
Abstract
Two Schiff bases, HL1 and HL2, were synthesized in two different reactions involving 2-hydroxynaphthaldehyde with 2-amino-6-methylbenzothiazole and 2-amino-6-florobenzothiazole respectively. Copper(II) and nickel(II) complexes of the Schiff bases were subsequently prepared in 1:1 metal-to-ligand stoichiometric reactions. The compounds were characterized extensively by 1H NMR, 13C NMR, Dept-90, UV–Vis, and IR spectroscopic techniques, magnetic susceptibility, TGA, DTG, and molar conductivity analysis. The spectroscopic results confirm bidentate nature of the Schiff bases and a four coordinate geometry for all the complexes: [CuL1ClH2O], [NiL1ClH2O], [Cu(L2)2], and [NiL2ClH2O]. Quantum chemical studies gave fully optimized geometries of the Schiff bases and metal complexes using the 6-31+g(d,p) basis set. The compounds were studied for their in vitro antibacterial activities against some selected Gram-positive and Gram-negative bacteria, using agar well diffusion. The metal complexes exhibited better antibacterial activities compared to the free ligand due to the effects of chelation, which improved the lipophilicity. Furthermore, the antioxidant potentials of the compounds were ascertained using DPPH radical scavenging and ferrous chelating assay. The copper complexes had the best antioxidant properties of all the tested compounds. The results of the biological analysis were in agreement with the theoretical data from quantum chemical calculations. The study presented biologically active coordination compounds with benzothiazole moiety that could be used as compounds of interest in the drug discovery processes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Heterocyclic compounds are group of compounds known to chemists for centuries and are considered very useful due to their diverse chemical reactivities and vast biological applications [1–6]. Their importance expands beyond chemistry as they are also part of biomolecules such as protein, DNA, RNA, and vitamins [2]. More specifically, nitrogen, sulfur, and oxygen containing heterocyclic compounds are of immense importance in drug discovery processes [1]; they are found in many therapeutic drugs such as metroindazole, thiabendazole, fluconazole, and riluzole [7].
Benzothiazole is a unique example of a heterocyclic compound with a thiazole ring fused to a benzene ring and has received a lot of interest due to its diversified molecular design and remarkable optical, liquid, and electronic properties [8]. The compound and its derivatives have been reported to possess a wide spectrum of biological applications such as antitumor [9, 10], antimicrobial [11, 12], schictosomicidal [13], anti-inflammatory [14, 15], anticonvulsant [16, 17], antidiabetic [18], antipsychotic [19, 20], and diuretic [8, 21]. Interestingly, the nature of the substituent on the benzothiazole moiety plays an important role in the overall biological properties of the compound. For example, phenyl-substituted benzothiazole derivatives have been reported to exhibit very intensive antitumor activity [22], especially those containing 2-substituted-6-nitro- and 6-aminobenzothiazole [23] and fluorobenzothiazoles [24].
Organic reactions involving the condensation of two or more molecules to obtain new compounds are common in drug design processes as compounds with new biological behaviours could be obtained [8]. Schiff bases are products of condensation reactions between primary amines and aldehydes or ketones synthesized under specific conditions. Schiff bases are important due to their ability to stabilize metal ions of various oxidation states, participation in numerous catalytic and industrial applications and broad spectrum biological activities [25, 26]. More specifically, Schiff bases of benzothiazole derivatives and their metal complexes have been reported to possess antibacterial [27], antifungal [28], antioxidant [29] anti-tumour, and anticancer [30] properties.
However, a detailed literature search has shown a dearth of information on syntheses, quantum chemical calculation, thermal, antioxidant, and antibacterial studies of 3-[(5-methyl-benzothiazol-2-ylimino)-methyl]-naphthalen-2-ol (HL1) and 3-[(5-floro-benzothiazol-2-ylimino)-methyl]-naphthalen-2-ol(HL2) and their Ni(II) and Cu(II) complexes [31–33]. Hence, this study reports the synthesis and characterization of metal (II) complexes of 3-[(5-methyl-benzothiazol-2-ylimino)-methyl]-naphthalen-2-ol (HL1) and 3-[(5-floro-benzothiazol-2-ylimino)-methyl]-naphthalen-2-ol (HL2). Since crystals of good quality could not be obtained, DFT studies will be used to investigate the proposed geometries of the Schiff bases and their metal complexes. The dipole moments, FMO energies, and reaction energies of the metal complexes will also be ascertained. Furthermore, the effectiveness of these compounds as broad-spectrum antibacterial agents (in vitro) will be verified via agar well diffusion assay and their antioxidant potentials evaluated via DPPH radical scavenging and ferrous chelating assay. The ligands and their metal complexes are new, being reported here for the first time, as a continuation of the research activities of our group on novel metal (II) complexes with potentials as broad spectrum antibacterial and antioxidant compounds [31–34].
Experimental
Materials
2-Hydroxy-1-napthaldehde, 2-amino-6-methylbenzothiazole, 2-amino-6-florobenzothiazole, 2,2-diphenyl-1-picrylhydrazyl (DPPH), copper(II) chloride dihydrate, nickel(II)chloride hexahydrate, 1,10-phenantroline, iron(II) sulphate heptahydrate, and triethylamine were purchased from BDH and Aldrich Chemicals Germany and were used as received. Ethanol and methanol were distilled before use according to standard procedures.
Physical measurements
Melting points (uncorrected) were determined using the Stuart scientific melting point SMP1 machine. Electronic and IR (KBr disc) spectra of the ketoamine and its metal complexes were recorded on Perkin-Elmer λ20 UV–Vis and Perkin-Elmer FT-IR Spectrum BX spectrophotometers, respectively. The elemental analysis was performed on an Elementar vario EL Cube set up for carbon, hydrogen, nitrogen, and sulfur (CHNS) analysis. The thermal analysis was done using SDTQ 600 thermal instrument with alumina crucibles and heated at a rate of 10 °C min−1 under nitrogen atmosphere. Furthermore, NMR measurements were done with a 300 MHz Bruker Advance III NMR spectrometer, room temperature magnetic moments, and molar conductance measurements were recorded on Sherwood Susceptibility balance MSB Mark 1 and Hanna conductivity model H19991300 meters, respectively. The percentage metal analyses were carried out using complexometric titration of the respective metal solutions against EDTA with murexide as an indicator.
Syntheses
Synthesis of 3-[(5-methyl-benzothiazol-2-ylimino)-methyl]-naphthalen-2-ol (HL1)
The ligand, HL1 was synthesized according to methods reported in literature [27, 31]. About 1.5 g (7.7 mmol) of 2-amino-6-methylbenzothiazole was added neat in bits to a stirring solution of 1.32 g (7.7 mmol) of 2-hydroxynaphthaldehyde in 20 mL of ethanol. A few drops of acetic acid were added and the solution was subsequently refluxed for 3 h. The resultant yellow coloured precipitates were filtered, dried and stored over silica gel.
CHNS (C19H14N2OS) Calcd: C, 71.67; H, 4.44; N, 8.80; S, 10.07. Found: C, 71.69; H, 4.54; N, 8.84; S, 10.10; Electronic spectra (kK): 30.68, 36.77 (kK = 1000 cm−1); IR(KBr)v/cm−1: 3397 (OH str), 3055 (Ar–CH str), 2922 (Aldehydic H–C=N), 2739 (CH3 str), 1620 (C=N str imine), 1246 (C–OH), 1601 (C=N str benzothiazole), 1557(C=C str), 1323 (CH3 bend); 1H-NMR (300 MHz, DMSO) δ (ppm): 14.426(s, H–C=N), 10.037(s, OH), 7.080–8.237(m, Ar–H), 2.367 (s, CH3); 13C-NMR (75 MHz, DMSO) δ (ppm): 160.78 (H–C=N imine), 137.73–118.84 (Ar–CH), 21.55(CH3); Dept-90 (75 MHz, DMSO) δ (ppm); 137.73–118.84 (Ar–CH), 160.78 (H–C=N imine); Molar mass: 318.14; Yield: (76.80%); Color: yellow; m.pt: 180–182 °C.
Synthesis of 3-[(5-floro-benzothiazol-2-ylimino)-methyl]-naphthalen-2-ol (HL2)
The ligand HL2 was synthesized according to methods reported in the literature [27, 33]. About 1.5 g (8.9 mmol) of 2-amino-6-florobenzothiazole was added to a stirring solution of 1.54 g (8.9 mmol) of 2-hydroxynaphthaldehyde dissolved in 20 mL ethanol. Few drops of acetic acid were then added and the solution was refluxed for 3 h. The resultant orange coloured precipitates were filtered, dried, and stored over silica gel.
CHNS (C18H11N2OSF) Calcd: C, 67.06; H, 3.45; N, 8.69; S, 9.95. Found: C, 67.08; H, 3.47; N, 8.71; S, 9.97; Electronic spectra (kK): 30.58, 36.50 (kK = 1000 cm−1); IR(KBr)v/cm−1: 3385 (str OH), 3061 (Ar–CH),1622 (str C=N imine), 1601 (str C=N benzothiazole), 1566 (str C=C), 1250 (C–OH); 1H-NMR (300 MHz, DMSO) δ (ppm): 14.325(s, H–C=N), 10.070(s, OH), 7.001–8.324(m, Ar–H). 13C-NMR (75 MHz, DMSO) δ (ppm): 161.33 (H–C=N imine), 139.14–107.52 (Ar–CH); Dept-90 (75 MHz, DMSO) δ (ppm); 139.14–107.52 (Ar–CH), 161.33 (H–C=N imine); Molar mass: 322.35; Yield: 74%; Color: Orange; m. pt: 140–142 °C.
Syntheses of metal complexes
The metal complexes were synthesized according to reported procedures [32–34]. About 0.34 g (1.0 mmol) of 3-[(5-methyl-benzothiazol-2-ylimino)-methyl]-naphthalen-2-ol (HL1) was dissolved in 20 mL methanol and subsequently added dropwise to an equimolar concentration of a metal(II) chloride salt in 10 mL of methanol, while stirring and maintaining a 50 °C solution temperature. The resulting homogeneous solution was then buffered with triethylamine to a pH of 9 and refluxed for 3 h. The precipitates formed were filtered under vacuum, washed with methanol and dried over silica gel. The same procedure was used in the syntheses of metal complexes of 3-[(5-floro-benzothiazol-2-ylimino)-methyl]-naphthalen-2-ol (HL2) (Scheme 1).
NiC19H15N2SO2Cl (NiL1): CHNS, Calcd (%): C, 53.12; H, 3.53; N, 6.52; S, 7.46%. Found (%): C, 53.31; H, 3.55; N, 6.70; S, 7.45; Electronic spectra (cm−1): 37,450, 30,120, 22,990 (ε = 220 L mol−1 cm−1); IR(KBr) v/cm−1: 3333b (str OH), 3057 m (Ar–CH), 2920 m (Aldehydic C–H), 1618(str C=N imine), 1603 (str C=N benzothizole), 1578 s, 1541 s (str C=C), 1248 s (C–OH), 457 m (M–O), 517(M–N); Conductance (Ω−1 cm2 mol−1):16.80; µ eff (B.M): 4.29; Molar mass: 429.61; yield: 47%; Colour: Red; M.pt: 240 °C; %Ni, Calc.(Anal), 13.67(13.69).
CuC19H15N2SO2Cl (CuL1): CHNS, Calcd (%): C, 52.52; H, 3.49; N, 6.45; S, 7.38%. Found (%): C, 52.57; H, 3.51; N, 6.70; S, 7.40%; Electronic spectra (cm−1): 29,560, 37,760, 23,700, 9600 (ε = 180 L mol−1 cm−1); IR(KBr) v/cm−1: 3511b (str OH), 3055 m (Ar–CH), 2938 m (Aldehydic C–H), 1616(str C=N imine), 1601 (str C=N benzothizole), 1576 s, 1541 s (str C=C), 1252 s (C–OH),459 m (M–O), 527(M–N); Conductance (Ω−1 cm2 mol−1):19.0; µ eff (B.M): 1.92; Molar mass: 434.47; yield: 87%; Colour: Brown; M.pt: 201–202 °C; %Cu, Calc.(Anal), 14.62(14.64).
CuC36H26N4S2O5F2 (CuL2): CHNS, Calcd (%): C, 56.82; H, 3.45; N, 7.37; S, 8.43%. Found (%): C, 56.87; H, 3.49; N, 7.40; S, 8.45%; Electronic spectra (cm−1): 36,760, 23,700, 9560 (ε = 198 L mol−1 cm−1); IR(KBr) v/cm−1: 3432b (str OH), 3057 m (Ar–CH), 2938 m (Aldehydic C–H), 1618(str C=N imine), 1601 (str C=N benzothizole), 1570 s, 1541 s (str C=C), 1252 s (C–OH), 467 m (M–O), 527(M–N); Conductance (Ω−1 cm2 mol−1):20.10; µ eff (B.M): 1.87; Molar mass: 760.33; yield: 80%; Colour: Brown; M.pt: 156 °C; %Cu, Calc.(Anal), 8.35(8.37).
NiC18H12N2SO2FCl (NiL2): CHNS, Calcd (%): C, 49.86; H, 2.80; N, 6.46; S, 7.39%. Found (%): C, 49.90; H, 2.87; N, 6.50; S, 7.40%. Electronic spectra (cm−1): 36,760, 22,940, 18,080 and 15,220(ε = 200 L mol−1 cm−1); IR(KBr) v/cm−1: 3385b (str OH), 3048 m (Ar–CH), 2922 m (Aldehydic C–H), 1620(str C=N imine), 1603 (str C=N benzothizole), 1578 s, 1541 s (str C=C), 1252 s (C–OH), 457 m (M–O), 516 (M–N); Conductance (Ω−1 cm2 mol−1):11.45; µ eff (B.M): 3.90; Molar mass: 433.55; yield: 60%; Colour: Red; M.pt: 250 °C; %Ni, Calc.(Anal), 13.54(13.55).

Formation of metal complexes of 3-[(5-methyl-benzothiazol-2-ylimino)-methyl]-naphthalen-2-ol (HL1) and 3-[(5-floro-benzothiazol-2-ylimino)-methyl]-naphthalen-2-ol (HL2)
Computational details
Geometries of the ligands and metal complexes were fully optimized in vacuo without symmetry constraint. Density functional theory (DFT) method was adopted and the calculations were carried out using the Beckethree-Parameter hybrid functional combined with the Lee–Yang–Parr correlation functional (B3LYP) [35, 36]. The B3LYP is considered one of the most popular and accurate DFT functionals [37]. It has been reported to produce good results at tolerable computational time for geometry optimization of some transition metal complexes [38–41]. The double-zeta polarized basis, 6-31+G(d,p) that places diffuse functions and d-type polarization functions on non-hydrogenic atoms, and p-type polarization functions on hydrogenic atoms was used for the non-metallic atoms. The metal atoms/ions were described by the LANL2DZ relativistic pseudopotential, which has proven dependable for quantum chemical calculations on several transition metal complexes [40–50].
All the calculations were carried out with the aid of Gaussian 09 software suite [51]. The optimized structures were confirmed to correspond to their respective ground state energy minima by the absence of imaginary frequency in the calculated vibrational spectra.
The formation of metal complexes was assumed to proceed via the general equation:
where M2+ is the divalent metal ion, L represents the ligand(s), and M–Lcomplex is the metal–ligand complex.
The reaction energy (ΔE) was calculated using the general equation:
where \(E_{{{\text{M}} - {\text{L}}_{\text{complex}} }}\) is the energy of the metal complex, E Ligand is the energy of the ligand, and E 2+M is the energy of the metal ion.
Biological studies
DPPH free-radical scavenging assay
2,2-Diphenyl-1-picrylhydrazyl (DPPH) is a stable free radical that has been widely used as a tool to estimate the free-radical scavenging ability of antioxidants. The reduction capacity of the DPPH radicals was determined by the decrease in its UV–vis absorbance which is induced by the presence of antioxidants, according to reported methods [52] with a few modifications. In the assay, the reacting mixtures consists of 0.4 mL of the test compounds (ligands and their metal complexes) or the standard drug (acetic acid) diluted to different concentrations (50, 100, and 200 μg mL−1) and 2.6 mL of a 0.025 g L−1 DPPH in DMSO. The mixture was vigorously shaken and left to stand at room temperature in the dark for 30 min. The absorbance was measured at 517 nm against a blank solution of DMSO [52]. The experiment was conducted in triplicate and the ability of the compounds to scavenge the DPPH radical was calculated using the following formula:
A o, is the absorbance of the control at 30 min; A s, is the absorbance of the sample at 30 min.
Ferrous ion chelating ability studies
The ferrous ion-chelating ability of the compounds was determined by a standard colorimetric method [53]. For this study, metal chelating activity of the compounds was monitored by the decrease in absorbance of the Fe2+–1,10-phenanthroline complex at 546 nm. Briefly, 0.4 mL of samples (50–200 μg mL−1, in methanol) was added to FeSO4·7H2O solution (1 mL, 400 μM). To initiate the reaction, 1 mL of 1,10-phenantrolin (5.0 mmol L−1, in methanol) was added to the solution. The mixture was vigorously shaken and kept at room temperature for 15 min. The absorbance of the resulting mixture was read at 546 nm. The control solution contained 2 mL of methanol with 1 mL of 1,10-phenantroline (50 mg in 100 mL of methanol) and 1 mL FeSO4·7H2O (400 μM). Ascorbic acid was used as the standard drug [54]. The experiment was conducted in triplicate and percentage ferrous ion-chelating ability was expressed as:
A c is the absorbance of control reaction, and A s is the absorbance of sample.
Antibacterial assay (agar well diffusion method)
The assay was carried out on the ligands and their metal (II) complexes in vitro using literature procedures [33]. The bacterial strains used in this work were identified clinical strains of Gram-negative bacteria (K. oxytoca, P. aeruginosa, E. coli) and Gram-positive bacteria (B. cereus and S. aureus) obtained from University College Hospital, Ibadan, Nigeria. The choice of bacterial strains were due to their clinical and pharmacological importance. The bacterial organisms were activated from nutrient slope and grown in nutrient broth at 37 °C for 24 h, after which the surfaces of Petri dishes were uniformly inoculated with 0.2 mL of 24-h-old test of 0.5 McFarland bacterial culture. Using a sterile cork borer, 6 mm wells were bored into the agar and 10 mg mL−1 solution of each test compound in DMSO was introduced to the well. The plates were thereafter allowed to stand on the bench for 30 min before incubation at 37 °C for 24 h, after which the inhibitory zone (in mm) was taken as a measure of their antibacterial activities. The antibacterial analysis was done in duplicates.
Statistical analysis
The biological applications results were given as mean ± S.D. of the two (for the antibacterial study) or three (for the antioxidant studies) parallel measurements. Analysis of variance was performed by reported procedures using SPSS 10.5 software. Significant differences between means were determined by Tukey’s HSD tests. p values of <0.05 were regarded as significant.
Results and discussion
The synthesized Schiff bases and metal complexes were found to be stable on exposure to air and were soluble in only coordinating solvents like DMF and DMSO. All efforts to grow crystals of the complexes were abortive; however, other experimental methods used to predict the geometry of complexes in the absence of crystals were adhered to [55–57].
Elemental analysis and molar conductivity measurements
The results of elemental analysis for the metal complexes were in good agreement with the calculated values showing that the complexes have 1:1 metal–ligand stoichiometric ratio for [NiL1ClH2O] [CuL1ClH2O] [NiL2ClH2O] complexes and 2:1 metal–ligand stoichiometric ratio for [Cu(L2)2]3H2O complex. The analytical data were in good agreement with the proposed molecular formula of the ligands and complexes. The molar conductivities of the complexes in 10−3 M DMSO solution at room temperature were measured. The low molar conductivity values (11.45–20.10 Ω−1 cm2 mol−1) of the complexes supported their non-electrolytic nature since 1:1 electrolytes in DMSO are expected to have values above 25 Ω−1 cm2 mol−1 [35, 58].
Magnetic measurements
The magnetic susceptibility of all the synthesized complexes was measured in solid state at room temperature. The occurrences of high spin (d8) nickel(II) centres were confirmed by the magnetic moment values of 4.29 B.M for NiL1 and 3.90 B.M for NiL2. Magnetic moments of nickel complexes in a cubic field falls between 2.8 and 4.2 B.M. Deviation from spin only magnetic moment of 2.83 B.M is attributed to orbital contribution to the magnetic moment, which depends very much on stereochemistry [41]. Octahedral Ni(II) complexes have magnetic moments between 2.9 and 3.3 B.M according to the literature. Generally, square planar complexes are diamagnetic, while tetrahedral complexes have moments in the range 3.2–4.1 B.M [31, 41]. The nickel(II) complexes (NiL1 and NiL2) were assigned slightly distorted tetrahedral geometries. The magnetic moments of CuL1 and CuL2 complexes were 1.92 B.M and 1.87 B.M, respectively. According to literature, mononuclear Cu(II) complexes have magnetic moment range of 1.9–2.20 B.M [59]. The Cu(II) complexes were assigned mononuclear four coordinate complexes.
Infrared spectra
The IR spectra of the complexes were compared to those of free ligands (HL1 and HL2) in order to determine the coordination sites that may be involved in complex formation. The important spectral features of the ligands involved strong absorption band around 1620 cm−1 for HL1 and 1622 cm−1 for HL2 , which were assigned to C=N (azomethine nitrogen) stretching vibration, confirming the formation of Schiff bases [28, 33]. This band was seen at lower frequencies in the spectra of the complexes indicating participation of the azomethine nitrogen of the ligands in coordination to the metal ions [60]. The infrared spectra of the ligands showed broad bands around 3385–3397 cm−1 due to O–H stretching vibrations. However the absence of this feature in the spectra of the complexes revealed the presence of deprotonated phenolic oxygen and its involvement in coordination with the metal ions. Furthermore, a shift to lower wavelengths (in the ranges of 02–20 cm−1) for the C–O stretching vibration frequency in the spectra of the complexes compared to the spectra of the ligands gave further credence to the previous notion [61, 62]. The bands around 2922–3061 cm−1 in the spectra of the complexes and Schiff bases were assigned to aromatic and aliphatic C–H stretching vibration of the Schiff bases. Additional weak bands were observed in the infrared spectra of all the complexes in the range of 516–527 and 457–467 cm−1 and were assigned to M–N and M–O [M = nickel(II) or copper(II)] stretching vibration, respectively. Conclusively, the results of the IR spectra of the compounds showed that the Schiff bases were coordinated in a bidentate fashion through azomethine (C=N) nitrogen and phenolic oxygen to the nickel and copper metal ions in the complexes.
Electronic spectra
The UV–visible spectra of the ligands were recorded in DMSO and they exhibited two bands in the region 30,581–30,675 and 36,496–36,765 cm−1 due to n–π* and π–π* transitions, respectively [63]. The bands were shifted to a lower energy in the spectra of the complexes due to coordination of the ligand to the metal ions. Additional bands around 18,080–18,100 and 22,900–22,940 cm−1 were observed in the spectra of the nickel(II) complexes (NiL1 and NiL2) and were assigned to 2T1(F) → 2T1(P) and 2T1(F) → 2A2 transitions, respectively. This was in agreement with other reported nickel(II) complexes of tetrahedral geometry in literature [64].
Copper(II) complexes of octahedral geometry are expected to show a single broad band due to 2Eg → 2T1g transition. However, the existence of Jahn–Teller effect due to asymmetrical filling of electrons into the 2Eg set of d orbitals, causes the splitting of 2Eg and 2T2g sets into 2B1g, 2A2g and 2B2g, 2Eg sets, respectively. These splittings are usually observed in the spectra of copper complexes as two or three absorption bands [65]. Hence, the electronic spectra of an octahedral copper(II) complexes may lie within one band or may resolve into two or three absorption band components. However, tetrahedral copper(II) complexes have less pronounced Jahn–Teller effect since it is the T2 set of orbitals that are asymmetrically filled. Hence, one of its absorption bands is usually observed below 10000 cm−1. Square planar copper(II) complexes usually show two absorption bands of high energy above 10000 cm−1. The appearance of an absorption band below 10000 cm−1 in the copper complexes (CuL1 and CuL2), gave credence to their tetrahedral geometric conformation. This was in agreement with other reported copper(II) complexes of tetrahedral geometry [41].
Quantum chemical studies
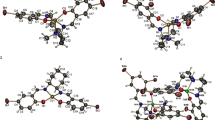
The quantum chemical studies were done to substantiate the assigned geometries to the compounds in the absence of single X-ray crystals and to obtain other electronic and geometric parameters, which may arise due to change in the substituent group on the benzothiazole unit of the ligands from methyl (in HL1) to fluro (in HL2). The optimized structures of the ligands and metal complexes were shown in Fig. 1. Selected bond lengths (in Å) around the coordination sites were labeled in the structures. The full geometry parameters are available upon request. The ligands adopted planar geometries but the metal–ligand bonds were not coplanar. The metal–ligand bond lengths in the optimized structures (Fig. 1) were in close agreement with reported values for similar bonds in the literature [66–68]. A comparison of the optimized structures of NiL1 and NiL2 complexes revealed that the metal–ligand bond lengths were not significantly affected by the change in the substituent group on the benzothiazole unit of the ligands from methyl (in HL1) to fluoro (in HL2). This suggests that the electronic effect associated with change in the substituent group was not extended to the coordination sites because the methyl and fluoro groups were not close to the sites. For the Ni(II) and Cu(II) complexes of HL1, Ni-ligand bonds was generally slightly shorter than their corresponding Cu-ligand bond. This may be due to a longer covalent radius of Cu2+ ion compared to Ni2+ ion. As a result, prediction of relative bond strengths of Ni-ligand and Cu-ligand based on these bond lengths may be subjective [69].

Optimized structures of the metal complexes showing selected bond lengths in Å
Furthermore, experimental results revealed that CuL2 contained Cu(II) ion and the ligand, HL2 , in a ratio of 1:2, hence, its parameters could not be compared absolutely with those of the other complexes. The lowest energy conformers in NiL1, NiL2 , and CuL1 corresponded to the structures that allowed the formation of H-bond between the coordinated water molecule and the nitrogen of the thiazole. The formation of this H-bond afforded a six-sided ring in each molecule, which promised to enhance the stability of the molecules. The H-bond in NiL1 (1.579 Å) was relatively stronger than that of CuL1 (1.616 Å). The same H-bond was 1.593 Å long in NiL2, suggesting a relatively weaker bond compared to NiL1.
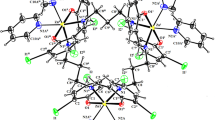
The frontier molecular orbital (FMO) electron density distributions of the complexes were shown in Fig. 2. The highest occupied molecular orbitals (HOMOs) revealed the sites of the molecule that are most likely to donate electrons to the appropriate orbitals of an electron-deficient species, while the lowest occupied molecular orbitals (LUMOs) represented the sites of the molecule that are most likely to accept electrons from suitable occupied orbitals of an electron-rich species [70]. All the studied complexes exhibit similar HOMO and LUMO electron density distributions. In all the metal complexes, the HOMOs were essentially distributed over the iminomethylnaphthalen-2-ol unit and extended to the chlorine and water ligands, and also the central metal in each case. The benzothiazole unit of the ligand was not involved in the HOMO electron density surface of the complexes. This suggests that prospective donor–acceptor π–π interactions that are driven by charge donations from the metal complexes to biological systems will be more favourable through the iminomethylnaphthalen-2-ol unit. In all the complexes, the LUMOs were delocalized over the entire organic ligand unit. The chlorine and water ligands were not involved in the LUMOs of NiL1, NiL2 , and CuL1. The central Cu atom was also excluded in the LUMO of CuL1 and CuL2. The methyl and fluoro substituents on the benzothiazole units of HL1 and HL2 , respectively, did not participate in the FMO electron density distributions.

HOMO and LUMO electron density isosurfaces of the metal complexes and spin density distributions of CuL1 and CuL2
The spin density distributions for Cu(II) complexes were shown in Fig. 2. Some fractions of the total spin resulting from the singly occupied molecular orbital of Cu(II) ion were delocalized to the ligands. In the case of CuL1, the Cl atom, water molecule and the iminomethylnaphthalen-2-ol unit of the organic ligand witness some fractions of the resulting spin from the central Cu(II) ion. The delocalization of the spin density in CuL1 was not extended to the benzothiazole unit. The spin density in CuL2 was mostly concentrated around the central Cu(II) and the coordinated N- and O-atoms of the ligands. Some C-atoms in the iminomethylnaphthalen-2-ol unit of the organic ligand also reflected a trace of the delocalized spin density. The quantitative values of atom-by-atom participation in the spin density distributions was estimated as reported in our previous paper [69]. These values were reported as the percentage ratio of the square of the spin density on an atom to the sum of squares of the spin densities on all atoms. For CuL1, the percentage contributions of the atoms at the coordination sites of the ligands are 9.33% (Cl atom), 6.19% (O-atom of the naphthalen-2-ol), 1.20% (O-atom of the water), and 0.86% (N-atom of the imino unit). The percentage of spin density retained by the central Cu(II) ion in CuL1 is 80.79%. These observations suggested that the Cu(II)–Cl bond has the highest covalent character. In CUL2, the N- and O-atoms of the ligands received 1.344 and 4.216% of the total spin, respectively, while the Cu(II) ion retained 87.93% of the total spin. This suggested that the Cu–O bonds in CuL2 exhibited more covalent character than the Cu–N bonds. In the overall, the metal–ligand bonds in CuL1 appeared to be more covalent than those in CuL2.
Some quantum chemically derived physical parameters of the complexes were listed in Table 1. All the synthesized complexes except CuL2 exhibited large dipole moments, which promise to favour dipole–dipole interactions between the metal complexes and active biological systems [71]. The trends of the HOMO and LUMO energies (E HOMO and E LUMO , respectively) can be summarized as CuL2 > CuL1; NiL1 > NiL2. This implied that NiL1 has better tendency of donating its HOMO electrons to appropriate accepting specie than NiL2, while CuL2 is more prospective than CuL1 in this regard. On the other hand, both CuL1 and NiL2 have higher tendencies of accepting charges into their LUMO orbitals than their corresponding CuL2 and NiL1 , respectively. The HOMO–LUMO energy gap (ΔE L–H) is a measure of relative stability of molecules. The results in Table 1 showed that NiL1 and CuL1 were more stable than their corresponding NiL2 and CuL2. This observation also relates with the order of H-bond strengths of the complexes. That is, NiL1 with strongest H-bond is the most electronically stable. The reaction energies (ΔE) for the formation of these complexes from their respective constituent ligands and metal ions were listed in Table 1. The results showed that the formation of NiL1 was energetically more favourable than CuL1. The formation of Ni(II) complex of HL1 was also energetically more feasible than that of HL2. The formation of CuL2 was the least energetically favourable. The trend of the values of ΔE also correlated with the relative strengths of H-bonds in the complexes.
The description of the bond strengths, and hence stability, of the complexes was extended further by calculating the hyperconjugation interaction energies between the filled and vacant orbitals in the systems. The natural bond orbital (NBO) analyses were carried out using the default NBO construct in Gaussian 09. The second order Fock matrix was solved for each system in order to evaluate the energy associated with donor–acceptor interactions in the metal complexes. The second order stabilization energy, E(2) is related to the weak departures from idealized natural Lewis structure (NLS). It is a measure of the delocalization effects due to a loss of occupancy from the localized NBO of the idealized NLS into an empty non-Lewis orbital [69]. The higher the E(2) value the stronger the interactions and the more stable the resulting bond. The E(2) associated with the delocalization from a donor (i) to an acceptor (j) was calculated as [69, 72]:
where \(\left\langle {\sigma_{i} \left| F \right|\sigma_{j} } \right\rangle^{2}\) or \(F_{ij}^{2}\) is the off diagonal Fock matrix element between the i and j NBOs, \(\varepsilon_{i}\) and a \(\varepsilon_{j}\) are the energies of i and j NBOs, and \(n_{i}\) is the occupancy or population of the i (donor) NBO. The values of the E(2) and the orbital interactions were listed in Table 2. The selected results in Table 2 were those that involved interactions between the metallic orbitals and the orbitals of ligand atoms at the coordination sites. Comparisons of similar orbital interactions in NiL1 and CuL1 reflected that the magnitudes of the E(2) in the former were significantly higher. This is in agreement with the deductions made from the H-bond strengths and reaction energies of the complexes. The results for NiL1 and NiL2 were very close in most cases, which again correlated with the observations from the geometries of the molecules (vide supra Fig. 1). The E(2) values for the LP(1)Nb→LP*(1)Ha interactions showed that result in H-bonding clearly revealed the trend of the strengths of H-bonds in the systems, in conformity with the results in Fig. 1. The BD*(1)Cl-M→LP*(5)M interactions, i.e. the interactions between the out-of-phase antibonding (BD*) orbital of Cl-M bond and the unfilled valence shell non-bonding (LP*) orbital of M were very strong in all the complexes. Based on the values of E(2), the interactions between the lone pair (LP) orbitals of N and O atoms with the LP* orbitals of Cu were not as strong as those in CuL1 complex. This again suggested stronger metal–ligand bond in CuL1 than CuL2 and the observation is in line with the trend of ΔE and ΔE L–H values (Table 1) for the complexes.
Thermal studies
The TG analysis of the ligands and their respective metal complexes were carried out in a temperature range of 25–800 °C and presented as supporting materials. The HL1 ligand exhibited a first estimated mass loss of 4.6% (calcd. 4.3%) at 116–184 °C, which could be attributed to the liberation of CH3 molecule. The remaining part of the ligand then undergoes a huge decomposition in the temperature range of 198–528 °C, producing a carbonaceous residue. The HL2 ligand undergoes similar decomposition pattern, but with a more profound first stage decomposition. The first step decomposition started at relatively low temperature of 85 °C and proceeded up to 196 °C. In this stage, approximately 2NH3 molecules were released (calc. 11.8%; found 10.6%). In the second stage, within 203–390 °C temperature range, the remaining part of the ligand decomposed into carbonaceous residue. The DTG curve showed the melting temperature for HL1 and HL2 at 187 and 192 °C, respectively.
The nickel complexes showed a broad and low weight loss in the temperature range of 30–90 °C for NiL1 and 31–146 °C for NiL2 with peak temperatures at 64 and 76 °C, respectively. This could be ascribed to the release of solvent molecules. However, both complexes showed peaks at approximately 168 °C due to the loss of one molecule of coordinated water. In hydrated complexes, water could be held by bonding to the metal ion or it may be held as lattice or crystal water. In the present nickel complexes, the DTG curve showed that dehydration occurred at relatively high temperature outside the range for lattice water. Hence, the water molecule could be considered to be involved in bonding due to the high temperature associated with this step [73]. The final step within the temperature range of 289–519 °C for NiL1 and 242–543 °C for NiL2 corresponded to the removal of the organic part of the ligand leaving behind the metal and some carbonaceous residue.
The thermogram of the copper complexes showed different decomposition steps from the nickel complexes. In CuL1, the first step of decomposition in the temperature range of 138–167 °C and with an estimated mass loss of 4.38% (calcd. 4.00%) corresponded to the loss of coordinated water. The second step occurred in the temperature range of 191–428 °C and involved two largely overlapping steps which were ascribed to the removal of the organic part of the ligand leaving metal and carbon and some carbonaceous residue. The overall weight loss amounts to 86.37% (calcd. 86.17%). The thermogram of CuL2 showed a broad DTG peak in the temperature range of 30–109 °C, which corresponded to three molecules of water (found/calcd. percentage mass loss: 7.6/7.1%). The second decomposition stage of the complex involved two largely overlapping steps in the 120–254 °C range. The DTG curves indicated that the decomposition can be resolved into two steps. The first was with a peak maximum of 208 °C, and the second showed a peak maximum at 222 °C, which was attributed to the decomposition of the organic ligand. Finally, the third stage involved a broad decomposition step which was associated with degradation of the previous residue to Cu and some carbonaceous residue.
Biological studies
Antibacterial studies
The basic principle for the design of any antimicrobial active compound is to inhibit the growth of causal microbes without any side effects on treatment. Antimicrobial activity of the ligands, metal complexes, and a standard drug were screened against Gram-positive and Gram-negative bacterial strains to verify the chance of detecting antibiotic principles in the tested materials. The complexes showed significantly enhanced antibacterial activities against the selected microbial strains in comparison to the free ligand (Fig. 3). The complexes were found to inhibit growth of all the tested bacterial strains at different variations in the order [CuL2] > [NiL1] > [NiL2] > [CuL1] for both Gram-negative and Gram-positive bacteria strains (Table 3). Similarly, the antibacterial result showed that CuL2 > CuL1 and NiL1 > NiL2, which was in agreement with the results obtained from dipole moment calculations of the complexes. CuL2 and NiL1 complexes had larger dipole moments as such have greater tendencies of donating electrons for a biomolecular interaction compared to NiL2 and CuL1 complexes, hence antibacterial activity of the complexes could be a result of interactions between the metal complexes and cell membrane of the bacteria strains. This interaction could result in cell death or increased lipophilicity of the complexes through the lipid membrane of the bacterial strain. Generally, the complexes exhibited better antibacterial activity compared to the ligands which could be explained on the basis of the overtone concept [74] and chelation theory [75, 76].

Histogram representation of antibacterial studies
The results of one way ANOVA carried out on the inhibitory values obtained from the antibacterial screening of the compounds showed that p < 0.05 at 95% confidence interval. Hence, the mean values were significantly different. This was further substantiated by a post hoc analysis carried out with Tukey’s HSD test using SPSS version 17.0.
Antioxidant studies
DPPH free radical scavenging activity
DPPH is a stable free radical, which can accept an electron or hydrogen radical and in turn gets converted into a stable diamagnetic molecule. DPPH has a strong absorption band at 517 nm due to the presence of odd electron in the molecule [77, 78]. In the present study, the ability of the ligands HL1 and HL2 and their complexes, to act as hydrogen donor or electron donor towards the transformation of DPPH into its reduced form was investigated. There were observable decrease in the absorbance of the DPPH radical, which was accompanied by a colour change from purple to yellow as the radical was scavenged by the ligands and their complexes (Fig. 4).

Histogram representation of DPPH radical scavenging activity of the compounds
The radical scavenging ability of the ligands could be due to the presence of phenolic OH group. The dissociation energy of the OH bond is considered to be one of the important parameters involved in the definition of the antioxidant potency of phenolic derivatives. Compounds with lower dissociation energy for their OH bonds tend to have better antioxidant properties compared to those with stronger dissociation energy for their OH bonds. HL2 had better antioxidant property compared to HL1 at all the concentrations tested. It was observed that Cu(II) complexes showed higher DPPH radical scavenging ability compared to the free ligands and nickel(II) complexes. From the results obtained, DPPH radical scavenging ability of complexes decreased in the following order: CuL2 > NiL1 > CuL1 > NiL2 (p < 0.05). Interestingly, increase in the concentration of the compounds caused a decrease in the DPPH radical scavenging ability. This could be attributed to increase in the amount of chromophores present in the test solutions as the concentration is increased, which in turn increases the absorption max of the solution. This occurrence is in order as most antioxidants are most effective at low concentrations and could become pro-oxidants at high concentrations.
Ferrous chelating ability
Iron is known as the most important lipid pro-oxidant due to its high reactivity among other transition metals. For example, ferrous ion (Fe2+) can facilitate the production of reactive oxygen species within living systems and therefore the ability of substances to chelate Fe2+ can be a valuable indication of its antioxidant capability. Ferrous ion chelation is considered as one of the most important antioxidant effects by retarding metal-catalyzed oxidation. The effective ferrous ion chelators may also provide protection against oxidative damage to cells by removing Fe2+ that may later participate in O2− generating Fenton type reactions as shown below [79].
Ferrozine or its methyl substituted compound (1,10-phenanthrolin) can quantitatively form complexes with Fe2+ when reacted together giving a red coloured solution. However, the intensity of the red colour of this complex decreases in the presence of other chelating agents; therefore one can assume that the complex formation is disrupted as a result of chelation. Hence, monitoring the intensity of the colour of the solution allows us to estimate the metal chelating activity of the coexisting chelators [80].
In this assay, the solution containing Fe2+ ion and 1,10-phenanthrolin were treated with the compounds (ligands and complexes), which have chelating activity and are, therefore, capable of capturing ferrous ion, thus preventing the formation of Fe-1,10-phen complex. As seen in Fig. 5, the ligands chelated with the Fe+2 ion. At concentrations of 50–200 μg, ferrous ion chelating ability of compounds decreased in the following order: CuL2 > CuL1 > NiL2 > NiL1 (p < 0.05). The results are presented in Fig. 5.

Histogram representation of ferrous ions chelating ability of the compounds
Conclusion
Two biologically active Schiff base ligands, HL1 and HL2, and their metal complexes have been synthesized and extensively characterised by the use of spectroscopic, thermal and magnetic studies. The synthesized Schiff bases were coordinated to Ni(II) and Cu(II) metal ions through their imine nitrogen and deprotonated phenolic oxygen in a bidentate fashion to give four set of complexes with tetrahedral geometries of [CuL1ClH2O], [NiL1ClH2O], [NiL2ClH2O], and [Cu(L2)2]3H2O. Results of spectral and thermal studies of the complexes were concordant with their formulation. The change in substituent group from methyl to fluoro on the benzothiazole moiety had no significant effect on the electronic, geometric, and magnetic properties of the complexes due to their distance from the imine nitrogen. The quantum chemical calculations gave extensive information about the optimised geometries of the complexes, their dipole moments, HOMO and LUMO energy gap, reaction energies and hydrogen bond strengths. The complexes were found to inhibit growth of all the tested bacterial strain and their antibacterial activity have the order [CuL2] > [NiL1] > [NiL2] > [CuL1] for both Gram-negative and Gram-positive bacterial strains. However, the fluoro substituted compounds showed better antibacterial activity compared to the methyl substituted compounds. The studied DPPH radical scavenging ability of complexes decreased in the following order: CuL2 > NiL1 > CuL1 > NiL2, while ferrous ion chelating ability of samples decreased in the following order: CuL2 > CuL1 > NiL2 > NiL1.
References
N.B. Patel, F.M. Shaikh, New 4-thiazolidinones of nicotinic acid with 2-amino-6-methylbenzothiazole and their biological activity. Sci. Pharm. 78, 753–765 (2010)
R. Abdul (2005) Synthesis and Biological Studies of Some Schiff bases Compounds and Their Transition Metal Complexes. A Ph.D. thesis submitted to the Bahuaddin Zakariya University, Multan Pakistan
P. Chaudhary, P. Sharma, A. Sharma, J. Varshney, Recent advances in pharmacological activity of benzothiazole derivatives. Int. J. Curr. Pharm. Res. 4, 5–11 (2010)
A. Rana, N. Siddiqui, S.A. Khan, Benzothiazoles: a new profile of biological activities. Int. J. Curr. Pharm. Res. 69, 10–17 (2007)
J. Malik, F.V. Manvi, B.K. Nanjwade, P. Purohit, New 2-amino substituted benzothiazoles: a new profile of biological activities. J. Pharm. Res. 11, 1687–1690 (2009)
J.K. Malik, F.V. Manvi, B.K. Nanjwade, S. Singh, P. Purohit, Review of the 2-amino substituted benzothiazoles: different methods of the synthesis. Der Pharm. Lett. 2, 347–359 (2010)
J.H. Block, J.H. Beale, Wilson and Gisvold, Textbook of Organic, Medicinal and Pharmaceutical Chemistry, 10th edn. (Lipincott William & Wilkins, New York, 1998)
L.S. Khokra, K. Arora, H. Mehta, A. Aggarwal, M. Yadav, Essential oil composition and antibacterial studies of Vitex negundo Linn. extracts. Int. J. Pharm. Sci. Res. 2, 1356–1377 (2011)
H.A. Bhuva, S.G. Kini, Synthesis, anticancer activity and docking of some substituted benzothiazoles as tyrosine kinase inhibitors. J. Mol. Graph. Model. 29, 32–37 (2010)
C.G. Mortimer, G. Wells, J.P. Crochard, E.L. Stone, T.D. Bradshaw, M.F.G. Stevens, A.D. Westwell, Antitumor benzothiazoles. 26. 2-(3,4-dimethoxyphenyl)-5-fluorobenzothiazole (GW 610, NSC 721648), a simple fluorinated 2-arylbenzothiazole, shows potent and selective inhibitory activity against lung, colon and breast cancer cell lines. J. Med. Chem. 49, 179–185 (2006)
S. Nagarjan, G. Crescenzo, D. Getman, H. Lu, J. Sikorski, J. Walker, J. McDonald, K. Houseman, G. Kocan, N. Kishore, P. Mehta, C. Shippy, L. Blystone, Discovery of novel benzothiazolesulfonamides as potent inhibitors of HIV-1 protease. Bioorg. Med. Chem. 11, 4769–4777 (2003)
S. Hout, N. Azas, A. Darque, M. Robin, C. Giorgio, M. Gasquet, J. Galy, P. David, Activity of benzothiazoles and chemical derivatives on Plasmodium falciparum. Parasitology 129, 525–542 (2004)
M. Maharan, S. William, F. Ramzy, A. Sembel, Synthesis and in vitro evaluation of new benzothiazole derivatives as schistosomicidal agents. Molecules 12, 622–623 (2007)
V. Kumar, T.S. Ngaraja, H. Shameer, E. Jayachandran, G.M. Sreenivasa, N-substituted-3-chloro-2-azetidinones: synthesis and characterization of new novel anti-inflammatory agents. J. Pharm. Sci. Res. 2, 83–92 (2009)
S. Hibi, Y. Okamoto, K. Tagami, H. Numata, N. Kobayashi, M. Shinoda, T. Kawahara, M. Murakami, K. Oketani, T. Inoue, H. Shibata, I. Yamatsu, Novel dual inhibitors of 5-lipoxygenase and thromboxane A2 synthetase: synthesis and structure-activity relationships of 3-pyridylmethyl-substituted 2-amino-6-hydroxybenzothiazole derivatives. J. Med. Chem. 37, 3062–3070 (1994)
N. Siddiqui, A. Rana, S. Khan, S. Haque, M. Arshad, S. Ahmed, W. Ahsan, Syntheis and preliminary screening of benzothiazol-2yl-thiadiazole derivatives for anticonvulsant activity. Acta Pharm. 59, 441–451 (2009)
N. Siddiqui, A. Rana, S. Khan, S. Haque, M. Alam, W. Ahsan, M. Arshad, Anticonvulsant and toxicity evaluation of newly synthesized 1-[2-(3,4-disubstitutedphenyl)-3-chloro-4-oxoazetidin-1-yl]-3-(6-substituted-1,3-benzothiazol-2-yl)ureas. Acta Chim. Slov. 59, 462–469 (2009)
A. Nitta, H. Fujii, S. Sakami, Y. Nishimura, T. Ohyama, M. Satoh, J. Nakaki, S. Satoh, C. Inada, H. Kozono, H. Kumagai, M. Shimamura, T. Fukazawa, H. Kawai, (3R)-3-amino-4-(2,4,5-trifluorophenyl)-N-{4-[6-(2-methoxyethoxy)benzothiazol-2-yl]tetrahydropyran-4-yl}butanamide as a potent dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. Bioorg. Med. Chem. Lett. 18, 5435–5438 (2008)
H. Diaz, R. Molina, R. Andrade, D. Coutino, J. Franco, S. Webster, M. Binniie, S. Estrada-Soto, M.I. Brajas, I.L. Rivera, G.N. Vazquez, Antidiabetic activity of N-(6-substituted-1,3-benzothiazol-2-yl)benzenesulfonamides. Bioorg. Med. Chem. Lett. 18, 2871–2877 (2008)
P. Arora, S. Das, M.S. Ranawat, N. Arora, M.M. Gupta, Synthesis and biological evaluation of some novel chrome-2-one derivatives for antipsychotic activity. J. Chem. Pharm. Res. 2, 317–323 (2010)
S.Y. Shahar, Z.H. Ansari, Synthesis and in vivo diuretic activity of biphenyl benzothiazole-2-carboxamide derivatives. Acta Pol. Pharm. Drug Res. 66, 387–392 (2009)
T.D. Bradshaw, M.S. Chua, H.L. Browne, V. Trapani, E.A. Sausville, M.F.G. Stevens, In vitro evaluation of amino acid prodrugs of novel antitumour 2-(4-amino-3-methylphenyl)benzothiazoles. Br. J. Cancer 86, 1348 (2002)
F. Delmas, A. Avellaneda, C.D. Gioegia, M. Robin, E.D. Clercq, D.P. Timol, J.P. Galy, Synthesis and antileishmanial activity of (1,3-benzothiazol-2-yl) amino-9-(10H)-acridinone derivatives. Eur. J. Med. Chem. 39, 685 (2004)
S.R. Pattan, S.N.N. Babu, J. Angadi, Synthesis and biological activity of 2-amino [5′-(4′-sulphonylbenzylidene)-2,4-thiazolidine dione]-7-(substituted)-6-fluoro benzothiazoles. Indian J. Heterocycl. Chem. 11, 333 (2002)
A.A. Osowole, A.C. Ekennia, B.O. Achugbu, Synthesis, spectroscopic characterization and antibacterial properties of some metal (II) complexes of 2-(6-methoxybenzothiazol-2-ylimino) methyl)-4-nitrophenol. Res. Rev. J. Pharm. Anal. 2, 1–5 (2013)
H.R.I. Tomi, J.H. Tomma, A.H.R. Al-Daraji, A.H. Al-Dujaili, Synthesis, characterization and comparative study the microbial activity of some heterocyclic compounds containing oxazole and benzothiazole moieties. J. Saudi Chem. Soc. 2012, 213–232 (2012)
H. Mahmood-ul, Z.H. Chohan, C.T. Supuran, Antibacterial Zn(II) compounds of Schiff bases derived from some benzothiazoles. Main Group Met. Chem. 25, 291 (2002)
S.P. Vartale, D.B. Kadam, N.K. Halikar, Synthesis of novel 4-thiazolidinone derivatives incorporated with benzothiazole and its antimicrobial activity. Der Pharma Chem. 3, 213–223 (2011)
R. Ali, N. Siddiqui, Biological aspects of emerging benzothiazoles: a short review. J. Chem 2013, 12 (2013). doi:10.1155/2013/345198
R. Caputo, M.L. Calabro, N. Micale, Synthesis and biological evaluation of new 2-amino-6-(trifluoromethoxy)benzoxazole derivatives, analogues of riluzole. Med. Chem. Res. 21, 2644–2651 (2012)
A.C. Ekennia, D.C. Onwudiwe, A.A. Osowole, Spectral, thermal stability ans antibacterial studies of copper complexes of N-methyl-N-phenyl dithiocarbamate. J. Sulfur Chem. 36, 96–104 (2015)
G.A. Kolawole, A.A. Osowole, Synthesis and characterization of some metal (II) complexes of isomeric unsymmetrical Schiff bases and their adducts with triphenylphosphine. J. Coord. Chem. 62, 1437–1444 (2009)
A.A. Osowole, R. Kempe, R. Schobert, Synthesis, spectral, thermal, in-vitro antibacterial and anticancer activities of some metal (II) complexes of 3-(-1-(4-methoxy-6-methyl)-2-pyrimidinylimino)methyl-2-napthol. Int. Res. J. Pure Appl. Chem. 2, 105–129 (2012)
A.C. Ekennia, D.C. Onwudiwe, C. Ume, E.E. Ebenso, Mixed ligand complexes of N-methyl-N-phenyldithiocarbamate: synthesis, characterisation, antifungal activity and solvent extraction studies of the ligand. Bioinorg. Chem. Appl. (2015). doi:10.1155/2015/913424
A.D. Becke, Density-functional exchange-energy approximation with correct asymptotic behaviour. Phys. Rev. A 38, 3098–3100 (1988)
C. Lee, W. Yang, R.G. Parr, Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988)
X. Xu, W.A. Goddard, The X3LYP extended density functional for accurate descriptions of nonbond interactions, spin states, and thermochemical properties. Proc. Natl. Acad. Sci. 101, 2673–2677 (2003)
I. Georgieva, N. Trendafilova, Bonding analyses, formation energies, and vibrational properties of M-R2dtc complexes (M = Ag (I), Ni (II), Cu (II), or Zn (II)). J. Phys. Chem. A 111, 13075–13087 (2007)
L. Chen, T. Liu, C. Ma, Metal complexation and biodegradation of EDTA and S, S-EDDS: a density functional theory study. J. Phys. Chem. A 114, 443–454 (2007)
Y. Niu, S. Feng, Y. Ding, R. Qu, D. Wang, J. Han, Theoretical investigation on sulfur-containing chelating resin-divalent metal complexes. Int. J. Quantum Chem. 110, 1982–1993 (2010)
S.I. Gorelsky, L. Basumallick, J. Vura-Weis, R. Sarangi, K.O. Hodgson, B. Hedman, K. Fujisawa, E.I. Solomon, Spectroscopic and DFT investigation of [M{HB(3,5-iPr2pz)3}(SC6F5)] (M = Mn, Fe Co, Ni, Cu, and Zn) model complexes: periodic trends in metal–thiolate bonding. Inorg. Chem. 44, 4947–4960 (2005)
T. Marino, M. Toscano, N. Russo, A. Grand, Structural and electronic characterization of the complexes obtained by the interaction between bare and hydrated first-row transition-metal ions (Mn 2 + , Fe 2 + , Co 2 + , Ni 2 + , Cu 2 + , Zn 2 +) and glycine. J. Phys. Chem. B 110, 24666–24673 (2006)
R. Terreux, M. Domard, C. Viton, A. Domard, Interactions study between the copper II ion and constitutive elements of chitosan structure by DFT calculation. Biomacromolecules 7, 31–37 (2006)
T.H. Dunning, P.Y. Hay, in Modern Theoretical Chemistry, 3rd edn., ed. by H.F. Schaefer (Plenum, New York, 1976)
P.J. Hay, W.R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 82, 270–283 (1985)
W.R. Wadt, P.J. Hay, Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J. Chem. Phys. 82, 284–298 (1985)
P.J. Hay, W.R. Wadt, Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J. Chem. Phys. 82, 299–310 (1985)
M.Y. Combariza, R.W. Vachet, J. Phys. Chem. A 108, 757–1763 (2004)
M.A. Carvajal, J.J. Novoa, S. Alvarez, Choice of coordination number in d10 complexes of group 11 metals. J. Am. Chem. Soc. 126, 1465–1477 (2004)
B.D. Alexander, T.J. Dines, Ab initio calculations of the structures and vibrational spectra of ethene complexes. J. Phys. Chem. A 108, 146–156 (2004)
M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, Gaussian 09, Revision D.01 (Gaussian, Inc., Wallingford CT, 2009)
W. Brands-Williams, M.E. Cuvelier, C.L.W. Berset, Use of a free radical method to evaluate antioxidant activity. LWT Food Sci. Technol. 18, 25–30 (1995)
T.C.P. Dinis, V.M.C. Madeira, L.M. Almeida, Action of phenolic derivatives (acetaminophen, salicylate, and 5-aminosalicylate) as inhibitors of membrane lipid peroxidation and as peroxyl radical scavengers. Arch. Biochem. Biophys. 315, 161–169 (1994)
A.A. Osowole, S.M. Wakil, M.O. Emmanuel, Synthesis, characterization, antioxidant and antimicrobial activities of some metal(II) complexes of the mixed-ligands, vitamin B2 and benzoic acid. Elixir Appl. Chem. 79, 30370–30374 (2015)
A.R.H. Pramanik, P.C. Paul, P. Mondal, C.R. Bhattacharjee, Mixed ligand complexes of cobalt(III) and iron(III) containing N2O2-chelating Schiff base: synthesis, characterisation, antimicrobial activity, antioxidant and DFT study. J. Mol. Struct. 1100, 496–505 (2015)
T. Prateek, C. Sulekh, B.S. Saraswat, S. Deepansh, Design, spectral characterization, DFT and biological studies of transition metal complexes of Schiff base derived from 2-aminobenzamide, pyrrole and furan aldehyde. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 143, 1–11 (2015)
D.C. Onwudiwe, A.C. Ekennia, Synthesis, characterisation, thermal, antimicrobial and antioxidant studies of some transition metal dithiocarbamates. Res. Chem. Intermed. (2016). doi:10.1007/s1116-016-2709-2
W.J. Geary, The use of conductivity measurements in organic solvents for the characterisation of coordination compounds. Coord. Chem. Rev. 7, 81–122 (1971)
M. Belcastro, T. Marino, N. Russo, M. Toscano, Interaction of cysteine with Cu2+ and group IIb (Zn2+, Cd2+, Hg2+) metal cations: a theoretical study. J. Mass Spectrom. 40, 300–306 (2005)
R.A. Lal, S. Adhikari, A. Kumar, M.L. Pal, Oxoperoxomolybdenum(vi) complexes derived from n-benzamidosalicylaldimine. J. Indian Chem. Soc. 75, 345–349 (1998)
S. Majumder, G.S. Panda, S.K. Choudhuri, Synthesis, characterization and biological properties of a novel copper complex. Eur. J. Med. Chem. 38, 893–898 (2003)
C.S. Marvel, S.A. Aspey, E.A. Dudley, Quadridentate and sexadentate chelates. Some preliminary studies in their preparation and thermal stability. J. Am. Chem. Soc. 78, 4905–4909 (1956)
S.N. Rao, K.N. Munshi, N.N. Rao, M.M. Bhadbhade, E. Suresh, Synthesis, spectral and X-ray structural characterization of [cis-MoO 2 (L)(solv)](L = salicylidene salicyloyl hydrazine) and its use as catalytic oxidant. Polyhedron 18, 2491–2497 (1999)
M.G. Abd El-Wahed, M.S. Refat, S.M. El-Megharbel, Metal complexes of antiuralethic drug: synthesis, spectroscopic characterization and thermal study on allopurinol complexes. J. Mol. Struct. 888, 416–429 (2008)
D. Nicholls, Comprehensive Inorganic Chemistry, vol. 3 (Wiley, Hoboken, 1973)
B. Tang, J.H. Ye, X.H. Ju, Computational study of coordinated Ni(II) complex with high nitrogen content ligands. ISRN Org. Chem. (2011). doi:10.5402/2011/920753
A. Nimmermark, L. Ohrstrom, Z. Reedijk, Metal-ligand bond lengths and strengths: are they correlated? A detailed CSD analysis. Z. Kristallogr. 228, 311–317 (2013)
T.H. Lu, C.S. Chung, T.J. Ashida, Chin. Chem. Soc. 38, 147–154 (1991)
C. Ravikumar, I.H. Joe, V.S. Jayakumar, Charge transfer interactions and nonlinear optical properties of push–pull chromophore benzaldehyde phenylhydrazone: a vibrational approach. Chem. Phys. Lett. 460, 552–558 (2008)
J.V. Burda, J. Sponer, P. Hobza, Ab initio study of the interaction of guanine and adenine with various mono- and bivalent metal cations (Li+, Na+, K+, Rb+, Cs+; Cu+, Ag+, Au+; Mg2+, Ca2+, Sr2+, Ba2+; Zn2+, Cd2+, and Hg2+). J. Phys. Chem. 100, 7250–7255 (1996)
A.C. Ekennia, D.C. Onwudiwe, L.O. Olasunkanmi, A.A. Osowole, E.E. Ebenso, Synthesis, biological and quantum chemical studies of Zn (II) and Ni(II) mixed ligand complexes derived from N,-disubstituted dithiocarbamate and benzoic acid. Bioinorg. Chem. Appl. 2015, 1–12 (2015)
J. Chocholousova, V. Spirko, P. Hobza, First local minimum of the formic acid dimer exhibits simultaneously red-shifted O–H···O and improper blue-shifted C–H···O hydrogen bonds. Phys. Chem. Chem. Phys. 6, 37–41 (2004)
V. Uivarosi, M. Badea, V. Aldea, L. Chirigiu, R. Olar, Thermal and spectral studies of palladium(II) and platinum(IV) complexes with dithiocarbamate derivatives. J. Therm. Anal. Calorim. 111, 1177–1182 (2013)
N. Raman, J.D. Raja, Synthesis, structural characterization and antibacterial studies of some biosensitive mixed ligand copper(II) complexes. Indian J. Chem. 46A, 1611–1614 (2007)
B.D. Corbin, E.H. Seeley, A. Raab, J. Feldmann, M.R. Miller, V.J. Torres, K.L. Anderson, B.M. Dattilo, P.M. Dunman, R. Gerads, R.M. Caprioli, W. Nacken, W.J. Chazin, E.P. Skaar, Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 319, 962–965 (2008)
N. Dharmaraj, P. Viswanathamurthi, K. Nataraj, Ruthenium(II) complexes containing bidentate Schiff bases and their antifungal activity. Transit. Met. Chem. 26, 105–109 (2001)
M.E. De-Leo, A. Tranghee, M. Passantino, A. Mordente, M.M. Lizzio, T. Galeotti, A. Zoli, Manganese superoxide dismutase, glutathione peroxidase, and total radical trapping antioxidant capacity in active rheumatoid arthritis. J. Rheumatol. 29, 2245–2246 (2002)
A. Mahajan, V.R. Tandon, Antioxidants and rheumatoid arthritis. J. Indian Rheumatol. Assoc. 12, 139–142 (2004)
B. Halliwell, J.M.C. Gutteridge, Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J. 219, 1–14 (1984)
A. Guder, H. Korkmaz, Investigation of antioxidant activity and total anthocyanins from blackberry (Rubus hirtus Waldst. and Kit) and cherry laurel (Laurocerasus officinalis Roem.). Asian J. Chem. 24, 4525–4531 (2012)
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ekennia, A.C., Osowole, A.A., Olasunkanmi, L.O. et al. Coordination behaviours of new (bidentate N,O-chelating) Schiff bases towards copper(II) and nickel(II) metal ions: synthesis, characterization, antimicrobial, antioxidant, and DFT studies. Res Chem Intermed 43, 3787–3811 (2017). https://doi.org/10.1007/s11164-016-2841-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-016-2841-z