
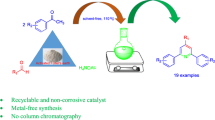
Abstract
Chitosan, a biodegradable green catalyst, was found to be an impressive system for one-pot four-component reaction of different aldehydes, dimedone, β‐ketoesters or acetoacetanilide, and ammonium acetate leading to 1,4-dihydropyridine derivatives via Hantzsch-type condensation under solvent-free condition. This methodology produces diverse superiorities such as operational simplicity, short reaction time, satisfied yield and recyclable catalyst. In this work, we attempted to develop several modifications for the classical Hantzsch conversion to report a novel eco-friendly method that is free from previously reported disadvantages.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Recently, multicomponent reactions (MCRs) have been found to be a powerful and versatile tool in the synthesis of many chemical compounds in biological and pharmacological scopes. The great capability of these reactions is to combine two or more components in a single step, without the isolation of any intermediates, to synthesize complex and substantial substances with high efficiency, simplicity, time saving and atom economy [1–5].
Nowadays, all over the world, green chemistry has become a concern for organic chemists in order to provide important biological and pharmaceutical compounds in a safe and eco-friendly manner to minimize environmentally pollutants as well as destructive consequences on the human body [6, 7]. Hence, in response to anxieties about environmental threats, organic chemists merge multicomponent reactions into green chemistry [8–11]. From this viewpoint, those reactions are more suitable which are operated without solvents or with non-hazardous solvents. Now, solvent-free multicomponent reactions have modernized conventional procedures via increasing yield, operational simplicity, cost saving and decreasing reaction times, energy consumption and reactor size. In regard to the aforementioned, the optimal option is a solvent-free reaction [12–15].
Chitosan is a bifunctional hetero bio-polymer catalyst possessing both glucosamine and acetyl glucosamine units prepared from alkali hydrolysis of chitin—obtained from the exoskeletons of crustaceans such as crabs, lobsters and shrimps, the radulas of mollusks as well as the beaks of cephalopods—the most plentiful available polysaccharide after cellulose in nature (Fig. 1) [16–18].

Chemical structure of chitosan
The presence of several –NH2 and –OH groups in chitosan disposes it either to directly catalyse miscellaneous sets of organic reactions such as transamidation [19], Strecker reaction [20], Biginelli reaction [21], Michael addition [22], and Aldol and Knoevenagel reactions [23–26], or to catalyse through metals supported on chitosan such as Suzuki–Miyaura and Heck cross-coupling reactions [27], metal-catalyzed reaction [28], oxidation [29], and [3 + 2] Huisgen cycloaddition [30]. Moreover, chitosan can be applied in medicine, drug delivery, agriculture, water treatment, fuel cells and so on [31].
One of the unique properties of chitosan is insolubility in both water and some organic solvents. This characteristic introduces it as a reusable catalyst that can be employed many times without any excess modification in organic synthetic designs.
1,4-Dihydropyridines are a valuable group of nitrogen-containing, six-member heterocyclic compounds that have attracted much attention because of numerous biological activities such as anti-HIV agents [32], anti-inflammatory and anti-diabetes [33], antitumor [34], analgesic [35] and the treatment of Alzheimer’s disease [36, 37]. Additionally, usage of them as a calcium channel blocker has enhanced their value among other heterocycles [38–41]. The structures of some biologically active 1,4-DHPs are illustrated in Fig. 2 [42, 43].

Representative 1,4-DHPs with biological properties
For the first time, the synthesis of 1,4-DHPs was by the famous, three-component condensation of aldehyde, ethylacetoacetate and ammonia carried out by Hantzsch in 1882 [44]. Since then, various changes have been made in the reactants, conditions and catalysts. The use of dimedone as C–H acid and amines or ammonium acetate as nitrogen sources have been performed. In addition, widespread catalysts and conditions have been used, such as phosphoric acid [45], l-proline [46], heteropolyacids [47], magnetite/chitosan [48], magnetic Fe3O4 nanoparticles [49], La2O3 [50], Yb(OTf)3 [51], ionic liquid(BMImSac) [52], solar thermal energy [53], I2 [54], ρ-toluenesulfonic acid [55], glycine [56], nano ZnO [57], FeF3 [58], Baker’s yeast [59] and hafnium(IV) bis(perfluorooctanesulfonyl) imide complex in fluorous media [60]. But most of these suffer from some drawbacks such as the use of a toxic solvent and catalyst, high cost, harsh reaction conditions and severe work-up procedures.
As part of our research on employing green catalysts to carry out MCRs in these reports [61, 62], chitosan has been assisted as a sustainable, reusable and biodegradable organocatalyst for the synthesis of 1,4-DHPs.
Experimental
General
All chemicals and reagents were purchased from Merck, Fluka and Aldrich and used as received without any purification. Melting points were measured using Electrothermal 9100 apparatus (UK). 1H NMR and 13C NMR spectra were recorded on a BRUKER DRX at 300 or 400 and 100 MHz in DMSO-d6, respectively. The mass spectra were recorded on an Agilent Technology (HP) mass spectrometer, operating at an ionization potential of 70 eV. Elemental analyses were performed using a Elementar, Vario EL III instrument. Chitosan of high molecular weight (from shrimp shells, MW: 310–375 kDa, degree of deacetylation >75 %) was provided from by Sigma-Aldrich and used as received.
General procedure for the synthesis of 1,4-dihidropyridine derivatives
Substituted aldehyde (0.5 mmol), ammonium acetate (1 mmol, 0.077 g), dimedone (0.5 mmol, 0.071 g), ethyl or methyl acetoacetate (0.6 mmol, 0.065 or 0.059 g) or acetoacetanilide (0.6 mmol, 0.106 g) in the presence of a catalytic amount of chitosan (0.015 g, more than 0.011 g deacetylated) were magnetically stirred without solvent at 60 °C for appropriate times as specified in Tables 3 and 4. The progress of the reaction was monitored by TLC 30 % EA: Hex. After completion of the reaction, the resulting solid products was dissolved in hot ethanol, filtered for removing the unsolvable catalyst and then the filtrate was concentrated and the crude was purified by recrystallization from ethanol to afford the pure 1,4-dihidropyridine drivatives (5a–5x). The catalyst was subsequently washed with 5 ml ethanol.
The structures of the synthesized products were confirmed by comparison of their melting points with reliable data reported in the literature and spectral techniques 1H, 13C NMR spectra, CHNS analysis and mass spectroscopy.
Ethyl 4-(4-methoxyphenyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (5b)
1H NMR (300 MHz, DMSO-d6): δ 0.86 (3H, s, CH 3), 1.01 (3H, s, CH 3), 1.14 (3H, t, J = 7.2 Hz, CH2–CH 3), 1.95-2.39 (4H, m, CH 2–CMe2–CH 2), 2.25 (3H, s, CH 3–C=), 3.68 (3H, s, OCH 3), 3.98 (2H, q, J = 7.2 Hz, OCH 2CH3), 4.75 (1H, s, C–CH(R)-C), 6.75 (2H, d, J = 8.7 Hz, CH(Ar)), 7.05 (2H, d, J = 8.7 Hz, CH(Ar)), 9.01 (1H, br, D2O-exchange).
Ethyl 4-(3-fluorophenyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (5c)
1H NMR (400 MHz, CDCl3): δ 1.00 (3H, s, CH 3), 1.10 (3H, s, CH 3), 1.22 (3H, t, J = 7.2 Hz, CH2–CH 3), 2.11-2.36 (4H, m, CH 2–CMe2–CH 2), 2.41 (3H, s, CH 3–C=), 4.07-4.12 (2H, m, OCH 2CH3), 5.09 (1H, s, C–CH(R)-C), 6.04 (1H, br, D2O-exchange), 6.81 (1H, t, J = 6.0 Hz), 7.00 (1H, d, J = 10 Hz), 7.12-7.20 (2H, m). 13C NMR (400 MHz, DMSO-d6): δ 14.6, 18.7, 26.8, 29.5, 32.6, 36.3, 50.6, 59.8, 103.5, 110.0, 112.9, 114.4, 123.9, 130.1, 146.1, 150.4, 162.5 (d, J CF = 243.5 Hz), 195.0. MS (EI, 70 eV): m/z (%) M+ 353 (12), 262 (100), 234 (23), 178 (6), 122 (3), 95 (1), 91 (6), 41 (5). Anal. Calcd. for C21H24FNO3: C (70.57), H (6.77), N (3.92); Found: C (70.27), H (7.40), N (3.80).
Ethyl 4-(4-hydroxyphenyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (5g)
1H NMR (300 MHz, DMSO-d6): δ 0.87 (3H, s, CH 3), 1.01 (3H, s, CH 3), 1.14 (3H, t, J = 7.2 Hz, CH2–CH 3), 1.95-2.44 (4H, m, CH 2–CMe2–CH 2), 2.30 (3H, s, CH 3–C=), 3.98 (2H, q, J = 7.2 Hz, OCH 2CH3), 4.80 (1H, s, C–CH(R)-C), 6.57 (2H, d, J = 8.4 Hz, CH(Ar)), 6.94 (2H, d, J = 8.4 Hz, CH(Ar)), 9.00 (1H, br, D2O-exchange), 9.08 (1H, br, D2O-exchange).
Ethyl 4-(3-methylphenyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (5i)
1H NMR (400 MHz, CDCl3): δ 0.98 (s, 3H, CH 3), 1.08 (s, 3H, CH 3), 1.24 (3H, t, J = 7.2 Hz, CH2–CH 3), 2.15-2.26 (4H, m, CH 2–CMe2–CH 2), 2.29 (3H, s, CH 3–C=), 2.37 (3H, s, CH 3–Ar), 4.04-4.12 (2H, m, OCH 2CH3), 5.04 (1H, s, C–CH(R)-C), 6.72 (1H, br, D2O-exchange), 6.93-6.96 (1H, m), 7.10–7.11 (2H, m), 7.13 (1H, m). 13C NMR (400 MHz, DMSO-d6): δ 14.6, 18.7, 21.6, 26.9, 29.6, 32.6, 36.2, 50.7, 59.5, 61.6, 104.3, 110.4, 125.1, 126.9, 128.2, 130.0, 137.0, 145.3, 148.1, 150.2, 167.3, 195.0. MS (EI, 70 eV): m/z (%) M + 353 (37), 262 (100), 234 (45), 178 (12), 122 (5), 91 (10), 65 (9), 41 (7). Anal. Calcd. for C22H27NO3: C (74.76), H (7.70), N (3.96); Found: C (74.77), H (7.10), N (4.06).
Ethyl 4-(4-cyanophenyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (5k)
1H NMR (400 MHz, CDCl3): δ 0.94 (3H, s, CH 3), 1.11 (3H, s, CH 3), 1.19 (3H, t, J = 7.2 Hz, CH2–CH 3), 2.15–2.38 (4H, m, CH 2–CMe2–CH 2), 2.44 (3H, s, CH 3–C=), 4.07 (2H, q, J = 7.2 Hz, OCH 2CH3), 5.12 (1H, s, C–CH(R)-C), 6.06 (1H, br, D2O-exchange), 7.45 (2H, d, J = 8.4 Hz, CH(Ar)), 7.53 (2H, d, J = 8.4, CH(Ar) Hz).
Ethyl 4-(2-naphthyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (5l)
1H NMR (400 MHz, CDCl3): δ 0.93 (3H, s, CH 3), 1.04 (3H, s, CH 3), 1.21 (3H, t, J = 7.2 Hz, CH2–CH 3), 2.14-2.40 (4H, m, CH 2–CMe2–CH 2), 2.47 (3H, s, CH 3–C=), 4.03–4.10 (2H, m, OCH 2CH3), 5.26 (1H, s, C–CH(R)-C), 6.41 (1H, br, D2O-exchange), 7.38–7.44 (2H, m), 7.52 (1H, d, J = 7.2 Hz), 7.70 (1H, s), 7.72 (1H, s), 7.75–7.78 (2H, m).
Ethyl 2,7,7-trimethyl-5-oxo-4-(1-phenylprop-1-en-2-yl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (5m)
1H NMR (400 MHz, CDCl3): δ 1.10 (3H, s, CH 3), 1.13 (3H, s, CH 3), 1.31 (3H, t, J = 7.2 Hz, CH2–CH 3), 1.90 (3H, s, CH 3–C=), 2.19–2.38 (4H, m, CH 2–CMe2–CH 2), 2.40 (3H, s, CH 3–C=), 4.07 (2H, q, J = 7.2 Hz, OCH 2CH3), 4.68 (1H, s, C–CH(R)-C), 5.90 (1H, br, D2O-exchange), 6.43 (1H, s, Ph-CH=C), 7.15–7.22 (3H, m), 7.27–7.31 (2H, m). 13C NMR (400 MHz, DMSO-d6): δ 14.8, 16.2, 18.7, 27.0, 29.7, 32.4, 46.8, 50.9, 59.5, 61.8, 102.8, 108.6, 125.3, 126.4, 128.6, 129.0, 145.6, 150.8, 167.7, 195.2. MS (EI, 70 eV): m/z (%) M+ 380 (3), 262 (100), 234 (24), 178 (6), 122 (2), 117 (4), 91 (6), 41 (2). Anal. Calcd. for C24H29FNO3: C (75.96), H (7.70), N (3.69); Found: C (76.20), H (7.36), N (3.79).
Ethyl 2,7,7-trimethyl-5-oxo-4-(thiophen-2-yl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (5n)
1H NMR (300 MHz, DMSO-d6): δ 0.95 (3H, s, CH 3), 1.04 (3H, s, CH 3), 1.20 (3H, t, J = 7.2 Hz, CH2–CH 3), 2.05-2.47 (4H, m, CH 2–CMe2–CH 2), 2.28 (3H, s, CH 3–C=), 4.08 (2H, q, J = 7.2 Hz, OCH 2CH3), 5.18 (1H, s, C–CH(R)-C), 6.67 (1H, d, J = 3.3 Hz), 6.83 (1H, t, J = 5.1 Hz), 7.19 (1H, dd, J 1 = 1.2 Hz, J 2 = 5.2 Hz) 9.26 (1H, br, D2O-exchange).
Ethyl 2,7,7-trimethyl-5-oxo-4-propyl-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (5o)
1H NMR (400 MHz, CDCl3): δ 0.85 (3H, t, J = 7.2 Hz, CH 3CH2–), 1.11 (3H, s), 1.13 (3H, s), 1.20-1.36 (4H, m, –CH 2–CH 2–), 1.30 (3H, t, J = 7.2 Hz), 2.19-2.37 (4H, m, CH 2–CMe2–CH 2), 2.33 (3H, s, CH 3–C=), 4.04 (1H, t, J = 10.0 Hz, C–CH(Pr)-C), 4.11-4.26 (2H, m, OCH 2CH3), 6.21 (1H, br, D2O-exchange).
Methyl 4-(4-nitrophenyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (5r)
1H NMR (300 MHz, DMSO-d6): δ 0.82 (3H, s, CH 3), 1.01 (3H, s, CH 3), 1.96–2.42 (4H, m, CH 2–CMe2–CH 2), 2.33 (3H, s, CH 3–C=), 3.53 (3H, s, OCH 3), 4.96 (1H, s, C–CH(R)-C), 7.42 (2H, d, J = 9.0 Hz), 6.96 (2H, d, J = 9.0 Hz), 9.29 (1H, br, D2O-exchange).
Methyl 4-(4-(dimethylamino)phenyl)-2,7,7-trimethyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (5s)
1H NMR (300 MHz, CDCl3): δ 0.88 (3H, s, CH 3), 1.02 (3H, s, CH 3), 2.01–2.45 (4H, m, CH 2–CMe2–CH 2), 2.28 (3H, s, CH 3–C=), 2.81 (6H, s, N–(CH 3)2), 3.54 (3H, s, OCH 3), 4.75 (1H, s, C–CH(R)-C), 6.56 (2H, d, J = 11.6, CH(Ar)), 6.96 (2H, d, J = 11.6, CH(Ar)), 9.01 (1H, br, D2O-exchange).
2,7,7-trimethyl-5-oxo-N,4-diphenyl-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (5t)
1H NMR (300 MHz, CDCl3): δ 0.90 (3H, s, CH 3), 1.09 (3H, s, CH 3), 2.15–2.38 (4H, m, CH 2–C(CH3)2–CH 2), 2.42 (3H, s, CH–CH 3), 4.96 (1H, s, C–CH(Ar)–C), 5.91 (1H, br, D2O-exchange), (1H,), (5H, d, J = 8.7 Hz, CH(Ar)), 7.36 (2H, t, J = 8.7 Hz, CH(Ar)), (2H, d,).
4-(4-methoxyphenyl)-2,7,7-trimethyl-5-oxo-N-phenyl-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (5x)
1H NMR (300 MHz, CDCl3): δ 0.86 (s, 3H, CH 3), 1.04 (s, 3H, CH 3), 2.11–2.30 (4H, m, CH 2–C(CH3)2–CH 2), 2.36 (3H, s, CH 3–CH), 2.38 (3H, s, CH 3–Ar), 4.90 (1H, s, C–CH(Ar)–C), 6.76 (1H, br, D2O-exchange), 6.88 (2H, d, J=), 7.03-7.08 (1H, m), 7.23-7.30 (4H, m), 7.40 (2H,). 13C NMR (300 MHz, CDCl3): δ 18.7, 27.0, 29.3, 32.6, 36.2, 37.1, 40.9, 50.6, 55.2, 108.3, 111.0, 119.9, 124.0, 128.9, 129.0, 137.7, 138.1, 141.1, 148.6, 158.7, 166.6, 195.4.
Result and discussion
In our work, we applied chitosan without any further modification for the Hantzch synthesis of 1,4-DHP derivatives. Fortunately, short reaction times and good to excellent yields were found; in addition, as in previous literature, recyclability and reusability of the catalyst were observed.
Catalytic studies
At the beginning of our ongoing research, we evaluated the reaction between benzaldehyde (1a), dimedone (2), ethyl acetoacetate (3a) and ammonium acetate (4) at different catalyst loading at room temperature. The results are presented in Table 1.

In the absence of the catalyst, only 45 % of corresponding product was obtained after 24 h (entry 1). To improve output of the reaction, chitosan was utilized in various loadings. Surprisingly, using just 0.010 g chitosan not only diminished the reaction time to one-third but also considerably enhanced the yield (entry 2). In the following, the reaction product was studied by catalyst loading variations (entries 2–4), and finally, due to the observed negligible difference between two last entries, we chose to apply 0.015 g chitosan as the optimum quantity of catalyst.
Effect of temperature and solvent
Next, considering the selected amount of catalyst (0.015 g), appropriate conditions of temperature and solvent were assessed (Table 2). As illustrated in Table 2, the reaction time in the solvent-free condition had a decreasing trend, when the temperature increased from room temperature up to 70 °C (entries 1–6). Considering green chemistry, we chose only safe solvents including H2O and EtOH. The reaction was carried out in both solvents at room temperature and reflux and no better results were found (entries 7–10).
A notable result was observed when H2O was utilized as the solvent as the desired corresponding product was not observed; instead, the reaction promoted to afford product 6 according as below (Scheme 1).

Unwanted reaction in water
The formation of product 6 was confirmed by 1H and 13C NMR spectra. Additionally, the melting point for product 6 was found to be 191–193 °C which was in accordance with the literature [45]. It is suggested that water-soluble ammonium acetate could not be treated with lipophilic other reactants dispersed in water, and so the reaction proceeded without involvement of ammonium acetate. On the other hand, regarding the higher activity of dimedone compared with ethyl acetoacetate, the latter did not participate in the reaction leading to the corresponding product 6.
Versatility of procedure
To demonstrate the repeatability of this strategy, a broad variety of aromatic aldehydes possessing electron-withdrawing and electron-releasing substitutions, heterocyclic and aliphatic, were employed and the results are summarized in Table 3, from which it can be seen than all the utilized aldehydes supplied the desired products with great yields in short reaction times.

To show the generality of our green protocol, in addition to the derivatives of β-ketoester (ethyl and methylacetoacetate), acetoacetanilide as a β-ketoeamide was selected to participate in this reaction and the results are shown in Table 4.

Reusability of chitosan
In terms of green chemistry, we investigated the reusability of chitosan as a heterogeneous catalyst. Hence, after completion of the reaction, 5 ml ethanol was added to the reaction mixture and the chitosan was separated by simple filtration, washed with ethanol and dried and was then reused up to nine times without any reduction in catalytic performance (Fig. 3). Ease of separation, recyclability and reusability of the catalyst are promising plus points of our protocol.

Reusability of chitosan for the preparation of 1,4-DHPs
Mechanistic evaluation
Here, a plausible mechanism for the Hantzsch synthesis of 1,4-dihydropyridine derivatives is depicted (Scheme 2). As obvious from the catalyst structure, each chitosan unit containing one amine group and two hydroxyl groups can activate both nucleophilic and electrophilic species participating in the pathway leading to 1,4-DHPs preparation. Mechanistically, H–H bonds between the hydroxyl and carbonyl groups in three steps including knoevenagel product formation (I), 1,4-addition (II) and cyclization (III) are key factors in this reaction. In addition, amine groups can also behave as a base, enabling enol and enamine to act powerfully (I,II). In addition to that, amine groups may speed up the dehydration reaction via proton abstraction (IV).

A plausible reaction pathway
Conclusion
In brief, we have introduced a new strategy for the preparation of a diversity of known and unknown 1,4-dihydropyridines via Hantzsch-type condensation using chitosan as an inexpensive green catalyst under solvent-free conditions. The structure of the synthesized products were confirmed by NMR, CHNS analysis and mass spectroscopy.
The promising aspects of this methodology are its efficiency, simplicity, clean reaction profile, compatibility with different functional groups, quick reaction, high yields of the reaction product and non-chromatographic purification. Moreover, from the green point of view, recyclability of the catalyst without loss of its activity, easy catalyst separation from the reaction mixture, low cost, small catalyst loading and eco-compatibility of the catalyst are other significant features of this green procedure.
References
V.A. Orru, E. Ruijter, Synthesis of Heterocycles via Multicomponent Reactions, I (Springer, Berlin, 2010)
C.O. Kappe, Curr. Opin. Chem. Biol. 6, 314 (2002)
V. Nair, C. Rajesh, A. Vinod, U.S. Bindu, A.R. Streekenth, S. Mathen, L. Balagopal, Acc. Chem. Res. 36, 899 (2003)
D.J. Ramon, M. Yus, Angew. Chem. Int. Ed. 44, 1602 (2005)
A.A. Domling, Chem. Rev. 106, 17 (2006)
P. Tundo, P. Anastas, D.S. Black, J. Breen, T. Collins, S. Memoli, J. Miyamoto, M. Polyakoff, W. Tumas, Pure Appl. Chem. 72, 1207 (2000)
P.T. Anastas, J.C. Wagner, Green Chemistry. Theory and practice (Oxford University Press, Oxford, 1998)
M.A.P. Martins, C.P. Frizzo, D.N. Moreira, L. Buriol, P. Machado, Chem. Rev. 109, 4140 (2009)
P.J. Walsh, H. Li, C.A. Parrodi, Chem. Rev. 107, 2503 (2007)
F. Toda, Acc. Chem. Res. 28, 480 (1995)
Z.L. Shen, S.J. Ji, T.P. Loh, Tetrahedron Lett. 46, 507 (2005)
K. Tanaka, Solvent-Free Organic Synthesis (Wiley-VCH, Weinheim, 2009)
K. Tanaka, F. Toda, Chem. Rev. 100, 1025 (2000)
G. Rothenberg, A.P. Downie, C.L. Raston, J.L. Scott, J. Am. Chem. Soc. 123, 8701 (2001)
M.S. Singh, S. Chowdhury, RSC Adv. 2, 4547 (2012)
S.P. Davis, Chitosan: Manufacture, Properties and Usage (Nova Science, New York, 2011)
R.G. Mackay, J.M. Tait, Handbook of Chitosan Research and Applications (Nova Science, New York, 2011)
A.E. Kadib, Chem. Sus. Chem 8, 217 (2015)
S.N. Rao, D.C. Mohan, S. Adimurthy, Green Chem. 16, 4122 (2014)
M.G. Dekamin, M. Azimoshan, L. Ramezani, Green Chem. 15, 811 (2013)
J. Lal, S.K. Gupta, D.D. Agarwal, Catal. Commun. 27, 38 (2012)
K. Khalil, H. Al-Matar, M. Elnagdi, Eur. J. Chem. 1, 252 (2010)
N. Sudheesh, S.K. Sharma, R.S. Shukla, A. Chrobok, J. Mol. Catal. A: Chem. 321, 77 (2010)
K.R. Reddy, K. Rajgopal, C.M. Maheswari, M.L. Kantam, New J. Chem. 30, 1549 (2006)
D. Kuhbeck, G. Saidulu, K.R. Reddy, D.D. Diaz, Green Chem. 14, 378 (2012)
P.K. Sahu, P.K. Sahu, S.K. Gupta, D.D. Agarwal, Ind. Eng. Chem. Res. 53, 2085 (2014)
B.C.E. Makhubela, A. Jardine, G.S. Smith, Appl. Catal. A Chem. 393, 231 (2010)
K. Martina, S.E.S. Leonhardt, B. Ondruschka, M. Curini, A. Binello, G. Cravotto, J. Mol. Catal. A: Chem. 334, 60 (2011)
T.C.O. Mac Leod, V. Palaretti, V.P. Barros, A.L. Faria, T.A. Silva, M.D. Assis, Appl. Catal. A Chem. 361, 152 (2009)
M. Chtchigrovsky, A. Primo, P. Gonzalez, K. Molvinger, M. Robitzer, Angew. Chem. Int. Ed. 48, 5916 (2009)
G.A. Dee, O. Rhode, R. Wachter, Cosmet Toilet. 116, 39 (2001)
C.K. Chu, V.S. Bhadti, K.J. Doshi, J.T. Etse, J.M. Gallo, F.D. Boudinot, R.F. Schinazi, J. Med. Chem. 33, 2188 (1990)
B. Mishra, R. Mishra, Pharmacist 2, 13 (2007)
R. Boer, V. Gekeler, Drugs Future 20, 499 (1995)
S. Gullapalli, P. Ramarao, Neuropharmacology 42, 467 (2002)
S. Yasar, M. Corrada, R. Brookmeyer, C. Kawas, Neurobiol. Aging 26, 157 (2005)
J. Marco-Counntelles, R. Leon, C. de los Rios, A. Guglietta, J. Terencio, M.G. Lopez, A.G. Garcia, M. Villarroya, J. Med. Chem. 49, 7607 (2006)
G.W. Zamponi, S.C. Stotz, R.J. Staples, T.M. Andro, J.K. Nelson, U. Hulubei, A. Blumenfeld, N.R. Natale, J. Med. Chem. 46, 87 (2003)
M. Ramesh, W.C. Matowe, A.R. Murthy, D. Vo, L. Dagnino, M.C. Li-Kwong-Ken, W.W. Michael, E.E. Knaus, J. Med. Chem. 41, 509 (1998)
D.R.H. Kumar, N. Naira, M. Gahate, Asian J. Chem. 21, 4357 (2009)
R. Miri, K. Javidnia, H. Sarkarzadeh, B. Hemmateenejada, BMC 14, 4842 (2006)
F. Bossert, H. Meyer, E. Wehinger, Angew. Chem. Int. Ed. Engl. 20, 762 (1981)
S. Cosconati, L. Marinelli, A. Lavecchia, E. Novellino, J. Med. Chem. 50, 1504 (2007)
A. Hantzsch, Ber. Dtsch. Chem. Ges. 14, 1637 (1881)
C.G. Evans, J.E. Gestwicki, Org. Lett. 11, 2957 (2009)
A. Kumar, R.A. Maurya, Tetrahedron 63, 1946 (2007)
A.G. Sathicq, G.P. Romanelli, A. Ponzinibbio, G.T. Baronetti, H.J. Thomas, Lett. Org. Chem. 7, 511 (2010)
A. Maleki, M. Kamalzare, M. Aghaei, J. Nanostruct. Chem. 5, 95 (2015)
M. Nasr-Esfahani, S.J. Hoseini, M. Montazerozohori, R. Mehrabi, H. Nasrabad, J. Mol. Catal. A: Chem. 382, 99 (2014)
S.U. Tekale, V.P. Pagore, S.S. Kauthale, R.P. Pawar, Chin. Chem. Lett. 25, 1149 (2014)
L.M. Wang, J. Sheng, L. Zhang, J.W. Han, Z.Y. Fao, H. Tian, C.T. Qian, Tetrahedron 61, 1539 (2005)
M. Li, W.S. Guo, L.R. Wen, Y.F. Li, H.Z. Yang, J. Mol. Catal. A: Chem. 258, 133 (2006)
R.A. Mekheimer, A.A. Hameed, K.U. Sadek, Green Chem. 10, 592 (2008)
S. Ko, M.N.V. Sastry, C. Lin, C.F. Yao, Tetrahedron Lett. 46, 5771 (2005)
S.R. Cherkupally, R. Mekalan, Chem. Pharm. Bull. 56, 1002 (2008)
S.K. Singh, K.N. Singh, J. Heterocycl. Chem. 47, 194 (2010)
F.M. Tamaddon, S. Moradi, J. Mol. Catal. A: Chem. 370, 117 (2013)
R. Surasani, D. Kalita, A.V.D. Rao, K. Yarbagi, K.B. Chandrasekhar, J. Fluorine Chem. 135, 91 (2012)
A. Kumar, R.A. Maurya, Tetrahedron Lett. 48, 3887 (2007)
M. Hong, C. Chai, W.B. Yi, J. Fluorine Chem. 131, 111 (2010)
S. Zhaleh, N. Hazeri, M.T. Maghsoodlou, Res. Chem. Intermed. doi:10.1007/s11164-016-2469-z
N. Hazeri, M.T. Maghsoodlou, F. Mir, M. Kangani, H. Saravani, E. Molashahi, Chin. J. Catal. 35, 391 (2014)
K.A. Undale, T.S. Shaikh, D.S. Gaikwad, D.M. Pore, Chimie 14, 511 (2011)
A. Khazaei, A. Moosavi-Zare, H. Afshar-Hezarkhania, V. Khakyzadeh, RSC Adv. 4, 32142 (2014)
L.J. Donelson, R.A. Gibbs, S.K. De, J. Mol. Catal. A: Chem. 256, 309 (2006)
A. Khazaei, M.A. Zolfigol, A.R. Moosavi-Zare, J. Afsar, A. Zare, V. Khakyzadeh, M.H. Beyzavi, Chin. J. Catal. 34, 1936 (2013)
K. Ahmeda, A.K. Jaina, B. Dubeya, B. Shrivastavab, P. Sharmab, S. Nadeemc, Der. Pharma. Chem. 7, 52 (2015)
Acknowledgment
The University of Sistan and Baluchestan is thanked for financial supporting of this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhaleh, S., Hazeri, N., Faghihi, M.R. et al. Chitosan: a sustainable, reusable and biodegradable organocatalyst for green synthesis of 1,4-dihydropyridine derivatives under solvent-free condition. Res Chem Intermed 42, 8069–8081 (2016). https://doi.org/10.1007/s11164-016-2579-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-016-2579-7