degree of mineralization was meaningfully improved by incorporation of Mo into magnetite, being about 34, 42, 66, and 68% higher than for unmodified magnetite when using Fe2.94Mo0.028O4, Fe2.89Mo0.069O4, Fe2.79Mo0.13O4, and Fe2.62Mo0.21O4, respectively. The differences in TOC removal significantly increased when the pH of the solution was changed to 10. Significant TOC removal efficiency of 71.5 and 72.4% was obtained when using Fe2.79Mo0.13O4 and Fe2.62Mo0.21O4 samples at pH 10. In similar studies, impregnation of magnetite with small quantities of other transition metals led to positive changes in the physicochemical characteristics for higher catalytic activity in the Fenton process. However, it should be noted that the amount of transition metal incorporated into the magnetite, as well as the reaction conditions including the dosage of Fenton reagents, nature and concentration of pollutant, composition and pH of the solution under treatment, and reaction duration, have distinctive effects on the process efficiency [7, 19]; For instance, in a comparative study, CuFe2O4 showed higher activity for removal of gallic acid in presence of H2O2 compared with Fe3O4 or FeTiO3 [31]. Synergy between octahedral Fe and Cu ions has been reported to be the main explanation for this enhanced activity, with Cu ions being introduced as the main active sites for generation of HO· radicals. In another similar study, Fe3−x Cu x O4 samples (0 ≤ x ≤ 0.25) were fabricated via coprecipitation method and employed as heterogeneous catalysts for electrodegradation of amaranth food dye through an electro-Fenton process [32]. Those authors reported that the highest (98 and 70%) degradation and mineralization efficiencies were obtained when using the Fe2.75Cu0.25O4 sample. The results of the current study strongly prove that Mo substitution improves the characteristics of magnetite for higher adsorption capacity and activity in Fenton oxidation of MB. Distinct enhancement was especially observed when using the Fe2.79Mo0.13O4 and Fe2.62Mo0.21O4 samples with the highest amounts of Mo in their structure. For the Fe2.62Mo0.21O4 sample, although the stoichiometric value of x = 0.3 was initially used, the actual structural value was x = 0.21. Therefore, similar to a Nb-substituted magnetite series [20], the value of x = 0.2 was found to be optimal for the Fe3−x Mo x O4 samples.
A number of factors could potentially be involved in the superior activity of the Fe3−x Mo x O4 NPs. Primarily, the increased surface area of the modified magnetite could host a larger number of surface OH groups, increasing the adsorption and resulting in higher degradation efficacy. Furthermore, Mo4+/Mo6+ redox pairs could be directly involved in the oxidation process through the Haber–Weiss mechanism (Eq. 6) and help regeneration of Fe2+ species (Eq. 7):
In addition, the large amount of oxygen vacancies resulting from cationic insufficiency and structural modification due to the unbalanced charge replacement (Mo4+ for Fe2+) could be activated by adsorbed H2O2 to act as reaction sites for degradation of MB molecules.
Kinetic studies
The MB oxidation rate through the Fenton reaction using the Fe3−x Mo x O4 NPs was determined under neutral condition. In previous reports using Nb-substituted [20] and Nb/Mo-codoped magnetite [28], the MB degradation kinetics well followed a pseudo-first-order model. Therefore, the data obtained for MB degradation in the Fenton reaction using the Fe2.79Mo0.13O4 sample were applied to study the oxidation rate parameters for a pseudo-first-order mechanism (Eq. 8) [33]:
where C 0 is the initial MB and C t is the MB concentration at reaction time t (mg L−1), and k app is the pseudo-first-order rate constant (min−1). Nevertheless, the degradation data did not fit well with this pseudo-first-order kinetics model, because the regression coefficient (R 2) values were not as high as in previous studies (Table 3). Therefore, a zeroth-order kinetics model was also studied to explore the most suitable model to describe the rate of degradation of MB by the Mo-substituted magnetite NPs (Eq. 9):
where k 0 is the zeroth-order rate constant (mg L−1 min−1). The data for the both kinetics models are presented in Table 3. Based on these R 2 values, it is evident that the zeroth-order kinetics model showed the best fit to the experimental kinetics data for Fenton degradation of MB by the Fe3−x Mo x O4 NPs (Fig. 7). Furthermore, the reaction rate constants were highest for the Fe2.62Mo0.21O4 sample, followed by Fe2.79Mo0.13O4, Fe2.89Mo0.069O4, and Fe2.94Mo0.028O4. These results therefore also validate the synergistic effect of the amount of Mo on the surface on MB degradation.

Zeroth-order kinetics for oxidation of MB catalyzed by Fe3−x Mo x O4 samples in Fenton reaction
Durability of samples
Durability and stability of synthesized catalysts under various experimental conditions are essential to preserve their effectiveness over several applications and also to abate environmental pollution from the utilized transition metals. Accordingly, a number of leaching experiments were carried out under the studied conditions. Based on the results, the concentration of Fe released was nondetectable in the solutions under all the studied pH conditions when using the Fe3−x Mo x O4 catalysts. In the case of Mo, although its tendency for incorporation was relatively low, the amount of Mo leached out was not measurable. The Fe2.62Mo0.21O4 sample was reemployed in three consecutive runs to assess its activity during reuse. The amount of MB (50 mg L−1) degradation during the three runs was found to be 94, 91, and 89% within 120 min, revealing an insignificant reduction in the activity of the catalyst. These results confirm the excellent durability of the Mo-substituted magnetite samples for practical applications in environmental purification and decomposition of toxic and organic pollutants. Furthermore, they show that MB oxidation was completed through the heterogeneous route while the role of leached ions and homogeneous Fenton reaction in the oxidation of MB was insignificant.
Conclusions
Four Mo-substituted magnetite (Fe3−x Mo x O4) NP samples with different amounts of Mo (x = 0.028–0.21) were synthesized and characterized. The structure and magnetic properties of the magnetite were retained after such modification. The decreased crystal size and increased surface area of the Mo-incorporated magnetite catalysts significantly improved their adsorption capacity. Based on XPS analysis, Mo4+ cations were mainly located at octahedral sites of Fe2+ cations. The activity of the catalysts for degradation of cationic MB dye solution in Fenton reaction was measured in neutral and basic conditions. Enhanced activity in the Fenton process was evident after substitution of magnetite with Mo, and the oxidation rate increased with increasing Mo content. This can be explained based on the role of the Mo4+/Mo6+ redox pair on the surface of the samples in the decomposition of H2O2 and regeneration of Fe2+ cations. Furthermore, the generated oxygen vacancies acted as active sites for enhanced ·OH radical production. Based on kinetics studies, the MB degradation through the Fenton reaction was well described by a zeroth-order kinetics model. The samples were well stable in the studied experimental conditions and during three successive applications. These results confirm the development of a powerful advanced oxidation system based on Mo-substituted magnetite/H2O2.
Abbreviations
- C :
-
Total concentration in bulk phase, mg L−1
- C 0 :
-
Initial concentration, mg L−1
- E 0 :
-
Oxidation potential, V
- Hci:
-
Coercivity, G
- k :
-
Kinetic rate constant
- m :
-
Mass, g
- M r :
-
Remanent magnetization, emu/g
- M s :
-
Saturation magnetization, emu/g
- q :
-
Adsorbed concentration
- q e :
-
Adsorbed concentration in equilibrium with fluid phase
- q t :
-
Adsorbed concentration at time t
- R 2 :
-
Regression coefficient
- t :
-
Time
- V :
-
Volume, L
- x :
-
Mole fraction
- λ :
-
Wavelength
- BET:
-
Brunauer–Emmett–Teller
- MB:
-
Methylene blue
- ·OH:
-
Hydroxyl radical
- PZC:
-
Point of zero charge
- TEM:
-
Transmission electron microscopy
- TOC:
-
Total organic carbon
- UV–Vis:
-
Ultraviolet–visible
- VSM:
-
Vibrating-sample magnetometry
- XPS:
-
X-ray photoelectron spectroscopy
- XRD:
-
X-ray diffraction
References
S. Rahim Pouran, A.R. Abdul Aziz, W.M.A.W. Daud, J. Ind. Eng. Chem. 21, 53–69 (2015)
A. Khataee, M. Taseidifar, S. Khorram, M. Sheydaei, S.W. Joo, J. Taiwan Inst. Chem. Eng. 53, 132–139 (2015)
J. Kruid, R. Fogel, J.L. Limson, Chemosphere 175, 247–252 (2017)
S. Rahim Pouran, A. Bayrami, A.R.A. Abdul Aziz, W.M.A.W. Daud, M.S. Shafeeyan, J. Mol. Liq. 222, 1076–1084 (2016)
J.M. Monteagudo, A. Durán, R. Culebradas, I. San Martín, A. Carnicer, J. Environ. Manage. 128, 210–219 (2013)
B.H. Diya’uddeen, S. Rahim Pouran, A.R. Abdul Aziz, W.M.A.W. Daud, RSC Adv. 5(83), 68159–68168 (2015)
S. Rahim Pouran, A.A. Abdul Raman, W.M.A.W. Daud, J. Clean. Prod. 64, 24–35 (2014)
A. Khataee, P. Gholami, M. Sheydaei, J. Taiwan Inst. Chem. Eng. 58, 366–373 (2016)
H. Aghdasinia, P. Arehjani, B. Vahid, A. Khataee, Res. Chem. Intermed. 42, 8083–8095 (2016)
A. Khataee, S. Sajjadi, S. Rahim Pouran, A. Hasanzadeh, J. Ind. Eng. Chem. (2017). doi:10.1016/j.jiec.2017.07.024
A. Khataee, S. Sajjadi, S. Rahim Pouran, A. Hasanzadeh, S.W. Joo, Electrochim. Acta 244, 38–46 (2017)
M. Taseidifar, A. Khataee, B. Vahid, S. Khorram, S.W. Joo, J. Mol. Catal. A 404–405, 218–226 (2015)
C. Su, J. Hazard. Mater. 322, 48 (2017)
G. Jenita Rani, M.A. Jothi Rajan, G. Gnana Kumar, Res. Chem. Intermed. 43(4), 2669–2690 (2017)
J.W. Moon, Y. Roh, R.J. Lauf, H. Vali, L.W. Yeary, T.J. Phelps, J. Microbiol. Methods 70(1), 150–158 (2007)
A. Gogoi, M. Navgire, K.C. Sarma, P. Gogoi, Chem. Eng. J. 311, 153–162 (2017)
A. Gangwar, S.K. Alla, M. Srivastava, S.S. Meena, E.V. Prasadrao, R.K. Mandal, S.M. Yusuf, N.K. Prasad, J. Magn. Magn. Mater. 401, 559–566 (2016)
Z. Qi, T.P. Joshi, R. Liu, H. Liu, J. Qu, J. Hazard. Mater. 329, 193–204 (2017)
M. Munoz, Z.M. de Pedro, J.A. Casas, J.J. Rodriguez, Appl. Catal. B 176–177, 249–265 (2015)
S. Rahim Pouran, A.R. Abdul Aziz, W.M.A.W. Daud, Z. Embong, Appl. Surf. Sci. 351, 175–187 (2015)
X. Liang, Y. Zhong, S. Zhu, L. Ma, P. Yuan, J. Zhu, H. He, Z. Jiang, J. Hazard. Mater. 199–200, 247–254 (2012)
L. Ai, C. Zhang, F. Liao, Y. Wang, M. Li, L. Meng, J. Jiang, J. Hazard. Mater. 198, 282–290 (2011)
C. Yoon, J.-H. Choi, Dyes Pigm. 101, 344–350 (2014)
B.J. Hwang, R. Santhanam, D.G. Liu, J. Power Sources 97, 443–446 (2001)
T. Yamashita, P. Hayes, Appl. Surf. Sci. 254, 2441–2449 (2008)
F. Magalhães, M.C. Pereira, S.E.C. Botrel, J.D. Fabris, W.A. Macedo, R. Mendonca, R.M. Lago, L.C.A. Oliveira, Appl. Catal. A 332(1), 115–123 (2007)
E. Paparazzo, J. Electron Spectrosc. 154(1–2), 38–40 (2006)
S. Rahim Pouran, A.R. Abdul Aziz, W.M.A.W. Daud, M.S. Shafeeyan, RSC Adv. 5(106), 87535–87549 (2015)
X. Liang, Z. He, Y. Zhong, W. Tan, H. He, P. Yuan, J. Zhu, J. Zhang, Colloids Surf. A 435, 28–35 (2013)
X. Liang, Y. Zhong, H. He, P. Yuan, J. Zhu, S. Zhu, Z. Jiang, Chem. Eng. J. 191, 177–184 (2012)
M.A. Fontecha-Cámara, C. Moreno-Castilla, M.V. López-Ramón, M.A. Álvarez, Appl. Catal. B 196, 207–215 (2016)
W.R.P. Barros, J.R. Steter, M.R.V. Lanza, A.C. Tavares, Appl. Catal. A 180, 434–441 (2016)
M.S. Shafeeyan, W.M.A.W. Daud, A. Shamiri, N. Aghamohammadi, Energy Fuels 29(10), 6565–6577 (2015)
Acknowledgements
The authors are grateful to the University of Malaya High Impact Research Grant (HIR-MOHE-D000037-16001) from the Ministry of Higher Education Malaysia. Support from the Iran Science Elites Federation is also acknowledged.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Rahim Pouran, S., Bayrami, A., Abdul Raman, A.A. et al. Comprehensive study on the influence of molybdenum substitution on characteristics and catalytic performance of magnetite nanoparticles. Res Chem Intermed 44, 883–900 (2018). https://doi.org/10.1007/s11164-017-3142-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-017-3142-x