
Abstract
A three-component, NBS-promoted synthesis of α,β-unsaturated isoxazol-5(4H)-ones by reaction of aromatic aryl or hetero-aryl aldehydes, hydroxylamine hydrochloride, and 1,3-dicarbonyl compounds (ethyl acetoacetate or ethyl 4-chloroacetoacetate), under mild reaction conditions at room temperature is described. This simple, efficient, and clean reaction is an expeditious means of obtaining the corresponding isoxazol-5(4H)-one derivatives in good to high yields. Geometrical properties and vibrational wavenumbers of 3-methyl-4-(thiophen-2-ylmethylene)isoxazol-5(4H)-one (MTISO) were predicted by use of density functional theory (DFT) by use of the B3LYP level with the 6-311++G(d,p) and 6-311++G(2d,p) basis sets. Results indicate that the B3LYP method enables satisfactory prediction of vibrational frequencies and structural data. The absorption spectra of MTISO in solvents of different polarity were studied at room temperature. The UV–visible spectrum of the compound was recorded and such electronic properties as the energies of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) were determined by the time-dependent DFT (TD-DFT) approach. The stability of the molecule arising from hyper-conjugative interaction and charge delocalization was studied by NBO analysis. A molecular electrostatic potential map (MEP) of the compound was also studied to predict reactive sites. Reactivity descriptors, Fukui functions, and electrophilic sites were found and are discussed. The thermal stability of MTISO was studied by thermogravimetric analysis (TGA).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The isoxazol-5(4H)-ones and their derivatives have applications in many areas, for example liquid crystalline materials [1], organic synthesis [2], light-conversion molecular devices [3], filter dyes in photographic films [4], and optical storage and nonlinear optical research [5]. Several substituted isoxazoles have been shown to have a broad range of biological properties, for example anti-obesity [6], analgesic [7], anti-inflammatory [8], and antitumor [9] activity. The isoxazole backbone is also a structural component of a variety of natural products, for example, muscimol [10], cycloserine [11], ibotenic acid, and isoxazol-4-carboxylic acid [12]. For this reason, much research has been focused on synthesis of isoxazoles and study of their properties.
In recent years, α,β-unsaturated isoxazol-5(4H)-ones have been prepared via a three-component condensation of β-oxoesters, hydroxylamine hydrochloride, and aromatic aldehydes by use of catalytic amounts of sodium benzoate [13], sodium sulfide [14], sodium silicate [15], DABCO [16], nano Fe2O3, clinoptilolite, and H3PW12O40 [17]. Other conditions have also been reported in the literature for this three-component reaction, including the use of sodium acetate and visible light [18], pyridine under ultrasonic irradiation [19, 20], pyridine under reflux [21, 22], and catalyst-free/grinding or heating [23]. We have synthesized several α,β-unsaturated isoxazol-5(4H)-one derivatives by using sodium ascorbate [24], sodium citrate [25], sodium saccharin [26], sodium tetraborate [27], sodium azide [28], boric acid [29], and potassium phthalimide [30] as catalysts. Although α,β-unsaturated 4H-isoxazol-5-ones have been synthesized, no results of three-component cyclocondensation of aryl aldehydes (1a–n), hydroxylamine hydrochloride (2), and β-oxoesters (3a–b), using NBS as a catalyst, have yet been reported in the literature. In the work discussed in this report, NBS was used as an organic catalyst for synthesis of α,β-unsaturated isoxazol-5(4H)-ones in aqueous media (Scheme 1).

Three-component synthesis of α,β-unsaturated isoxazol-5(4H)-ones promoted by NBS
In this study, FT-IR, 1H NMR, 13C NMR, and UV spectral investigation of MTISO were performed by use of density functional theory (DFT). NBO analysis was performed to provide valuable information about a variety of intermolecular interactions. The molecular structure, E HOMO (energy of the highest occupied molecular orbital), E LUMO (energy of the lowest unoccupied molecular orbital), LUMO–HOMO energy gap (ΔE), global hardness (η), softness (S), electronegativity (χ), dipole moment (µ), polarizabilities (〈α〉), anisotropy of the polarizabilities (〈Δα〉), first-order hyperpolarizabilities (〈β〉), Fukui functions, molecular electrostatic potential maps (MEP), and thermodynamic data (molecular energy (E), heat capacity (\(C_{p,m}^{0}\)), entropy (\(S_{m}^{0}\)) and enthalpy (\(H_{m}^{0}\))) were calculated and are discussed.
Experimental
General
Mid-IR spectra in the range 4,000–400 cm−1, with spectral resolution of 2 cm−1, were acquired by use of a Perkin–Elmer RXI Fourier-transform spectrophotometer with the KBr pellet technique (solid phase); results from 16 scans were averaged. Ultraviolet absorption spectra in the range 200–800 nm were acquired by use of a Perkin–Elmer Lambda 25 spectrophotometer. NMR spectra were recorded at ambient temperature on a Bruker Avance DRX 400 MHz with CDCl3 or DMSO-d 6 as solvent. Melting points were measured on a Buchi 510 melting point apparatus and are uncorrected. Chemicals were obtained from Merck and Fluka and were used without further purification, with the exception of NBS, which was purified by recrystallization from water. The progress of reactions was monitored by thin-layer chromatography (TLC) on silica gel 60 GF254 aluminium sheets, with ethyl acetate–petroleum ether (1:3) as mobile phase. The spots were exposed by UV light and iodine vapor. To determine thermal stability, thermal analysis (TGA and DTA) was performed with a Perkin–Elmer instrument. Analysis was performed in an atmosphere of nitrogen at a heating rate of 25°/min for 10 min in the temperature range 25–750 °C for TGA.
Typical procedure for synthesis of α,β-unsaturated isoxazol-5(4H)-ones (4a–t)
A mixture of equimolar quantities of hydroxylamine hydrochloride (0.07 g, 1 mmol) and ethyl acetoacetate (0.130 g, 1 mmol) in 5 mL water was stirred for 5 min. Thiophene-2-carbaldehyde (0.112 g, 1 mmol) and NBS (5 mol%) were then added and the reaction mixture was stirred at room temperature for 95 min. The solid products were isolated by simple filtration, washed with water (5 mL), then recrystallized from ethanol. Synthesis of compounds 4a–t has been reported in our previous papers, so the products are known; their identity was confirmed by comparison of their physical and spectral data with those available in our recent papers [24–30].
Spectral data for 3-methyl-4-(thiophen-2-ylmethylene)isoxazol-5(4H)-one: 1H NMR (400 MHz, CDC13): δ 2.32 (s, 3H), 7.29 (t, J = 4.8 Hz, 1H), 7.65 (s, 1H), 7.95 (d, J = 4.8 Hz, 1H), 8.13 (d, J = 3.6 Hz, 1H); 13C NMR (100 MHz, CDC13): δ 11.5, 114.6, 128.9, 136.5, 139.2, 139.6, 141.5, 160.7, 168.7.
Calculations
The molecular structure of MTISO and the corresponding vibrational harmonic frequencies were calculated by use of the B3LYP method, combined with the 6-311++G (d, p) and 6-311++G(2d,p) basis sets, in the Gaussian 03 software package [31], without any constraint on geometry. B3LYP uses Becke’s three-variable hybrid functional method [32] with the Lee–Yang–Parr correlation functional (LYP) [33, 34]. Assignment of calculated wavenumbers was aided by the animation option of Gaussview software, which gives a visual presentation of the vibrational modes [35]. In this investigation we found that the calculated frequencies were slightly greater than the fundamental frequencies. Such discrepancies are usually corrected either by computing anharmonic corrections explicitly [36] or by introducing a scale factor. In this study selective scaling factors were used: 0.9961 for wavenumbers <1,700 cm−1, 0.9556 for wavenumbers >1,700 cm−1 at 6-311++G(d, p), and 0.9963 for wavenumbers <1,700 cm−1 and 0.9959 for wavenumbers >1,700 cm−1 at 6-311++G(2d,p) [37]. Detailed interpretation of the vibrational spectra of MTISO was performed on the basis of calculated potential energy distribution (PED). Assignments of the experimental frequencies are based on the observed band frequency and intensity changes in the infrared spectra, and confirmed by establishing one-to-one correlation between observed and theoretically calculated frequencies.
The electronic properties were also calculated by use of the B3LYP method of the time-dependent DFT (TD-DFT) [38], on the basis of the optimized structure. TD-DFT has been proved to be a powerful and effective computational tool for study of ground and excited state properties by comparison with available experimental data. Hence, we used TD-DFT to obtain wavelengths and compared the calculated results with experimental UV absorption values. To understand intramolecular delocalization and hyperconjugation, NBO calculations [38] were performed by use of NBO 3.1 software as implemented in the Gaussian 03W package at the DFT/B3LYP level. The molecular electrostatic potential (MEP) surface was plotted over the optimized geometry to elucidate the reactivity of the MTISO molecule. The HOMO and LUMO were calculated by use of the B3LYP/6-311++G(2d,p) method. The Lorentzian function was used for deconvolution of IR spectra, by use of the Genplot package.
Results and discussion
Optimization of reaction conditions and scope of the reaction
To initiate our study, we checked the three-component reaction of 4-hydroxybenzaldehyde (1h), hydroxylamine hydrochloride (2), and ethyl acetoacetate (3a) as model reaction. The effects of the amounts of catalyst and solvents were investigated; the experimental results are summarized in Table 1. Yields of product 4h were 80, 91, and 95 %, respectively, when 1, 2.5, and 5 mol% NBS were used (Table 1, entries 2–4). On the basis of reaction time (100 min) and yield (95 %), 5 mol% of NBS provided the best result and was thus selected as the optimum quantity for promotion of the reaction. Use of larger amounts of NBS resulted in only trace amounts of the product (Table 1, entries 5 and 6).

We also screened a variety of solvents (EtOH, acetone, hexane, 1,4-dioxane, cyclohexane, THF, 1:1 (v/v) H2O–EtOH, and 1:1 (v/v) H2O–acetone) for this reaction, with 5 mol% NBS as catalyst (Table 1, entries 7–14). This investigation revealed that reaction in water resulted in the best reaction time and yield, so water was selected as the optimum solvent. Water is also an environmentally benign solvent. When the reaction was performed under solvent-free conditions the yield in 120 min was lower (Table 1, entry 15). Increasing the reaction temperature to 50, 70, and 100 °C had no significant effect on the yield of the product or on reaction rate.
After optimization of the reaction conditions, we turned our attention toward the scope of the reaction, by using a variety of aryl aldehydes with different substituents on the benzene ring, and π-excessive heterocyclic aldehydes (1j–l), and ethyl acetoacetate (3a) or chloroethyl acetoacetate (3b) to construct α,β-unsaturated isoxazol-5(4H)-ones (4a–t). As is apparent from Table 2, with substituted benzaldehydes containing electron-donating groups at the m- or p-position of the benzene ring and with heterocyclic aldehydes this three-component reaction proceeded efficiently to give the corresponding products in good to excellent yields and with reasonable reaction times (entries 1–19 in Table 2).
Although we did not investigate the mechanism of this reaction, on the basis of the literature [13–30] we propose the mechanism outlined in Scheme 2 for formation of α,β-unsaturated isoxazol-5(4H)-ones (4a–t). According to this mechanism, NBS effectively catalyzes the Knoevenagel condensation between an aldehyde and an active methylene compound (3). Intramolecular O-attack cyclization of 10 to 11, proton exchange between 11 and 12, then deethanolization lead to formation of the target products.

A plausible mechanism for the synthesis of α,β-unsaturated isoxazol-5(4H)-ones (4a–t)
Geometrical data
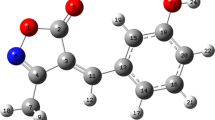
The optimized geometrical data for MTISO, calculated at B3LYP/6-311++G(d,p) and B3LYP/6-311++G(2d,p) levels of theory are presented in Table 3, in accordance with atom numbering scheme given in Fig. 1.

Theoretical optimized geometric structure and atom numbering for MTISO
The optimized structure can only be compared with other similar systems for which the crystal structures have been solved [39, 40]. For title compound, it was found that the carbon–carbon bond lengths of the molecular skeleton was intermediate between typical C–C single (1.54 Å) and C=C double (1.34 Å) bonds. The π electrons were delocalized throughout the molecule, and the methyl group in the isoxazolone ring was involved in molecular conjugation. For MTISO, all of the torsion angles were close to 180° or 0°, which indicates that the molecular framework of title compound is planar. The torsion angles C4–C9–C11–C16 and C3–C4–C9–C11 (−179.91° and 0.06°) show that the thiophene group and isoxazole group are nearly coplanar. The C2–C3 (1.41 Å) and C9–C4 (1.43 Å) bond lengths are shorter than those of C11–C12 (1.48 Å), C11–C16 (1.45 Å), and C16–C17 (1.49 Å) (Table 3), because of delocalization of the electron density of the C=C of thiophene. The bond angles between C3=C4–C9 (132.52°) and C9–C11–C16 (125.39°) are larger than 120°, indicating the presence of hyper-conjugation.
The difference between the C11–C12 and C11–C16 bond lengths arises because of the different polarity of the groups attached to them. Cheng et al. [40] reported N–O and C=N bond lengths of approximately 1.44 Å and 1.28 Å, respectively. In our study C=N bond lengths were computed 1.29 Å and N–O bond lengths as approximately 1.43 Å, in good agreement with experimental data and literature values. It seems the B3LYP/6-311++G(2d,p) method correctly predicted bond lengths, bond angles, and torsion angles; this is useful for investigation of the characteristics of structurally related isoxazoles.
The potential energy profile as a function of the dihedral angle between the isoxazole and thiophene rings (C11=C9–C4–C3) was obtained by use of the DFT method; a plot of the potential energy surface (PES) for this molecule is shown in Fig. 2. During the calculations, all the geometrical parameters were simultaneously relaxed and the dihedral angle was varied in steps of 10° from 360° and reduced up to zero. The optimized structure was obtained at 0.06° for the C11=C9–C4–C3 dihedral angle; the corresponding minimum energy was −950.5347 Hartree, which implies that the structure obtained was at the global minimum.

PES scan for the dihedral angle C11=C9–C4–C3, obtained by use of the DFT/B3LYP/6-311++G(d,p) method
Vibrational analysis
MTISO consists of 20 atoms which have 54 normal modes. These normal modes were assigned in accordance with the detailed motion of the individual atoms. The observed FTIR spectrum of MTISO is shown in Fig. 3.

FTIR spectrum of 3-methyl-4-thiophen-2-ylmethylene-4H-isoxazol-5-one
The observed IR bands, calculated frequencies, IR intensities, Raman activity, and normal mode descriptions (characterized by PED) of the stable conformer of MTISO are depicted in Table 4. For clear observation of these bands, deconvolution techniques can be used for the IR spectrum. A deconvoluted IR spectrum of MTISO is depicted in Fig. 4. The ultimate purpose of this part of the study was assignment of vibrational wave numbers. Vibrational analysis of MTISO was performed on the basis of the characteristic vibration modes of the C–O, C=O, C=N, N–O, C–S, CH, and CH3 groups of the compound.

Deconvoluted IR spectrum of MTISO (in the 1,800–400 cm−1 region)
The C–O stretching vibrations of isoxazole produce strong bands in the 1,126–1,084 cm−1 region of the spectrum [40]. According to our deconvolution results, the IR spectrum of MTISO contains medium FT-IR bands arising from C–O stretching vibrations at 1,130 cm−1 (Fig. 4) and 1,166 cm−1 (B3LYP). Normally, carbonyl group vibrations occur in the region 1,780–1,680 cm−1 [41]. In this study, the C=O stretching vibration is assigned at 1,736 cm−1 in the FT-IR spectrum, with very strong intensity. The bands observed at 512 and 531 cm−1 (DFT) were assigned to C=O in-plane bending vibrations.
Brancatelli et al. [42] reported a value 1,571 cm−1 for the isoxazol-5(4H)-one C=N stretching mode. Deconvoluted IR spectra of MTISO indicate the presence of three bands at 1,612, 1,602, and 1,576 cm−1 in IR spectra, and the bands at 1,629, 1,613, 1,531 cm−1 given by calculation are assigned as υC=N (Table 4). In The N–O stretching skeletal bands are expected in the range 878 cm−1 [39]. A band observed at 876 cm−1 in the FT-IR spectrum is assigned to the N–O stretching vibration mixed with C–CH3 stretching.
C–H stretching of methyl group occurs at lower frequencies than for that in the aromatic ring (3,100–3,000 cm−1). For MTISO B3LYP calculations give υaCH3 at 3,116, 3,115, and 3,070 cm−1 and υ sCH3 at 3,020 cm−1. The bands at 3,077, 2,960, 2,926, and 2,856 cm−1 in the IR spectrum are assigned as υaCH3 and υsCH3. For many molecules the symmetric deformation appears in the range 1,380 ± 25 cm−1 with intensity varying from medium to strong [43]. For MTISO, the scissoring modes of the CH3 group were calculated to be at 1,476, 1,426, and 1,403 cm−1 (B3LYP); the measured values (IR spectrum) were 1,509, 1,420, and 1,408 cm−1. The rocking vibrations of the CH3 group are usually observed in the region 1,070–1,010 cm−1 [44]. The bands at 1,030, 998, and 981 cm−1 in the IR spectrum, and three bands at 1,061, 1,037, and 1,005 cm−1 (B3LYP) were assigned as ρCH3 mode for MTISO. The twisting modes were not observed in the FT-IR spectrum because they appear at very low frequency. The band at 166 cm−1 (B3LYP) was assigned as the twisting mode τCH3.
The thiophene ring C–H stretching vibrations were calculated to be at 3,243, 3,216, and 3,195 cm−1 and observed at 3,549, 3,465, and 3,420 cm−1 in the FT-IR spectrum. The C–H out-of-plane bending modes of thiophene derivatives are observed in the region 600–900 cm−1. Aromatic C–H out-of-plane bending of the thiophene ring is assigned to medium to weak bands observed at 986, 932, 876, and 624 cm−1. Aromatic C–H in-plane bending modes of thiophene and its derivatives are observed in the region 1,360–1,080 cm−1 [44]. According to our deconvolution results, C–H in-plane bending modes of the thiophene ring are predicted at 1,216, 1,092, 1,072, and 1,037 cm−1.
Identification of C–S stretching vibration is difficult and uncertain. Four peaks recorded at 608.8, 753.5, 839.5, and 872.8 cm−1 have been assigned to C–S stretching [45] on the basis of potential energy distributions (PED) obtained from DFT calculations. Coruh et al. [46] assigned this mode at 834 cm−1 in the FT-IR spectrum. In our study, experimental C–S stretching vibrations were observed at 866 and 776 cm−1.
It is apparent from Fig. 5 that correlation between the theoretical and experimental FT-IR spectra is excellent, i.e. experimental fundamentals correlate well with B3LYP results. The correlation plot is expressed by the linear equation, y = 55.063 + 0.9387x, where y is the experimental wavenumber and x the calculated wavenumber (cm−1). The correlation coefficient (R 2 = 0.9989) also shows there is good agreement between experimental and calculated results.

Correlation of calculated and experimental IR frequencies for MTISO
NBO analysis
Natural bond orbital (NBO) analysis provides a description of the structure of a conformer as a set of localized bonds, antibonds, and Rydberg extra valence orbitals. NBO analysis was used to determine the interaction between bond orbitals, electron delocalization, bond-bending effect, and intramolecular charge transfer (ICT). The second-order Fock matrix was used to evaluate donor–acceptor interactions in the NBO analysis [47]. The interactions result in loss of occupancy from the localized NBO of the idealized Lewis structure into an empty non-Lewis orbital. For each donor (i) and acceptor (j) the stabilization energy E (2) associated with the delocalization i → j is determined as:
where q i is the donor orbital occupancy, E i and E j are diagonal elements, and F ij is the off-diagonal NBO Fock matrix element [48]. In NBO analysis, a large E (2) value is indicative of strong interaction between electron donors and electron acceptors, and the greater the extent of conjugation of the whole system, the greater is the interaction given by NBO (Table 5).
Second-order perturbation theory analysis of the Fock matrix in NBO revealed strong intermolecular hyper-conjugative interactions as a result of orbital overlap between n(O) and n(S) with the π*(C–O), π*(C–N), and π*(C–C) bond orbitals, resulting in ICT stabilization of the system.
The strong intermolecular hyper-conjugative interaction πC3–C4→π*(C1–C2 and C9–C11) of C3–C4 in MTISO increases the ED (0.24 and 0.17 e) and thus weakens the respective bonds, leading to stabilization of 30.54 and 34.60 kcal mol−1. The hyper-conjugative interaction in S5 of n2(S5)→π*(C1–C2 and C3–C4) increases the ED (0.24 and 0.28 e) and weakens the respective bonds leading to stabilization of 40.66 and 30.61 kcal mol−1. There is a strong intermolecular hyper-conjugative interaction in MTISO from O13 of n2(O13)→σ*(C11–C12), σ*(C12–O14) which increases the ED (0.07 and 0.10 e) and weakens the respective bonds, leading to stabilization of 32.14 and 50.63 kcal mol−1, and also hyper-conjugative interaction for O14 of n2(O14)→π*(C16–N15, C12–O13) which increases the ED (0.14 and 0.19 e) and weakens the respective bonds, C16–N5 and C12–O13, leading to stabilization of 23.35 and 60.77 kcal mol−1. The increased electron density on the oxygen atoms leads to elongation of the respective bonds and reduction of the corresponding stretching wavenumber. The intramolecular interaction is a result of orbital overlap between the π(C–C) and π*(C–C) bond orbitals, which results in ICT and stabilization of the system. These interactions are observed as an increase in electron density (ED) in the C–C, C–N, and C–O antibonding orbitals, which weakens the respective bonds. The electron density (ED) is transferred from the n(O) to the antibonding π* orbital of the C–C, C–O, and N–C bonds, explaining both the elongation and the red shift [49].
Charge distribution
The Mulliken [50] atomic charge distribution of the MTISO is shown in Fig. 6. Mulliken atomic charges were computed by use of the DFT/B3LYP method with the 6-311++G(d,p) and 6-311++G(2d,p) basis sets. Calculation of atomic charges is important in the application of quantum mechanical calculations to molecular systems [51]. The charge distribution of MTISO shows that carbon atoms attached to hydrogen atoms are negative whereas other carbon atoms are positively charged. The oxygen atoms are more negative, and all the hydrogen atoms have positive charges. This results in positive charges on C3, C4, C12, H6, H7, H8, H10, H18, H19, and H20, which shows the direction of delocalization and that the natural atomic charges are more sensitive than Mulliken’s net charges to changes in the molecular structure. Negatively charged lone pairs on the S5, O13, O14, and N15 atoms show that charge is transferred from S, O, and N to C (O13, O14 → C12, N15 → C16, and S5 → C4).

Comparative Mulliken’s plot at the B3LYP/6-311++G(d,p) and B3LYP/6-311++G(2d,p) levels for MTISO
The charge changes with basis set as a result of polarization. For example, the charges on the H6 and H7 atoms are 0.248 e and 0.73 e at the B3LYP/6-311++G(d,p) level and 0.192 e and 0.149 e at the B3LYP/6-311++G(2d,p) level. The charge distribution on the C9 and C11 atoms is increased at the levels of the 6-311++G(d,p) and 6-311++G(2d,p) basis set whereas the opposite trend is observed for the C3 atom. Considering all the basis sets and the levels used for atomic charge calculation, the oxygen, nitrogen, and sulfur atoms have substantial negative charges, and are donor atoms. The hydrogen atoms are positively charged, and are acceptor atoms.
NMR spectral studies
The 1H and 13C NMR spectra of MTISO are shown in Figs. 7 and 8, respectively. The 1H NMR and 13C NMR experimental chemical shifts, isotropic shielding constants, and spectral assignments are listed in Table 6. The atoms are numbered in accordance with Fig. 1.

Experimental 1H NMR spectrum of MTISO

Experimental 13C NMR spectrum of MTISO
Aromatic carbon atoms give signals in overlapped areas of the spectrum, with chemical shifts from 100 to 200 ppm [52]. The signals from the eight aromatic carbon atoms are in the range 114.58–168.69 ppm.
The O and N atoms are highly electronegative and reduce the electron density on carbon atoms of the isoxazole ring. The chemical shifts of C12 and C16 are thus attributed to the downfield NMR signals at 177.69 and 170.75 ppm, respectively. Carbon atoms C2, C9, C3, C4, and C1 are observed up-field, with chemical shifts of 128.92, 136.46, 139.22, 139.59, and 141.52 ppm, respectively; this indicates the effect of the electronegative sulfur atom is negligibly small, because their signals are observed in the normal range. Normally, the proton chemical shift of organic molecules varies substantially with the electronic environment of the proton. Shielding of hydrogen atoms attached to or near electron-withdrawing atoms or groups is reduced, moving the resonance to higher frequency, whereas electron-donating atoms or groups increase shielding and move the resonance to lower frequency [53]. The chemical shifts of aromatic protons of organic compounds are usually observed in the range 7.00–8.00 ppm, whereas aliphatic protons resonate at high field. The signals of the four aromatic protons (1H) are observed at 7.29–8.12 ppm. We predicted H8 at 8.12 ppm in the lowest field of the aromatic region, and H7 at 7.29 ppm in the highest field of this region. The aliphatic protons were observed at 2.32 ppm.
Molecular electrostatic potential (MEP) surface
To investigate the chemical reactivity of the molecule, the molecular electrostatic potential (MEP) surface was plotted over the optimized electronic structure of MTISO by using the density functional B3LYP level with the 6-311++G(2d,p) basis set. The MEP surface is related to electron density and is a very useful aid to understanding sites of electrophilic and nucleophilic reactions and hydrogen-bonding interactions [54]. Figure 9 shows the computationally observed MEP surface map, with point charges fitted to the electrostatic potential V(r).

Molecular electrostatic potential surfaces (left) and the point charges and electric potential values (right) on the atoms of MTISO
The negative (red) regions of the MEP were related to electrophilic reactivity and the positive (blue) regions to nucleophilic reactivity. In Fig. 9, this molecule has several possible sites for electrophilic attack on the nitrogen and oxygen atoms (O13, O14, and N15). The average maximum negative electrostatic potential value for these electrophilic sites, calculated at the B3LYP/6-311++G(2d,p) level, are approximately −22.377 a.u. The point charges fitted to these electrostatic potentials are −0.568 (O13), −0.215 (O14), and −0.384 (N15). Regions of maximum positive charge are located on the hydrogen atoms, indicating possible sites for nucleophilic attack. The MEP map shows that the negative potential sites are on electronegative atoms and the positive potential sites are around the hydrogen atoms. The MEP provides a visual representation of the chemically active sites and comparative reactivity of the atoms.
HOMO–LUMO band gap
Gauss-Sum 2.2 software [55] was used to calculate group contributions to the molecular orbitals (HOMO and LUMO) and prepare the density of the state (DOS), shown in Fig. 10. The DOS spectra were created by convoluting the molecular orbital information with Gaussian cures of unit height.

Calculated TDOS diagrams for MTISO
The energy gap between the HOMO and the LUMO is a critical property determining molecular electrical transport behavior [56]. Conjugated molecules are characterized by HOMO–LUMO separation which is the result of substantial ICT from the end-capping electron-donor groups to the efficient electron-acceptor groups through a π-conjugated path. Therefore, ED transfer occurs from the more aromatic part of the π-conjugated system at the electron-donor side to its electron-withdrawing part. The HOMO, HOMO-1 (An orbital below the HOMO orbital), LUMO, and LUMO+1 (An orbital above the LUMO orbital) are shown in Fig. 11.

The atomic orbital compositions of the frontier molecular orbital for MTISO
The energy of the HOMO is directly related to the ionization potential, and the energy of the LUMO is related to the electron affinity. The HOMO–LUMO energy gap explains charge transfer interactions within the molecule. The frontier orbital gap for MTISO, obtained by use of the TD-DFT method with the 6-311++G(2d,p) basis set, is found to be 3.5784 eV. Excitation of intramolecular electron displacement occurs from the thiophene group to the isoxazole moiety, with the thiophene and isoxazole rings acting as electron donors and electron acceptors, respectively.
UV–visible spectral analysis
On the basis of a fully optimized ground state structure, electronic spectra were computed for MTISO in the gas phase and in solution in ethyl acetate and DMSO, by use of the TD-DFT method [57] and the IEF-PCM model [58], using the B3LYP/6-311++G(d,p) functional, in an attempt to understand the nature of the electronic transitions. The positions of experimental and calculated absorption peaks (λ max’s), vertical excitation energies (E), and oscillator strengths (f) are presented in Table 7.
UV–visible electronic spectra of 3-methyl-4-thiophen-2-ylmethylene-4H-isoxazol-5-one in ethanol, DMSO, 1,4-dioxan, CHCl3, and ethyl acetate were recorded in the range 200-800 nm. Representative spectra are shown in Fig. 12. A clear red shift of the absorption maxima was observed when changing from low-polarity to high-polarity solvents. It is suggested for MTISO that π–π* transitions are more polar in the excited states than in the ground states. In another study it was concluded that a spectral red shift in high-polarity solvents was mainly because of greater reduction of the energy of the excited states than that of the ground states [59].

UV–visible absorption spectra of MTISO in different solvents
Common types of electronic transition in organic compounds are π–π*, n–π*, and π*(acceptor)–π(donor). The strong band for MTISO in DMSO, attributed to the π → π* transition, was observed experimentally at 371 nm and calculated to be at 359 nm. The bands observed at 319 and 298 nm are because of n–π* transitions. The lowest-energy absorption of MTISO corresponded to the first dipole-allowed π–π* electronic transition from the HOMO orbital to the LUMO orbital, and could be attributed to ICT.
Thermal analysis
Thermal analysis of MTISO was performed by use of a Perkin-Elmer simultaneous thermogravimetric and differential thermal (TG/DT) analyzer. The sample was scanned in the temperature range 25–750 °C at 25 °/min, and the final temperature was maintained for 10 min. The thermogravimetric (TG) and differential thermal gravimetry (DTG) curves are shown in Fig. 13. As the temperature was increased to 200 °C, sample weight decreased slightly as a result of evaporation of water. Weight loss started at approximately 177 °C. Two major weight losses were observed above 177 °C. The first, 50 % loss, occurred between 177 and 215 °C. The major weight loss (65 %) occurred over a large temperature range (215–750 °C), in which almost all the compound was decomposed into gaseous products (10 % charred material remaining). In the DTG curve, two peaks were identified at 183 °C and 389 °C, indicating that weight loss by thermal decomposition occurred in two steps (Fig. 13b).

a TGA and b DTG curves of MTISO
Non-linear optical properties
On the basis of the finite-field approach, the non-linear optical properties dipole moment, polarizability, anisotropy polarizability, and first order hyperpolarizability of MTISO were calculated at the B3LYP level with the 6-311++G(d, p) and 6-311++G(2d, p) basis sets, for more reliability. The numerical values obtained are listed in Table 8.
The first hyperpolarizability is a third-rank tensor that can be described by a 3 × 3 × 3 matrix. The 27 components of the 3D matrix can be reduced to 10 components as a result of the Kleinman symmetry [60]. The components of β are defined as the coefficients in the Taylor series expansion of the energy in the external electric field. When the electric field is weak and homogeneous, this expansion becomes:
where E 0 is the energy of the unperturbed molecule, F i is the field at the origin, µ i , α ij , β ijk , and γ ijkl are the components of dipole moment, polarizability, first hyperpolarizability, and second hyperpolarizability, respectively [61]. The total static dipole moment, mean polarizability (α tot), anisotropy polarizability (Δα), and average value of the first hyperpolarizability (〈β〉) can be calculated by use of the equations:
It is well known that high values of the dipole moment, molecular polarizability, and hyperpolarizability are important for more active non-linear optical (NLO) properties. The calculated dipole moments for MTISO are 6.590 and 6.426 D. Calculated polarizabilities αij have non-zero and zero values and are dominated by the diagonal components. Total polarizability (α tot) was calculated as –12.338 × 10−24 and –12.263 × 10−24 esu. The first hyperpolarizability values (〈β〉) for MTISO are 9.2366 × 10−31 and 8.9007 × 10−31 esu. Urea a prototypical molecule used to study the NLO properties of molecular systems. It has, therefore, frequently been used as a threshold value for comparative purposes. The first hyperpolarizability for MTISO is 7.11 times the magnitude for the standard NLO material urea (0.13 × 10−30 esu) [62].
Thermodynamic functions
On the basis of vibrational calculation at the B3LYP/6-311++G(2d,p) level of theory, the standard statistical thermodynamic functions heat capacity (\(C_{p,m}^{0}\)), entropy (\(S_{m}^{0}\)), enthalpy (\(H_{m}^{0}\)), and molecular energy (E) for MTISO were obtained from theoretically calculated frequencies; the results are listed in Table 9.
It is apparent these thermodynamic functions increase with temperature in the range 100–800 K; this is because molecular vibrational intensities increase with temperature [63]. The corresponding quadratic equations obtained by use of B3LYP/6-311++G(d,p) are:
These equations can be used to compute other thermodynamic quantities, because of the relationships among thermodynamic functions, and to estimate the directions of chemical reactions by use of the second law of thermodynamics. All thermodynamic calculations were performed for the gas phase and cannot be used for solutions.
The related figure is shown in Fig. 14.

Correlation graph of thermodynamic properties and temperatures for the MTISO molecule, obtained by use of B3LYP/6-311++G(2d,p)
Reactive descriptors of MTISO
Global reactivity
The energies of the frontier molecular orbitals (E HOMO, E LUMO), energy band gap (E HOMO − E LUMO), electronic chemical potential (µ), electronegativity (χ), global hardness (η), global softness (S), and electrophilicity index (ω) [64] of MTISO are listed in Table 10.
On the basis of E HOMO and E LUMO, these were calculated by use of the equations:
The electrophilicity index has recently been used as a descriptor of biological activity. A quantitative structure–activity relationship (QSAR) study performed by multiple linear regression found that the HOMO and LUMO energies are the most important descriptors for describing the drug–receptor interactions of molecules [65–67]. The usefulness of this new index for understanding the toxicity of pollutants in terms of their reactivity and site selectivity has recently been demonstrated [65]. The computed electrophilicity index of MTISO describes the biological activity of drug–receptor interaction.
Local reactivity descriptors
The most relevant local descriptor of reactivity is the Fukui function, the derivative of the electronic chemical potential with respect to external potential as a result of the compensating nuclear charges in the system. For a system of N electrons, independent calculations are made for corresponding N + 1, N − 1, and N total electrons present in the anion, cation, and neutral state of molecule. Mulliken population analysis yields gross charges \(q_{k} \left( {N + 1} \right),q_{k} \left( {N - 1} \right),q_{k} \left( N \right)\) for all atoms k. In a finite-difference approximation, the condensed Fukui functions are given by the equations:
The condensed-to-atom quantity, \(\omega_{k}^{\alpha }\) corresponding to local electrophilicity index, ω(r), was obtained as described previously [67].
where α = +, −, and 0 refer to nucleophilic, electrophilic, and free radical reactions, respectively. Fukui functions, local softness, and local electrophilicity indices for selected atomic sites in MTISO have been listed in Table 11.
The order of nucleophilic reactivity for MTISO was C16 > C12 by use of the 6-311++G(2d,p) and 6-311++G(d,p) basis sets. Major nucleophilic reactivity was observed for this kind of attack in comparison with electrophilic attack. In contrast, the order of electrophilic reactivity for MTISO was N15 > O13 > C11 > S5 > C1 > C4 > H6 > H7 > O14 > H10 > C9 > H20 > C3 > H18 > H19 > C2 > H8 > C17 and N15 > O13 > S5 > C1 > C4 > C11 > H6 > C2 > O14 > H10 > C9 > H7 > H20 > H18 > H19 > H8 > C17 > C3 by use of the 6-311 ++G(d,p) and 6-311 ++G(2d,p) basis sets, respectively. If one compares the three kinds of attack it is possible to observe that electrophilic attack is more reactive than free radical and nucleophilic attack.
The advantage of this methodology over that reported previously is apparent from Table 12. According to Table 12, this catalyst is superior to some previous catalysts in terms of efficiency (entries 1–7, 9, and 10), reaction times (1–3 and 9), and amount of catalyst (entries 1, 4–8, 10, and 12). In addition, there is no need for bases such as pyridine, heating, or special devices such as ultrasound.

Conclusions
We have described a convenient method for synthesis of α,β-unsaturated isoxazol-5(4H)-ones (4a–t) by reaction of aryl aldehydes (1a–n), hydroxylamine hydrochloride (2), and two active methylene compounds (3a–b) in the presence of NBS. The attractive features of this procedure are: simplicity, relatively short reaction times, mild reaction media, environmental friendly character, and good to high yields. FT-IR spectral analysis shows that the predicted vibrational frequencies are in good agreement with the experimental values. NBO analysis confirms ICT (π*(C–O), π*(C–N), and π*(C–C)) is occurring as a result of orbital overlap between n(O)and n(S). TD-DFT calculations result in very close agreement with experimental absorption spectra both in the gas phase and in solvent. Molecular orbital coefficient analysis reveals that electronic transitions are mainly π → π*. The MEP map shows that negative potential sites are on the electronegative atoms whereas positive potential sites occur around the hydrogen atoms. These sites give information about regions of the compound which can participate in noncovalent interactions. NLO properties predicted for MTISO are much greater than those of urea. The compound is a good candidate as second-order NLO material. This article should be helpful in the design and synthesis of new materials.
References
J. Han, H. Guo, X.G. Wang, M.L. Pang, J.B. Meng, Chin. J. Chem. 25, 129 (2007)
M. Pineiro, T.M.V.D. Pinho-e-Melo, Eur. J. Org. Chem. 31, 5287 (2009)
S. Biju, M.L. Reddy, R.O. Freire, Inorg. Chem. Commun. 10, 393 (2007)
E. Aret, H. Meekes, E. Vlieg, G. Deroover, Dyes Pigment. 72, 339 (2007)
X.H. Zhang, Y.H. Zhan, D. Chen, F. Wang, L.Y. Wang, Dyes Pigment. 93, 1408 (2012)
B. Kafle, N.G. Aher, D. Khadka, H. Park, H. Cho, Chem. Asian J. 6, 2073 (2011)
H. Kano, I. Adachi, R. Kido, K. Hirose, J. Med. Chem. 10, 411 (1967)
T. Karabasanagouda, A.V. Adhikari, M. Girisha, Indian J. Chem. 48B, 430 (2009)
A. Mor, S. Ahn, P. Izmirly, S. Reddy, J. Greenberg, C.O. Bingham, P.B. Rosenthal, Indian Phytopathol. 59, 370 (2006)
J. Getal, J. Antibiot. Antibiot. 28, 91 (1975)
K. Bowden, G. Crank, W.J. Ross, J. Chem. Soc. C. 172 (1968). doi:10.1039/J39680000172
C.H. Stammer, A.N. Wilson, C.F. Spencer, F.W. Bachelor, F.W. Holly, K. Folkers, J. Am. Chem. Soc. 79, 3236 (1957)
Q. Liu, Y.N. Zhang, Bull. Korean Chem. Soc. 32, 3559 (2011)
Q. Liu, X. Hou, Phosphorus, Sulfur Silicon. Relat. Elem. 187, 448 (2012)
Q. Liu, R.T. Wu, J. Chem. Res. 35, 598 (2011)
M. Mirzadeh, G.H. Mahdavinia, Eur. J. Chem. 9, 425 (2012)
S. Fozooni, N. Gholam Hosseinzadeh, H. Hamidian, M.R. Akhgar, J. Braz. Chem. Soc. 24, 1649 (2013)
F. Saikh, J. Das, S. Ghosh, Terahedron Lett. 54, 4679 (2013)
K. Ablajan, H. Xiamuxi, Synth. Commun. 42, 1128 (2012)
Q.F. Cheng, X.Y. Liu, Q.F. Wang, L.S. Liu, W.J. Liu, Q. Lin, X.J. Yang, Chin. J. Org. Chem. 29, 1267 (2009)
K. Ablajan, H. Xiamuxi, Chin. Chem. Lett. 22, 151 (2011)
Y.Q. Zhang, J.J. Ma, C. Wang, J.C. Li, D.N. Zhang, X.H. Zang, J. Li, Chin. J. Org. Chem. 28, 141 (2008)
Y.Q. Zhang, C. Wang, M.Y. Zhang, P.L. Cui, Y.M. Li, X. Zhou, J.C. Li, Chin. J. Org. Chem. 28, 914 (2008)
H. Kiyani, Org. Chem. Indian J. 13, 97 (2013)
H. Kiyani, F. Ghorbani, Heterocycl. Lett. 3, 145 (2013)
H. Kiyani, F. Ghorbani, Heterocycl. Lett. 3, 359 (2013)
H. Kiyani, F. Ghorbani, Open. J. Org. Chem. 1, 5 (2013)
H. Kiyani, F. Ghorbani, Elixir Org. Chem. 58A, 14948 (2013)
H. Kiyani, F. Ghorbani, Res. Chem. Intermed. (2013). doi:10.1007/s11164-013-1411-x
H. Kiyani, F. Ghorbani, J. Saudi Chem. Soc. (2013). doi:10.1016/j.jscs.2013.11.002
M.J. Frisch et al., Gaussian 03, Revision C.01 (Gaussian, Inc., Wallingford, 2004), p. 255
A.D. Becke, J. Chem. Phys. 98, 5648 (1993)
C. Lee, W. Yang, R.G. Parr, Phys. Rev. B 37, 785 (1988)
B. Miehlich, A. Savin, H. Stoll, H. Preuss, Chem. Phys. Lett. 157, 200 (1989)
R. Dennington, T. Keith, Millam Gaussview Version 5. Semichem Inc., Shawnee Mission KS (2009)
A.P. Scott, L. Radom, J. Phys. Chem. 100, 16502 (1996)
M. Karabacak, M. Cinar, M. Kurt, Spectrochim. Acta 74A, 1197 (2009)
E.D. Glendening, A.E. Reed, J.E. Carpenter, F. Weinhold, NBO Version 3.1 (Gaussian Inc., Pittsburgh, 2003)
C. Gündoğdu, S. Alp, Y. Ergün, B. Tercan, T. Hökelek, Acta Cryst. E67, 1321 (2011)
Q. Cheng, X.Y. Xu, L.S. Liu, L. Zhang, Acta Cryst. E65, 3012 (2009)
J. Mohan, Organic Spectroscopy—Principle and Applications, 2nd edn. (Narosa Publishing House, New Delhi, 2000)
G. Brancatelli, G. Bruno, F. Nicolò, M. Cordaro, G. Grassi, F. Risitano, A. Scala, J. Mol. Struct. 998, 157 (2011)
R.M. Silverstein, G.C. Basseler, C. Morill (eds.), Spectroscopic Identification of Organic compounds (Wiley, New York, 1981)
T. Kupka, R. Wrzalik, G. Pasterna, K. Pasterny, J. Mol. Struct. 616, 17 (2002)
D.K. Singh, S.K. Srivastava, A.K. Ojha, B.P. Asthana, J. Mol. Struct. 892, 384 (2008)
A. Coruh, F. Yilmaz, B. Sengez, M. Kurt, M. Cinar, M. Karabacak, Struct. Chem. 22, 45 (2011)
M. Szafran, A. Komasa, E.B. Adamska, J. Mol. Struct. (Theochem) 827, 101 (2007)
N. Günay, H. Pir, D. Avcı, Y. Atalay, J. Chem. 2013, 1 (2012)
J. Choo, S. Kim, H. Joo, Y. Kwon, J. Mol. Struct. (Theochem) 587, 1 (2002)
R.S. Mulliken, J. Chem. Phys. 23, 1833 (1995)
R. Meenakshi, L. Jaganathana, S. Gunasekaranb, S. Srinivasan, Mol. Simul. 36, 425 (2010)
H.O. Kalinowski, S. Berger, S. Braun, Carbon-13 NMR Spectroscopy (Wiley, Chichester, 1988)
N. Subramania, N. Sundaraganesan, J. Jayabharathi, Spectrochim. Acta A 76, 259 (2010)
E. Scrocco, J. Tomasi, Adv. Quant. Chem. 11, 115 (1978)
N.M. O’Boyle, A.L. Tenderholt, K.M. Langner, J. Comp. Chem. 29, 839 (2008)
K. Fukui, Science 218, 747 (1982)
R.E. Stratmann, G.E. Scuseria, M.J. Frisch, J. Chem. Phys. 109, 8218 (1998)
B. Mennucci, J. Tomasi, J. Chem. Phys. 106, 5151 (1997)
A. Airinei, M. Homocianu, D. Dorohoi, J. Mol. Liq. 157, 13 (2010)
D.A. Kleinman, Phys. Rev. 126, 1977 (1962)
A. Chandran, S. Mary, H.T. Varghese, C.Y. Panicker, T.K. Manojkumar, C.V. Alsenoy, G. Rajendran, ISRN Anal. Chemistry. 2012, 1 (2011)
C. Adant, M. Dupuis, L. Bredas, Int. J. Quantum Chem. 56, 497 (2004)
M.A. Palafox, Int. J. Quantum Chem. 77, 661 (2000)
J. Padmanabhan, R. Parthasarathi, V. Subramanian, P.K. Chattaraj, J. Phys. Chem. A 111, 1358 (2007)
A.P. Garrido, A.M. Helguera, A.A. Guillén, M.N.D.S. Cordeirom, A.G. Escudero, Bioorg. Med. Chem. 17, 896 (2009)
R. Parthasarathi, J. Padmanabhan, V. Subramanian, U. Sarkar, B. Maiti, P.K. Chattraj, Internet Electron. J. Mol. Des. 2, 798 (2003)
R.G. Parr, W. Yang, J. Am. Chem. Soc. 106, 4049 (1984)
Acknowledgments
Financial support by Damghan University is acknowledged.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article

Cite this article
Kiyani, H., Kanaani, A., Ajloo, D. et al. N-bromosuccinimide (NBS)-promoted, three-component synthesis of α,β-unsaturated isoxazol-5(4H)-ones, and spectroscopic investigation and computational study of 3-methyl-4-(thiophen-2-ylmethylene)isoxazol-5(4H)-one. Res Chem Intermed 41, 7739–7773 (2015). https://doi.org/10.1007/s11164-014-1857-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-014-1857-5