
Abstract
Efficient synthesis of 2,4,5-triarylimidazole derivatives with simple workup is described using benzil, different aldehydes, and ammonium acetate in presence of fluorinated graphene oxide (A-MFGO) as effective catalyst that can be easily recovered and reused for several times without significant loss in activity. The simple preparation of the catalyst, short reaction time, mild conditions, high yield, and low cost make this procedure economically attractive and highly suitable for synthesis of 2,4,5-triarylimidazole derivatives.
Graphical Abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Graphene oxide (GO), which can be obtained from graphite by decoration with oxygen functional groups, easily decomposes and may convert to smaller components under heat treatment, a routine condition for fluorination of carbon. We selected GO to make a composite in this work because of its high surface area, nontoxicity, and ability to be functionalized with different chemical groups. The most important feature of GO is that it can be properly prepared on large scale from normal graphite at low cost. GO is similar to benzene and polycyclic aromatic hydrocarbons in that it comprises sp2 and sp3 carbon atoms [1–3]. GO can be densely decorated with hydroxyl, epoxy, carbonyl, and carboxyl functionalities on the surface and edges of the sheet, providing a variety of opportunities for chemical modification and transformation [4–6]. Recently, we envisaged that GO could be a promising starting material for cost-effective, large-scale preparation of fluorinated graphene (FG) if oxygen-containing functionalities could be successfully transformed into C–F bonds.
Fluorination stands out among the most effective chemical techniques to change the physiochemical properties of carbon materials, since further substitution can be promptly accomplished [7, 8]. The C–F bond exhibits excellent oxidative and thermal stability [9]. In addition, fluorine is the most electronegative element in the Periodic Table, while C–F bonds possess high polarity and low surface free energy. Because of the unique nature of covalent fluorine atoms, FG has different properties compared with GO. FG with fluorine atoms covalently functionalized on GO therefore represents an innovative two-dimensional carbon material [10]. Experimental methods for synthesis of FG can be divided into two approaches: (1) direct fluorination of graphene sheets using XeF2 and plasma (CF4 and SF6) as fluorination agents [11, 12], and (2) exfoliation of graphite fluoride by mechanical cleavage or ultrasonication in sulfolane, isopropanol, or ionic liquid [13, 14]. Recently, fluorination of GO utilizing hydrogen fluoride, a corrosive reagent, was described [15]. However, an effective, convenient, environmentally friendly, and solution-based synthetic route to FG is required for mass production to study its properties and practical applications. FG has attracted interest for technological applications, for instance, for use in lubricants [16]. Therefore, many experimental and theoretical studies [17, 18] describe its synthesis or application in organic synthesis as a catalyst. FG has been utilized in biomedical applications. FG is in vogue once more due to the recent scientific and technological interest in graphene [19].
Imidazoles are a class of nitrogen-containing heterocyclic compounds that include pharmaceutical agents with antibacterial [20], antitumor [21], and antiinflammatory actions [22], herbicides [23], fungicides [24], as well as inhibitors of mammalian 15-LOX [25] and B-Raf kinase [26]. Imidazole moieties form the fundamental structural skeleton of numerous significant biological molecules such as histamine and histidine [27, 28]. Besides, substituted imidazoles are used in synthesis of several ionic liquids [29] applied in the new approach known as green chemistry. They are also used in photography as photosensitive compounds [30]. Due to their wide range of pharmacological, biological, and industrial activities, synthesis of imidazoles has received extensive attention in organic synthesis [31–36]. Over the years, numerous methods for synthesis of highly substituted imidazoles have been published in literature. Generally, synthesis of 2,4,5-trisubstituted imidazoles via three-component cyclocondensation of 1,2-diketone, α-hydroxyketone or α-ketomonoxime with aldehyde and ammonium acetate can be catalyzed by various catalysts, including ytterbium trifoliate [37], ytterbium perfluorooctanesulfonate [38], InCl3·3H2O [39], acetic acid [40], glyoxylic acid [41], oxalic acid [42], ionic liquids [43], silica sulfuric acid [44], cellulose sulfuric acid [45], ceric(IV) ammonium nitrate [46], sodium bisulfate [47], anhydrous FePO4 [48], and nanocrystalline magnesium oxide [49]. Many of these methods suffer from one or more disadvantages, such as longer reaction time, poor yield, expensive reagents and catalyst, difficult workup, or excessive use of reagents and catalyst, finally resulting in generation of large amounts of dangerous waste. It is therefore important to develop more convenient procedures for synthesis of these compounds. Hence, development of simple, efficient, high-yielding, environmentally friendly approaches using new catalysts for synthesis of imidazoles remains a challenge for organic chemists. Changing conventional, toxic Brønsted and Lewis acid catalysts for environmentally benign, easily recoverable solid catalysts is an active area of research. In recent years, employment of solid acids as catalysts has received great attention in diverse fields of organic synthesis [50, 51]. Solid acid catalysts offer various advantages, such as catalyst reuse, environmental acceptability compared with liquid acid catalysts, and easy separation of products. Exploration of the design and activity of such novel specific catalysts could greatly improve the efficiency of a wide range of organic syntheses.
One of our aims is to introduce a new safe, efficient, easy to handle, reusable catalyst for synthesis of 2,4,5-triarylimidazole derivatives. In this study, a simple and inexpensive method was used for synthesis of A-MFGO by employing graphene oxide (Scheme 1). We also describe an excellent procedure for preparation of 2,4,5-triarylimidazole derivatives using A-MFGO as a solid acid catalyst that is both efficient and reusable. The method reported herein is not cumbersome, thus we consider that the methodology is a good addition to the list of procedures available for synthesis of 2,4,5-triarylimidazole derivatives (Scheme 2).

Synthesis procedures for catalyst A-MFGO

Synthesis of some useful 2,4,5-triarylimidazole derivatives using A-MFGO
Experimental
Materials and methods
All chemicals used in this work were purchased from Merck or Fluka and used without further purification. Melting points were recorded using an Electrothermal 9100 apparatus. Fourier-transform infrared (FTIR) spectroscopy (Bruker ALPHA) was used to determine the quality and composition of the materials. Reactions were monitored by thin-layer chromatography (TLC). 1H and 13C NMR spectra were measured on a Bruker (300 MHz) spectrometer using dimethyl sulfoxide (DMSO)-d6 as solvent and tetramethylsilane (TMS) as internal standard. Elemental (C, H, N) analyses were performed using a Heraeus CHN-O-Rapid analyzer. Field-emission scanning electron microscopy (FESEM) images were captured using a Hitachi S4160 cold field-emission instrument. X-ray diffraction (XRD) analysis was performed using an INEL Equinox 3000 diffractometer. Energy-dispersive X-ray spectroscopy (EDS) measurements were performed using a SAMx analyzer. X-ray photoelectron spectroscopy (XPS) was performed on a VG Scientifics ESCALAB 250 spectrometer with an Al Kα irradiation source (1486.7 eV) at 15 kV and 10 mA.
Synthesis of catalyst
Synthesis of graphene oxide (GO)
GO was synthesized from graphite powder and purified according to literature procedure [52]. Graphite (10 g) and sodium nitrate (7.5 g) were mixed together, followed by addition of 98% (621 g) sulfuric acid under constant stirring; the mixture was stirred while being cooled in an ice-water bath. KMnO4 (45 g) was added gradually to the above solution for about 1 h, while keeping the temperature lower than 10 °C for 2 h to prevent overheating and explosion. The mixture was allowed to stand for 5 days at room temperature with gentle stirring to obtain highly viscous liquid, which was added to H2SO4 aqueous solution (1000 mL) with stirring in an ice bath; the resultant mixture was further stirred for 2 h. After removing the ice bath, H2O2 (10%, 30 g), slowly and with caution, was added to the above liquid, and the mixture was stirred for 2 h. Then, the resultant mixture was allowed to stand for 2 days in order to precipitate. The resultant mixture was purified by filtration and washed with 3 wt% H2SO4 and 0.5 wt% H2O2 aqueous solution. The mixture was filtered for further purification. The filter cake was washed twice with distilled water, then the resulting GO was dried and stored for subsequent use.
Synthesis of fluorinated graphene oxide (FGO)
FGO was synthesized by a simple reaction between GO dispersion and HF. GO powder (1 g) and N-methyl-2-pyrrolidinone (NMP) (100 mL) was sonicated at room temperature for 15 min to obtain completely exfoliated, monolayer GO dispersion. After sonication, GO suspension was transferred to an autoclave. Afterwards, HF (20 mL) was added and maintained at 180 °C for 30 h. The autoclave was naturally cooled to room temperature. The final product was obtained by successively washing with excess tetrahydrofuran (THF) and deionized water to remove impurities and dried in an oven at 60 °C for 3 h.
Modification of FGO with diamine (MFGO)
Butylethylimidazolium bromide (4 g) ionic liquid (IL) was dissolved in NMP (20 mL), then triphenyl phosphate (TPP) (4 mL) was added to a 50-mL, round-bottomed flask [53]. FGO (1 g) and 1,6-diaminohexane (2 g) were sequentially added to the above mixture. The whole solution was heated at 100 °C for 48 h. The resulting product was isolated by filtration of the mixture through sintered glass and washed with methanol (3 × 35 mL), followed by drying at 60 °C for 24 h to obtain MFGO.
Functionalized MFGO with 1,3-propane sultone (A-MFGO)
A-MFGO was obtained by reaction of MFGO (0.5 g) with 1,3-propane sultone (1 g) in DMSO (20 mL). The mixture was refluxed at 160 °C for 4 days. After reaction completion, the reaction mixture was cooled to room temperature. The resulting product was isolated by filtration of the mixture over sintered glass and washed with methanol (3 × 35 mL). Then, the solid product was added to HCl aqueous solution (40 mL, 1:3), and the mixture was stirred at ambient temperature for 20 min and left for 2 days. Afterwards, the separated solid was filtered using sintered glass, followed by washing three times with water to remove residual acid. Finally, the catalyst was removed by filtration and drying at 60 °C for 24 h.
Synthesis of 2,4,5-triarylimidazole derivatives
A mixture of benzil (1.05 g, 5.0 mmol), 3-nitrobenzaldehyde (0.755 g, 5.0 mmol), and ammonium acetate (1.54 g, 20.0 mmol) in ethanol (15 mL) was taken, and A-MFGO as catalyst (0.15 g) was added at room temperature. The resulting mixture was stirred for 2 h at reflux temperature. Upon reaction completion (monitored by TLC), dichloromethane (10 mL) was added with stirring for 15 min at ambient temperature. The catalyst was recovered by filtration and washed with dichloromethane for reuse in the next cycle (up to five runs) (Fig. 4). Then, the filtrate was poured into cold water (20 mL) and stirred at room temperature for 10 min. The crude product was obtained after addition of water to the organic layer. Two phases of solution were extracted, then the solvent was evaporated. The solid product was washed with water, dried, and recrystallized from ethanol to obtain 2-(3-nitrophenyl)-4,5-diphenylimidazole (4c) (0.998 g, yield 95%). The same procedure was used for all other compounds. All 2,4,5-triarylimidazole derivatives were fully characterized by usual spectroscopic techniques.
Spectral data for products
Spectroscopic data
2,4,5-Triphenylimidazole ( 4a )
M.p.: 271–273 °C; IR (KBr) \(\overline{\upsilon}\) (cm−1): 1601 (C=N), 3038 (Ar–H), 3446 (NH) cm−1; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.24–7.58 (m, 13H, Ar–H), 8.09–8.11 (m, 2H, Ar–H), 12.71 (s, NH); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 125.7, 127.0, 127.6, 128.3, 128.6, 128.7, 128.9, 129.1, 129.2, 130.7, 130.8, 131.5, 131.6, 135.7, 137.6, 145.9, 146.0; Anal. calcd. for C21H16N2 (296.37): C, 85.11; H, 5.44; N, 9.45. Found: C, 84.91; H, 5.42; N, 9.41.
2-(2-Nitrophenyl)-4,5-diphenylimidazole ( 4b )
M.p.: 234–237 °C; IR (KBr) \(\overline{\upsilon}\) (cm−1): 1352 and 1526 (NO2), 1626 (C=N), 3069 (Ar–H), 3427 (NH) cm−1; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.25–7.50 (m, 10H, Ar–H), 7.66 (td, J 1 = 8.0, J 2 = 1.2 Hz, 1H, Ar–H), 7.81 (td, J 1 = 7.6, J 2 = 1.2 Hz, 1H, Ar–H), 7.95 (dd, J 1 = 8.0, J 2 = 0.8 Hz, 1H, Ar–H), 8.01 (dd, J 1 = 8.0, J 2 = 1.2 Hz, 1H, Ar–H), 12.99 (s, NH); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 123.9, 124.5, 127.2, 127.5, 128.5, 128.7, 129.2, 129.7, 130.0, 130.3, 131.1, 132.6, 135.2, 138.0, 141.5, 148.8; Anal. calcd. for C21H15N3O2 (341.36): C, 73.89; H, 4.43; N, 12.31. Found: C, 73.85; H, 4.41; N, 12.21.
2-(3-Nitrophenyl)-4,5-diphenylimidazole ( 4c )
M.p.: 316–319 °C; IR (KBr) \(\overline{\upsilon}\) (cm−1): 1348 and 1522 (NO2), 1629 (C=N), 3064 (Ar–H), 3424 (NH) cm−1; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.27–7.60 (m, 10H, Ar–H), 7.81 (t, J = 8.0 Hz, 1H, Ar–H), 8.23–8.25 (m, 1H, Ar–H), 8.53–8.55 (m, 1H, Ar–H), 8.98 (t, J = 2.0 Hz, 1H, Ar–H), 13.14 (s, NH); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 119.9, 123.1, 127.3, 127.6, 128.6, 128.8, 128.9, 129.2, 129.7, 130.9, 131.0, 131.1, 131.6, 132.2, 132.3, 135.2, 138.1, 143.7, 143.9, 148.8; Anal. calcd. for C21H15N3O2 (341.36): C, 73.89; H, 4.43; N, 12.31. Found: 73.85; H, 4.41; N, 12.21.
2-(4-Nitrophenyl)-4,5-diphenylimidazole ( 4d )
M.p.: 237–240 °C; IR (KBr) \(\overline{\upsilon}\) (cm−1): 1343 and 1518 (NO2), 1639 (C=N), 3050 (Ar–H), 3423 (NH) cm−1; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.32 (d, J = 8.8 Hz, 2H, Ar–H), 7.61 (d, J = 8.4 Hz, 1H, Ar–H), 7.85 (dd, J 1 = 7.2, J 2 = 2.0 Hz, 2H, Ar–H), 7.97 (d, J = 8.8 Hz, 2H, Ar–H), 8.17–8.44 (m, 7H, Ar–H), 13.75 (s, NH); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 123.3, 124.2, 124.4, 124.5, 124.8, 126.8, 128.4, 129.2, 129.4, 131.1, 135.9, 146.0, 146.6, 147.9, 149.4, 151.1, 163.3; Anal. calcd. for C21H15N3O2 (341.36): C, 73.89; H, 4.43; N, 12.31. Found: C, 73.86; H, 4.41; N, 12.25.
2-(3-Methoxyphenyl)-4,5-diphenylimidazole ( 4e )
M.p.: 225–227 °C; IR (KBr) \(\overline{\upsilon}\) (cm−1): 1240 (C–O), 1592 (C=N), 2964 (-CH), 3059 (Ar–H), 3416 (NH) cm−1; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 3.85 (s, 3H, CH3), 6.95–6.98 (m, 1H, Ar–H), 7.23–7.57 (m, 11H, Ar–H), 7.67–7.71 (m, 2H, Ar–H), 12.71 (s, NH); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 55.7, 110.6, 114.7, 118.1, 127.0, 127.5, 128.3, 128.7, 129.0, 129.2, 130.3, 131.5, 132.1, 135.6, 137.5, 145.8, 160.0; Anal. calcd. for C22H18N2O (326.39): C, 80.96; H, 5.56; N, 8.58. Found: C, 80.94; H, 5.53; N, 8.54.
2-(4-Methoxyphenyl)-4,5-diphenylimidazole ( 4f )
M.p.: 235–238 °C; IR (KBr) \(\overline{\upsilon}\) (cm−1): 1249 (C–O), 1659 (C=N), 2960 (-CH), 3059 (Ar–H), 3422 (NH) cm−1; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 3.83 (s, 3H, CH3), 7.06 (dd, J 1 = 6.8, J 2 = 2.0 Hz, 2H, Ar–H), 7.23–7.67 (m, 7H, Ar–H), 7.80–8.05 (m, 5H, Ar–H), 12.54 (s, NH); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 55.7, 114.6, 123.6, 126.9, 127.2, 127.6, 128.1, 128.6, 128.8, 129.1, 129.5, 129.6, 129.7, 130.0, 130.1, 131.7, 132.7, 135.8, 136.0, 137.3, 146.1, 159.9; Anal. calcd. for C22H18N2O (326.39): C, 80.96; H, 5.56; N, 8.58. Found: C, 80.92; H, 5.53; N, 8.54.
2-(3-Chlorophenyl)-4,5-diphenylimidazole ( 4g )
M.p.: 211–213 °C; IR (KBr) \(\overline{\upsilon}\) (cm−1): 778 (C–Cl), 1603 (C=N), 3060 (Ar–H), 3423 (NH) cm−1; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.26–7.58 (m, 12H, Ar–H), 8.07 (dt, J 1 = 8.0, J 2 = 1.2 Hz, 1H, Ar–H), 8.17 (t, J = 2.0 Hz, 1H, Ar–H), 12.87 (s, NH); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 124.2, 125.1, 127.2, 127.6, 128.4, 128.8, 129.1, 131.2, 132.7, 134.1, 135.4, 137.9, 144.4; Anal. calcd. for C21H15ClN2 (330.81): C, 76.24; H, 4.57; N, 8.47. Found: C, 76.22; H, 4.54; N, 8.42.
2-(4-Bromophenyl)-4,5-diphenylimidazole ( 4h )
M.p.: 209–212 °C; IR (KBr) \(\overline{\upsilon}\) (cm−1): 604 (C–Br), 1635 (C=N), 3064 (Ar–H), 3421 (NH) cm−1; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.25–7.72 (m, 10H, Ar–H), 7.82–8.06 (m, 4H, Ar–H), 12.82 (s, NH); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 121.9, 127.1, 127.5, 127.6, 128.4, 128.7, 128.9, 129.1, 129.2, 129.9, 130.0, 130.1, 131.3, 131.4, 132.2, 132.7, 135.4, 136.0, 137.8, 144.8, 144.9; Anal. calcd. for C21H15BrN2 (375.26): C, 67.21; H, 4.03; N,7.47. Found: C, 67.20; H, 4.00; N, 7.45.
4,5-Diphenyl-2-(thiophen-2-yl)imidazole ( 4i )
M.p.: 256–259 °C; IR (KBr) \(\overline{\upsilon}\) (cm−1): 849 (C–S), 1658 (C=N), 3057 (Ar–H), 3424 (NH) cm−1; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.16–7.34 (m, 3H, Thiophene H), 7.39–7.96 (m, 10H, Ar–H), 12.81 (s, NH); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 127.1, 127.6, 128.0, 128.2, 128.3, 128.4, 128.7, 128.8, 129.2, 130.0, 130.1, 131.2, 131.3, 132.7, 134.3, 134.4, 135.3, 136.0, 137.3; Anal. calcd. for C19H14N2S (302.39): C, 75.47; H, 4.67; N, 9.26. Found: C, 75.44; H, 4.64; N, 9.22.
2-(5-Methylfuran-2-yl)-4,5-diphenylimidazole ( 4j )
M.p.: 260–263 °C; IR (KBr) \(\overline{\upsilon}\) (cm−1): 1022 (C–O), 1629 (C=N), 2918 (-CH), 3056 (Ar–H), 3445 (NH) cm−1; 1H NMR (400 MHz, DMSO-d6) δ (ppm): 2.39 (s, 3H, CH3), 6.27 (dd, J 1 = 3.2, J 2 = 1.2 Hz, 1H, Furan H), 6.87 (d, J = 3.2 Hz, 1H, Furan H), 7.23–7.54 (m, 10H, Ar–H), 12.71 (s, NH); 13C NMR (100 MHz, DMSO-d6) δ (ppm): 13.9, 127.0, 127.6, 127.7, 128.2, 128.6, 128.7, 129.1, 130.0, 130.1, 131.2, 131.3, 132.7, 135.5, 136.1, 137.3, 139.1, 139.3, 144.6; Anal. calcd. for C20H16N2O (300.35): C, 79.98; H, 5.37; N, 9.33. Found: C, 79.94; H, 5.36; N, 9.32.
Results and discussion
Synthesis of catalyst
The catalyst A-MFGO was synthesized by a simple and inexpensive method as shown in Scheme 1. A-MFGO was evaluated using various methods as follows:
XRD analysis
The XRD patterns of GO, FGO, MFGO, and A-MFGO samples are shown in Fig. 1. GO shows a peak at 11.3° corresponding to d-spacing of 7.77 Å due to incorporation of oxygen-containing functional groups on graphite sheets (Fig. 1a). The XRD pattern of FGO (Fig. 1b) exhibits a new peak at 24.5°, corresponding to d-spacing of 3.61 Å, while the GO peak at 11.3° disappeared, indicating successful fluorination of GO. The decrease in the d-spacing between the layers in FGO is attributed to removal of some oxygen functionalities from GO after fluorination. Figure 1c, d presents the XRD patterns of MFGO and A-MFGO. Both modified samples (MFGO and A-MFGO) showed peaks at 24.5° and 24.3°, with d-spacing of 3.62 and 3.66 Å, respectively. These d-spacing values are lower than the value of 7.77 Å for GO due to the modification, proving successful modification of GO by introduction of 1,6-diaminohexane and 1,3-propane sultone groups. Furthermore, this indicates that the reaction occurred on the edges of GO [54, 55].

XRD patterns of a GO, b FGO, c MFGO, and d catalyst
FTIR spectral analysis
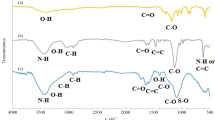
The FTIR spectrum was measured to distinguish between the functional groups on GO and the three products (Fig. 2). The FTIR spectrum of GO (Fig. 2a) exhibited a broad band at 3419 cm−1 due to O–H bond stretching. The peak at 1721 cm−1 is assigned to C=O bond stretching at the edges of GO. Other oxygen-containing groups were also observed, for example, C–O bond in carboxylic acid at 1383 cm−1, C–O bond in epoxy group at 1222 cm−1, and C–O bond in alcohol group at 1050 cm−1. These oxygen functional groups indicate that oxidation of graphite was successful. The two peaks at 1222 and 1050 cm−1, corresponding to C–O in epoxy and alcohol group, respectively, disappeared in the FTIR spectrum of FGO (Fig. 2b), while a new peak corresponding to C–F bond was observed at 1167 cm−1, indicating successful fluorination of GO. The FTIR spectrum of MFGO (Fig. 2c) exhibited slightly different absorption behavior. The peak at 1636 cm−1 is attributed to C=O bond in amide group. Besides, the peak of –CH2– group is observed at 2920 cm−1. These peaks confirm the reaction between –COOH groups on GO sheet and 1,6-diaminohexane. Finally, the FTIR data of A-MFGO (Fig. 2e) are clearly similar to those for MFGO, with the addition of a peak at 1035 cm−1, assigned to S=O bond in –SO3H group, and another peak at 741 cm−1, assigned to C–S bond, indicating successful attachment of –SO3H group onto GO sheet [56–58].

FTIR spectra of a GO, b FGO, c MFGO, and d catalyst
SEM and EDX analysis
Figure 3 shows SEM images of GO, FGO, MFGO, and A-MFGO. SEM is a useful technique to study the surface morphology of samples. In the SEM image of GO in Fig. 3a, a model folded morphology can be observed. After fluorination, less accumulation of FGO sheets is observed in Fig. 3b. To check the composition of the FGO, EDX analysis was also carried out (Fig. 4). The EDX spectrum revealed presence of C, O, and F elements in FGO, confirming presence of fluorine atoms on the surface of the FGO sheets and thus successful fluorination of GO. As shown in Fig. 3c, MFGO showed less accumulation, clearly revealing the efficiency of the modification method for prevention of restacking of GO sheets. Figure 3d shows a SEM image of A-MFGO, which reveals large particles with low rank, indicating presence of bulk carbon. This also suggests that –NH2 groups on MFGO sheet and 1,3-propane sultone reacted [59–61].

SEM images of a GO, b FGO, c MFGO, and d catalyst

EDX spectrum of FGO
XPS analysis
The XPS results for the catalyst A-MFGO are shown in Fig. 5. The XPS survey spectrum of A-MFGO reveals the presence of elements including sulfur, carbon, oxygen, nitrogen, and fluorine (Fig. 5a). The observation of the fluorine peak demonstrates that fluorine atoms were introduced into the graphene oxide. The total survey of the A-MFGO also displays presence of nitrogen and sulfur elements. The C 1s region of the XPS spectrum of A-MFGO could be divided into eight components at 284.6, 285.4, 285.8, 286.8, 287.1, 287.8, 288.2, and 288.8 eV (Fig. 5b), associated with C–C, C–N, C–CF, C–S, C=O, O=C–N, C–F, and O=C–O groups, respectively. Two fluorine groups are observed at 285.8 eV (C–CF) and 288.2 eV (C–F), while peaks of C–OH and C–O–C group did not appear. All these observations support successful fluorination of GO. The peaks at 286.8 eV (C–S), 285.4 eV (C–N), and 287.8 eV (O=C–N) from the surface of the GO sheets indicate successful functionalization of GO. Moreover, the XPS spectrum (Fig. 5c) showed a peak at 168.2 eV corresponding to –SO3H group, indicating incorporation of sulfonic acid group [62–65].

a XPS survey spectrum of catalyst, b XPS carbon region spectrum for catalyst, and c sulfur region of catalyst with curve fitting results
Optimization of reaction conditions
Firstly, the reaction between benzil (5 mmol), 3-nitrobenzaldehyde (5 mmol), and ammonium acetate (20 mmol) was selected as model reaction (4c) to identify the optimum reaction conditions (Scheme 2). Various solvents and catalysts were utilized, and the results are presented in Tables 1 and 2. Ethanol with catalytic amount of sulfuric acid was clearly the best choice for this reaction (Table 1, entry 3). The efficiency of the various catalysts was also investigated. The best yield was obtained with A-MFGO (Table 2, entry 6). In absence of catalyst, product formation was not observed (Table 2, entry 1). The amount of A-MFGO was also evaluated using the model reaction. The maximum yield was obtained with 0.15 g A-MFGO (Table 3, entry 4). Further increase to 0.2 g did not change the yield (Table 3, entry 5).

After successful optimization of the reaction conditions, the series of 2,4,5-triarylimidazole derivatives 4a–j were synthesized (Table 4). Under these conditions, the reaction between a variety of aromatic aldehydes and benzil (Scheme 2) was carried out in presence of A-MFGO as catalyst to synthesize 2,4,5-triarylimidazole derivatives. Excellent product yield was observed, and reaction workup was simple. The results obtained are summarized in Table 4. The structures of all products obtained were characterized well by spectral analyses (FTIR, 1H NMR and 13C NMR). Moreover, the melting points were recorded and compared with those reported in literature, as given in Table 4 [66–72]. The FTIR spectra of compounds 4a–j demonstrated peaks at 1592–1659 and 3416–3446 cm−1 for (C=N) and (N–H) groups, respectively. The 1H NMR spectra exhibited bands at 12.54–13.75 ppm for N–H proton of imidazole moiety.

Finally, a mechanism for the synthesis of the 2,4,5-triarylimidazole derivatives using the catalyst is proposed in Scheme 3. The initial step involves H-bond formation between catalyst and aldehyde, which activates the carbonyl group of the aldehyde to give hydroxylamine intermediate [I]. Dehydration of hydroxylamine leads to formation of imine intermediate [II]. Nucleophilic attack of another NH3 on imine forms diamine intermediate [III]. This stage is followed by nucleophilic attack of diamine intermediate [III] on activated carbonyl group of benzil to give cycle intermediate [IV], which later undergoes release of a water molecule [V] and proton transfer to produce 2,4,5-triarylimidazole derivatives [VI].

Proposed mechanism for synthesis of 2,4,5-triarylimidazole derivatives
Reusability of catalyst
The reusability of the catalyst A-MFGO was also investigated using the model reaction. After completion of the reaction, the catalyst was filtered and washed with dichloromethane. Recovered catalyst could be dried and further reused in another reaction. The recovered catalyst could be reused five times without addition of fresh catalyst (Fig. 6) with the same purity as the first run, indicating that A-MFGO shows high catalytic activity.

Reusability of catalyst in reaction of benzil with 3-nitrobenzaldehyde
Conclusions
A highly efficient approach with mild reaction conditions and operationally simple protocol for synthesis of 2,4,5-triarylimidazole derivatives via condensation of benzil, various aromatic aldehydes, and ammonium acetate as ammonia source in ethanol is presented, using A-MFGO as inexpensive, recyclable, and effective catalyst. The catalyst could be reused several times without activity loss. The easy workup, high yield, simple operation, short reaction time, and safer reaction conditions of this method using a reusable catalyst with simple preparation are the highlights of this method. This research also provides interesting ideas regarding fluorination and chemical modification of graphene oxide.
References
G. Eda, Y.Y. Lin, C. Mattevi, H. Yamaguchi, H.A. Chen, I.S. Chen, C.W. Chen, M. Chhowalla, Adv. Mater. 22, 505 (2010)
M. Li, S.K. Cushing, X. Zhou, S. Guo, N.Q. Wu, J. Mater. Chem. 22, 23374 (2012)
A. Ahmadi, B. Ramezanzadeh, M. Mahdavian, RSC Adv. 6, 54102 (2016)
D. Chen, H. Feng, J. Li, Chem. Rev. 112, 6027 (2012)
G. Xu, T. Shi, M. Li, F. Yu, Y. Chen, Res. Chem. Intermed. 41, 8499 (2015)
Q. Xu, M. Zeng, Z. Feng, D. Yin, Y. Huang, Y. Chen, C. Yan, R. Li, Y. Gu, RSC Adv. 6, 31484 (2016)
Y. Ahmad, E. Disa, M. Dubois, K. Gurin, V. Dubois, W. Zhang, P. Bonnet, F. Masin, L. Vidal, D.A. Ivanov, A. Hamwi, Carbon 50, 3897 (2012)
A. Maio, D. Giallombardo, R. Scaffaro, A.P. Piccionellob, I. Pibiri, RSC Adv. 6, 46037 (2016)
M.G. Campbell, T. Ritter, Chem. Rev. 115, 612 (2015)
F. Karlicky, K.K.R. Datta, M. Otyepka, R. Zboril, ACS Nano 7, 6434 (2013)
R. Stine, W.K. Lee, K.E. Whitener, J.T. Robinson, P.E. Sheehan, Nano Lett. 13, 4311 (2013)
H. Zhang, L. Fan, H. Dong, P. Zhang, K. Nie, J. Zhong, Y. Li, J. Guo, X. Sun, ACS Appl. Mater. Interfaces 8, 8652 (2016)
H. Chang, J. Cheng, X. Liu, J. Gao, M. Li, J. Li, X. Tao, F. Ding, Z. Zheng, Chem. Eur. J. 17, 8896 (2011)
M. Dubois, K. Guerin, Y. Ahmad, N. Batisse, M. Mar, L. Frezet, W. Hourani, J.L. Bubendorff, J. Parmentier, S.H. Garreau, L. Simon, Carbon 77, 688 (2014)
P. Gong, Z. Wang, Z. Li, Y. Mi, J. Sun, L. Niu, H. Wang, J. Wang, S. Yang, RSC Adv. 3, 6327 (2013)
X. Ye, L. Ma, Z. Yang, J. Wang, H. Wang, S. Yang, ACS Appl. Mater. Interfaces 8, 7483 (2016)
A. Zajac, P. Pelikan, J. Minar, J. Noga, M. Straka, P. Banacky, S. Biskupic, J. Solid State Chem. 150, 286 (2000)
P. Lazar, E. Otyepkova, F. Karlicky, K. Cepe, M. Otyepka, Carbon 94, 804 (2015)
P.M. Sudeep, J.T. Tijerina, P.M. Ajayan, T.N. Narayananc, M.R. Anantharaman, RSC Adv. 4, 24887 (2014)
M. Antolini, A. Bozzoli, C. Ghiron, G. Kennedy, T. Rossi, A. Ursini, Bioorg. Med. Chem. Lett. 9, 1023 (1999)
L. Wang, K.W. Woods, Q. Li, K.J. Barr, R.W. McCroskey, S.M. Hannick, L. Gherke, R.B. Credo, Y.H. Hui, K. Marsh, R. Warner, J.Y. Lee, N.Z. Mozng, D. Frost, S.H. Rosenberg, H.L. Sham, J. Med. Chem. 45, 1697 (2002)
V.G. Silva, R.O. Silva, S.R.B. Damasceno, N.S. Carvalho, R.S. Prudencio, K.S. Aragao, M.A. Guimaraes, S.A. Campos, L.M.C. Veras, M. Godejohann, J.R.S.A. Leite, A.L.R. Barbosa, J.V.R. Medeiros, J. Nat. Prod. 76, 1071 (2013)
J.T. Li, B.H. Chen, Y.W. Li, X.L. Sun, IJAPBC 1, 287 (2012)
M. Kidwai, S. Saxena, S. Rastogi, Bull. Korean Chem. Soc. 26, 2051 (2005)
D.S. Weinstein, W. Liu, K. Ngu, C. Langevine, D.W. Combs, S. Zhuang, C. Chen, C.S. Madsen, T.W. Harper, J.A. Robl, Bioorg. Med. Chem. Lett. 17, 5115 (2007)
A.K. Takle, M.J.B. Brown, S. Davies, D.K. Dean, G. Francis, A. Gaiba, A.W. Hird, F.D. King, P.J. Lovell, A. Naylor, A.D. Reith, J.G. Steadman, D.M. Wilson, Bioorg. Med. Chem. Lett. 16, 378 (2006)
S. Li, M. Hong, J. Am. Chem. Soc. 133, 1534 (2011)
T. Kottke, K. Sander, L. Weizel, E.H. Schneider, R. Seifert, H. Stark, Eur. J. Pharmacol. 654, 200 (2011)
P. Wasserscheid, W. Keim, Angew. Chem. Int. Ed. Eng. 39, 37872 (2000)
B. Maleki, H. Eshghi, A. Khojastehnezhad, R. Tayebee, S.S. Ashrafi, G.E. Kahoo, F. Moeinpour, RSC Adv. 5, 64850 (2015)
C.C. Zeng, N.T. Zhang, C.M. Lam, R.D. Little, Org. Lett. 14, 1314 (2012)
C. Kurumurthy, G.S. Kumar, G.M. Reddy, P. Nagender, P.S. Rao, B. Narsaiah, Res. Chem. Intermed. 38, 359 (2012)
X. Deng, Z. Zhou, A. Zhang, G. Xie, Res. Chem. Intermed. 39, 1101 (2013)
N.N. Lu, S.J. Yoo, L.J. Li, C.C. Zeng, R.D. Little, Electrochim. Acta 142, 254 (2014)
M. Dabiri, S.K. Movahed, D.I. MaGee, Res. Chem. Intermed. 41, 3335 (2015)
K. Pradhan, B.K. Tiwary, M. Hossain, R. Chakraborty, A.K. Nanda, RSC Adv. 6, 10743 (2016)
L.M. Wang, Y.H. Wang, H. Tian, Y.F. Yao, J.H. Shao, B. Liu, J. Fluor. Chem. 127, 1570 (2006)
M.G. Shen, C. Cai, W.B. Yi, J. Fluor. Chem. 129, 541 (2008)
S.D. Sharma, P. Hazarika, D. Konwar, Tetrahedron Lett. 49, 2216 (2008)
J. Wang, R. Mason, D.V. Derveer, K. Feng, X.R. Bu, J. Org. Chem. 68, 5415 (2003)
K. Shelke, G. Kakade, B. Shingate, M. Shingare, Rasayan J. Chem. 1, 489 (2008)
N.D. Kokare, J.N. Sangshetti, D.B. Shinde, Synthesis 18, 2829 (2007)
S.A. Siddiqui, U.C. Narkhede, S.S. Palimkar, T. Daniel, R.J. Lahoti, K.V. Srinivasan, Tetrahedron 61, 3539 (2005)
A. Shaabani, A. Rahmati, J. Mol. Catal. A Chem. 249, 246 (2006)
K.F. Shelke, S.B. Sapkal, G.K. Kakade, B.B. Shingate, M.S. Shingare, Green Chem. Lett. Rev. 3, 27 (2010)
K.F. Shelke, S.B. Sapkal, M.S. Shingare, Chin. Chem. Lett. 20, 283 (2009)
J.N. Sangshetti, N.D. Kokare, S.A. Kotharkar, D.B. Shinde, Monatsh. Chem. 139, 125 (2008)
F.K. Behbahani, T. Yektanezhad, A.R. Khorrami, Heterocycles 81, 2313 (2010)
J. Safari, S.D. Khalili, M. Rezaei, S.H. Banitaba, F. Meshkani, Monatsh. Chem. 141, 1339 (2010)
Y.R. Girish, K.S.S. Kumar, K.N. Thimmaiah, K.S. Rangappa, S. Shashikanth, RSC Adv. 5, 75533 (2015)
R. Ramesh, P. Vadivel, S. Maheswari, A. Lalitha, Res. Chem. Intermed. 42, 7625 (2016)
W.S. Hummers, R.E. Offeman, J. Am. Chem. Soc. 80, 1339 (1958)
S. Mallakpour, E. Kowsari, J. Polym. Sci. Part A Polym. Chem. 43, 6545 (2005)
G. Shao, Y. Lu, F. Wu, C. Yang, F. Zeng, Q. Wu, J. Mater. Sci. 47, 4400 (2012)
H.O. Dogan, D. Ekinci, U. Demir, Surf. Sci. 611, 54 (2013)
W. Yan, Y. Huang, Y. Xu, L. Huang, Y. Chen, J. Nanosci. Nanotechnol. 12, 2270 (2012)
K. Samanta, S. Some, Y. Kim, Y. Yoon, M. Min, S.M. Lee, Y. Park, H. Lee, Chem. Commun. 49, 8991 (2013)
M. Liu, G. Liu, Y. Zhou, K. Han, H. Ye, J. Mol. Catal. A Chem. 408, 85 (2015)
A. Dimiev, D.V. Kosynkin, L.B. Alemany, P. Chaguine, J.M. Tour, J. Am. Chem. Soc. 134, 2815 (2012)
S.K. Yadav, J.H. Lee, H. Park, S.M. Hong, T.H. Han, C.M. Koo, Bull. Korean Chem. Soc. 35, 2139 (2014)
M. Nurhadi, J. Efendi, S.L. Lee, T.M.I. Mahlia, S. Chandren, C.S. Ho, H. Nur, J. Taiwan Inst. Chem. Eng. 46, 183 (2015)
M.Y. Lin, C.E. Chang, C.H. Wang, C.F. Su, C. Chen, S.C. Lee, S.Y. Lin, Appl. Phys. Lett. 105, 073501-1 (2014)
W. Zheng, R. Tan, S. Yin, Y. Zhang, G. Zhao, Y. Chen, D. Yin, Catal. Sci. Technol. 5, 2092 (2015)
M. Saikia, L. Saikia, RSC Adv. 6, 15846 (2016)
R. Xing, Y. Li, H. Yu, Chem. Commun. 52, 390 (2016)
A.A. Marzouk, V.M. Abbasov, A.H. Talybov, S.K. Mohamed, World J. Org. Chem. 1, 6 (2013)
S. Samai, G.C. Nandi, P. Singh, M.S. Singh, Tetrahedron 65, 10155 (2009)
M.M. Heravi, K. Bakhtiari, H.A. Oskooie, S. Taheri, J. Mol. Catal. A Chem. 263, 279 (2007)
H.N. Roy, M.M. Rahman, P.K. Pramanick, Indian J. Chem. 52B, 153 (2013)
A. Patel, S.Y. Sharp, K. Hall, W. Lewis, M.F.G. Stevens, P. Workman, C.J. Moody, Org. Biomol. Chem. 14, 3889 (2016)
J. Safari, S. Naseh, Z. Zarnegar, Z. Akbari, J. Taibah Univ. Sci. 8, 323 (2014)
M. Kidwai, P. Mothsra, V. Bansal, R. Goyal, Monatsh. Chem. 137, 1189 (2006)
Acknowledgements
The authors wish to gratefully thank the Research Affairs Division at Amirkabir University of Technology (AUT), Tehran, for financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hanoon, H.D., Kowsari, E., Abdouss, M. et al. Highly efficient and simple protocol for synthesis of 2,4,5-triarylimidazole derivatives from benzil using fluorinated graphene oxide as effective and reusable catalyst. Res Chem Intermed 43, 4023–4041 (2017). https://doi.org/10.1007/s11164-016-2847-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-016-2847-6