
Abstract
Petra/Osiris/Molinspiration analysis (POM) is a promising new bioinformatical approach to establish structure and activity correlations. In the present study, we have reported the POM analyses of Raltegravir analogues that have aimed to figure out the structural features of HIV-integrase inhibitory activity. The resulting model exhibited two controllable bidentate O, O-pockets taken into consideration contributions from the steric and electrostatic fields. The POM analysis has provided interesting insights into the understanding the steric and electronic structural requirements for HIV-IN inhibitory activity. Furthermore, all the molecules were subjected to the toxicity assessment using Molinspiration and Osiris calculations. Among the various HIV-IN inhibitors, compound 27 (Raltegravir) displayed optimum drug-like characteristic activity with low toxicity. The mechanism of HIV-integrase inhibition by different Raltegravir derivatives is also discussed. This study also concluded that the bioactivity of DKA analogues should be discussed on the basis of catalytic activity of bimetallic complexes, not just on the basis of DKA or Raltegravir/HIV-integrase interaction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
It is estimated that approximately 39–40 million people are living with HIV/AIDS worldwide, with infection and death rates of around 3–4 million per year [1]. There are 20–22 FDA-approved experimental drugs for the treatment of HIV infection (Fig. 1) which are either used alone or in combination with other drugs. Multidrug cocktails consisting of a protease inhibitor or a non-nucleoside reverse transcriptase inhibitor in combination with two nucleoside reverse transcriptase inhibitors is the current standard for HIV therapy (HAART). While HAART is undeniably effective, it can fail to control HIV replication in patients due to several limitations, such as side effects and development of resistance, including multidrug resistance and cross-resistance [2].

Structural insight of clinical HIV-1 integrase inhibitors (pink indicates the pharmacophore sites) and Raltegravir derivatives 1–28
Therefore, it is not only essential to continue to develop new antiretroviral drugs with potency against a broad range of viral mutants including those which cause resistance to multidrug classes, but it should be understood not only how they work but also to determine the way to control their bioactivity on the basis of physico-chemical and structural parameters.
Summa et al. [2] successfully reported the discovery of Raltegravir (MK-0518), the first HIV-integrase inhibitor approved by FDA for the treatment of HIV infection. The pharmacological profile of Raltegravir has enabled its progression toward the end of phase III clinical trials for the treatment of HIV-1 infection, and culminated with the FDA approval as the first HIV-integrase inhibitor for the treatment of HIV-1 infection. However, the catalytic analysis of the mechanism of HIV-integrase inhibition has not been determined.
In order to get more efficient HIV-integrase inhibitors, Merck Research Laboratories (MRL) chemically modified 5, 6-dihydroxypyrimidine-4-carboxamides and N-methyl-4-hydroxypyrimidinone-carboxamides, which resulted in the formation of an improved product, Raltegravir (MK-0518).
It is interesting to note here that extensive structure–activity relationship studies on the carboxamide moiety led MRL to the identification of the p-fluorobenzyl as the optimal amide residue and of the gem-dimethyl as the optimal 2-substituent for the dihydroxy pyrimidine core. Parallel efforts led to the identification of the related N-methylpyrimidone scaffold showing equal or enhanced activity for the HIV-integrase [2]. However, MK-0518 in clinical practice is being slowly abandoned due to adverse effects, such as diarrhea, dizziness, headache, nausea, tiredness, trouble sleeping, and weakness [3].
Both positive and negative aspects of Raltegravir (MK-0518) encouraged us to execute a bioinformatic search for Raltegravir analogues (Fig. 2), and our effort is considerably supported by Petra/Osiris/Molinspiration (POM) analyses.

The structure of Raltegravir (27) and analogues as HIV-integrase inhibitors
Results and discussion
POM molecular virtual screening
The structures of the Raltegravir derivatives and the biological activities data were obtained from Summa et al. [2]. Three-dimensional structure building and all modeling were performed using the ACD free program package on a personal computer equipped with a Pentium IV processor, which allows one to draw chemical structures including organics, organometallics, polymers, and Markush structures. It also includes features such as the calculation of molecular properties. Toxicity risks, physic-chemical evaluation and pharmaco-molecular properties calculations were done with Molinspiration and Osiris freeware programs.
Petra/Osiris/Molinspiration analysis (POM) is one of the well-known approaches that has been used regularly to produce two-dimensional models to identify and to indicate the type of pharmacophore site that affects biological activity with a change in the chemical substitution [4–22]. The advantages of POM are the ability to predict the biological activities of the molecules and to represent the relationships between steric/electrostatic properties as well as biological activity in the form of a pharmacophore site, which gives key features on not only the ligand–receptor interaction but also on the topology of the receptor [4–22]. The Raltegravir analogues are a family of natural and/or synthetic compounds with different pharmacological activities, one of which is HIV-integrase (IN) inhibitory activity. [2]. Hence, to find out the structural features for the HIV-IN inhibitory activity, we have carried out POM analysis of Raltegravir analogues.
POM analysis dataset
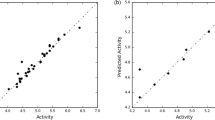
Structures and associated antiviral activities of compounds 1–28 are given in Fig. 1 and Table 1, respectively. QSAR models were randomly derived from a training set of 28 molecules. The most active compound, 27, was used as a template molecule for comparison.
The docking analyses and X-ray diffraction have already determined the method of interaction between Raltegravir analogues and HIV-1 integrase [23–26]. However, it is extremely important to understand why and how it works.
Osiris calculations [27]
Structure-based design is now fairly routine, but many potential drugs fail to reach the clinic because of ADMET liabilities (absorption, distribution, metabolism, excretion, and toxicity in pharmacokinetics). One very important class of enzymes, responsible for many ADMET problems, is the cytochromes P450. Inhibition of these or production of unwanted metabolites can result in many adverse drug reactions. To assess the possible toxicity risks associated with Raltegravir analogues, Osiris, a freely available online program, was used [28]. Toxicity risks (mutagenicity, tumorigenicity, irritation, reproduction) and physico-chemical properties (m i Log P, solubility, drug-likeness, and drug-score) of compounds 1–29 were calculated by the methodology developed by Osiris. Toxicity risk alerts are an indication that the created structure may be harmful concerning the risk category specified.
The data evaluated in Table 1 indicate that all structures are supposed to be non-mutagenic, non-irritating, and with no reproductive effects when run through the mutagenicity assessment system comparable with Raltegravir, except cfor ompound 9.
The log P value of a compound, which is the logarithm of its partition coefficient between n-octanol and water, is a well-established measure of the compound’s hydrophilicity. Low hydrophilicities and therefore high log P values may cause poor absorption or permeation. It has been shown that, for compounds to have a reasonable probability of being well absorbed, their log P value must not be greater than 5.0. On this basis, all the compounds are having log P values under the acceptable criteria.
The aqueous solubility of a compound significantly affects its absorption and distribution characteristics. Typically, a low solubility goes along with a bad absorption and therefore the general aim is to avoid poorly soluble compounds. Our estimated log S value is a unit stripped logarithm (base 10) of a compound’s solubility measured in mol/l. More than 80 % of the drugs on the market have an (estimated) log S value greater than −4. In the case of compounds 1–28, values of log S are under acceptable criteria. Furthermore, Table 1 shows the drug likenesses of compounds 1–28 which are in a comparable zone with that of standard drugs. We have calculated overall drug score (DS) for the compounds 1–28. The drug score combines drug likeness, m i Log P, log S, molecular weight, and toxicity risks in one handy value may then be used to judge the compound’s overall potential to qualify for a drug. This value is calculated by multiplying contributions of the individual properties with Eq. 1 [27]:
where S = 1/1 + e ap+b, DS is the drug score, S i is the contribution calculated directly from of m i Log P, log S, molecular weight, and drug likeness (pi) via the second equation, which describes a spline curve. Parameters a and b are (1, −5), (1, 5), (0.012, −6) and (1, 0) for m i Log P, log S, molecular weight, and drug likeness, respectively. t i is the contribution taken from the four toxicity risk types. The t i values are 1.0, 0.8, and 0.6 for no risk, medium risk, and high risk, respectively. The reported compounds 1–28 showed moderate to good drug scores as compare to Raltegravir.
Molinspiration calculations
CLog P (octanol/water partition coefficient) is calculated by the methodology developed by Molinspiration as a sum of fragment-based contributions and correction factors (Table 2). The method is very robust and is able to process practically all organic and most organometallic molecules. The topological polar surface area (TPSA) is calculated based on the methodology published by Ertl et al. [29] as a sum of fragment contributions. O- and N-centered polar fragments are considered. TPSA has been shown to be a very good descriptor characterizing drug absorption, including intestinal absorption, bioavailability, Caco-2 permeability, and blood–brain barrier penetration. Prediction results (TPSA, GPCR ligand, and ICM) of the compounds 1–28 have been evaluated (Table 2).
TPSA values are important properties for the prediction of per oral bioavailability of drug molecules. The TPSA is calculated from the surface areas that are occupied by oxygen and nitrogen atoms and by hydrogen atoms attached to them. Thus, the TPSA is closely related to the hydrogen bonding potential of a compound. Molecules with TPSA values of 140 Å or more are expected to exhibit poor intestinal absorption. Table 2 shows that all the compounds are within this limit. Note that all except a few of the compounds have only one violation of the Rule of 5. Two or more violations of the Rule of 5 suggest the probability of problems in bioavailability. All the compounds have only one violation of the Rule of 5 with the majority of the compounds having zero violations. The drug likenesses of compounds 1–28 are tabulated in Table 2. Drug likeness may be defined as a complex balance of various molecular properties and structural features which determine whether a particular molecule is similar to the known drugs. These properties, mainly hydrophobicity, electronic distribution, hydrogen bonding characteristics, molecule size and flexibility, and the presence of various pharmacophores features influence the behavior of the molecule in a living organism, including bioavailability, transport properties, affinity to proteins, reactivity, toxicity, metabolic stability, and many others. Activity of all nine compounds and standard drugs were rigorously analyzed under four criteria of known successful drug activity in the areas of GPCR ligand activity, ion channel modulation, kinase inhibition activity, and nuclear receptor ligand activity. The results are shown for all compounds in Table 2 by means of numerical assignment. Likewise, all compounds have consistent negative values in all categories and numerical values conforming and comparable to that of the standard drug used for comparison. Therefore, it is readily seen that all the compounds are expected to have near similar activity to the standard drug used based upon these four rigorous criteria (GPCR ligand, ion channel modulator, kinase inhibitor, and nuclear receptor ligand) (Fig. 3).

Plausible mechanism of 1–28 drugs/HIV-IN interaction
The new docking analyses information of Raltegravir derivatives
A literature survey [30] revealed that Raltegravir interacts with HIV-1 integrase due to H-bonding and mild polar and hydrophobic interactions (Fig. 4) but did not explain the consequence and the real chemical mechanism of this Raltegravir/HIV-integrase complication. Here, we have obtained some insight into this phenomenon, which remained unknown for a long period of time: whether Raltegravir interacts with the catalytic site of HIV-integrase and as a consequence leads to the inhibition of vital processes of viral reproduction or results in a bimetallic [(Raltegravir)Mg-integrase] complex that continues to be active as a new biological catalyst.

Docking of Raltegravir in catalytic domain of HIV-1 integrase [30]
To decipher these questions, we need first to correct an important point: the Mg2+ is always hexaccordinated [31–40]. Of course, Mg2+ can sometimes be tetra- or penta-coordinated [41, 42], but these unsaturated structures are realisable just with a steric effect of ligands and in just a few limited cases (Fig. 5).

Examples of tetra-, penta- and hexa-coordinated magnesium(II) complexes
A thorough analysis of the two Mg2+ cations showed that there is not only D64, D116, and E152 and the three oxygen atoms of O,O,O-ligand (Raltegravir), which are involved in the two coordination spheres of the two Mg2+ but there are also two other aquo (water) ligands. The presence of these two coordinated water molecules in a bimetallic moiety is not innocent at all. We have previously indicated the catalytic redox behavior of [(polypyridyl)-ruthenium-(X)2] complexes and their potential as therapeutical agents [43]. We have attributed the anti-tubercular activity of ruthenium(II) complexes to the “Ru-(H2O)2” moiety. Our conclusion was based on the general catalytic redox of this type of complexes. On the other hand, we noted that, if it works for active monometallic complexes, this redox reaction is faster in the case of a bimetallic system because there is a synergic effect between the two metal atoms. Many biologists continue to look on the metal–aquo moiety as an inert moiety, which we think is incorrect; in fact, it is a crucial part of the puzzle.
The Ullmann C–C coupling is the classical example of Cu-catalyzed biaryl coupling; the dicopper complex C-01 was shown to couple 2,4-di-tert-butylphenol to the corresponding bisphenol derivative (Scheme 1), while, with 2,6-substituted phenols, oxidative coupling at the para-position was observed to yield the diphenoquinone derivatives. Similar reactivity is observed for various other dicopper systems [44].

Realisation of classical Ullmann coupling reaction by using a copper(I) bimetallic system [44]
Nature itself uses dinuclear metal species at the active sites of metalloenzymes to activate and utilize small molecules under ambient conditions, taking advantage of the cooperativities between metal centers. For example, a carboxylate-bridged non-heme diiron active site is responsible for O2 utilization in a subfamily of non-heme enzymes [45].
A dinuclear Ni/Ru aqua complex bridged by a diamine ligand (Scheme 2) was synthesized and found to be able to activate and heterolytically cleave molecular H2 in water under ambient conditions [46]. This complex is a model complex mimicking a hydrogenase function.

Molecular H2 activation by a mixture Ru/Ni bimetallic-aqua complex
In another case, a dinuclear copper(II) complex supported by a dithio-containing amine ligand (Scheme 3) could activate CO2 and catalyze CO2 reduction electrochemically to oxalate [47].

Molecular CO2 activation by a bimetallic copper–solvato complex
Dioxygen activation has also been achieved using a pre-oriented di-iron complex (Scheme 4), which was supported by a multidentate binucleating ligand [48]. Although they have been studied less than monometallic systems, these bimetallic systems showed potential to more precisely mimic nature and deliberately utilize the cooperative effect between multiple metal centers.

Molecular O2 activation by a bimetallic iron–carboxylato complex
Cooperation between multiple metal centers played a key role in the metal–small-molecule interactions even though mononuclear metal complexes are used in these systems. It would then be anticipated that two metals could be reorganized and brought into close proximity initially just as those in nature (Fig. 3; Scheme 5).

Postulated mechanism for oxidative damage of the HIV virus
Conclusion
A series of critical notes on catalytic processes of bimetallic complexes, focussing on specific antiviral activity of diketo acid derivatives (DKA) interfacing with small molecules activation, are provided. For this field, the critical reviews cover topics such as the activation of “inert” oxygen–hydrogen bonds of aquo ligand, bimetallic complexes design, and organometallic M–O–M species. This is a good example, which shows how bimetallic compounds may perform the highly selective activation of O–H bonds of aquo ligand and, in particular, how synergic relationships between various metals are crucial to this therapeutic approach. It was found that there exists very good information concerning the crucial role of metal–OH2, metal–OH, and metal=O as intermediaries in catalytic oxygen transfer to organic molecules (redox catalysis). This is supported by the extensive research documented in the literature [49–53].
This is in perfect agreement with our hypothetical mechanism (Fig. 3, Scheme 5), concerning the catalytic continuity of bimetallic drug/integrase complexes of Mg2+. It means that Raltegravir proceeds with interaction with an HIV-integrase enzyme via coordination to its two magnesium atoms but does not stop at this step. The complex continues to work as a catalyst to activate water molecules and destroy the genetic material of the HIV virus. It is as if HIV kills itself. Thus, it is concluded that the bioactivity of DKA analogues should be discussed on the basis of the catalytic activity of bimetallic complexes, not just on the basis of DKA or Raltegravir/HIV-integrase interaction. The focus should be on the catalytic redox properties of their different transition metal complexes and also how they may be applied in various industrial catalytic applications.
References
UNAIDS, AIDS epidemic update: December 2000 (UNAIDS/WHO, Geneva, 2000)
V. Summa, A. Petrocchi, F. Bonelli, B. Crescenzi, M. Donghi, M. Ferrara, F. Fiore, C. Gardelli, O.G. Paz, D.J. Hazuda, P. Jones, O. Kinzel, R. Laufer, E. Monteagudo, E. Muraglia, E. Nizi, F. Orvieto, P. Pace, G. Pescatore, R. Scarpelli, K. Stillmock, M.V. Witmer, M. Rowley, Discovery of Raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV–AIDS infection. J. Med. Chem. 51, 5843 (2008)
Side effects of Raltegravir. http://www.drugs.com/sfx/raltegravir-side-effects.html
B. Ozcelik, J. Sheikh, I. Orhan, H. Juneja, B. Bennani, S.M.T. Cavalcanti, A.C.L. Leite, T. Ben Hadda, Outstanding effect of the conformational restriction of isoquinolines: hints for the development of optimized antimicrobial agents. Res. Chem. Intermed. 39, 2955 (2012). doi:10.1007/s11164-012-0808-2
V.H. Masand, D.T. Mahajan, K.N. Patil, T. Ben Hadda, V. Rastija, Integrating GUSAR and QSAR analyses for anti-malarial activity of synthetic prodiginines against multi drug resistant strain. Med. Chem. Res. 22, 2284 (2012). doi:10.1007/s00044-012-0223-7
T. Ben Hadda, R. Mouhoub, R. Jawarkar, V. Masand, I. Warad, POM analyses of antitrypanosomal activity of 2-iminobenzimidazoles : favorable and unfavorable parameters for drugs optimization. Med. Chem. Res. 22, 2284 (2013)
T. Ben Hadda, M.A. Ali, V. Masand, S. Gharby, T. Fergoug, I. Warad, Tautomeric origin of dual effects of N1-nicotinoyl-3-(4′-hydroxy-3′-methyl phenyl)-5-[(sub)phenyl]-2-pyrazolines on bacterial and viral strains: POM analyses as new efficient bioinformatics’ platform to predict and optimize bioactivity of drugs. Med. Chem. Res. 22, 1438 (2013)
A.M. Alafeefy, S.I. Alqasoumi, A.E. Ashour, V. Masand, N.A. Al-Jaber, T. Ben Hadda, M.A. Mohamed, Quinazoline–tyrphostin as a new class of antitumor agents, molecular properties prediction, synthesis and biological testing. Eur. J. Med. Chem. 53, 133 (2012)
T. Ben Hadda, J. Fathi, I. Chafchaouni, V. Masand, Z.H. Chohan, R. Jawarkar, T. Fergoug, Computational POM and 3D-QSAR evaluation of experimental in vitro HIV-1 integrase inhibition of amide-containing di-ketoacids. Med. Chem. Res. 22, 1456 (2013)
D.T. Mahajan, V.H. Masand, K.N. Patil, T.B. Hadda, R.D. Jawarkar, S.D. Thakur, Vesna Rastija, CoMSIA and POM analyses of anti-malarial activity of synthetic prodiginines. Bioorg. Med. Chem. Lett. 22, 4827 (2012)
T. Ben Hadda, A. Kerbal, B. Bennani, G. Al Houari, M. Daoudi, A.C.L. Leite, V.H. Masand, R.D. Jawarkar, Z. Charrouf, Molecular drug design, synthesis and pharmacophore site identification of spiroheterocyclic compounds: Trypanosoma crusi inhibiting studies (Chem. Res, Med, 2012). doi:10.1007/s00044-012-0010-5
V.H. Masand, D.T. Mahajan, K.N. Patil, K.D. Chinchkhede, R.D. Jawarkar, T. Ben Hadda, A.A. Alafeefy, I.G. Shibi, k-NN, quantum mechanical and field similarity based analysis of xanthone derivatives as a-glucosidase inhibitors (DOI, Med Chem Res, 2012). doi:10.1007/s00044-012-9995-z
T.B. Hadda, T. Fergoug, I. Warad, POM theoretical calculations and experimental verification of antibacterial potential of 5-hydroxy-4-(substituted-amino)-2(5H)-furanones. Res. Chem. Intermed. (2012). doi:10.1007/s11164-012-0729-0
J. Sheikh, T. Hadda, Antibacterial, antifungal and antioxidant activity of some new water-soluble b-diketones. Med. Chem. Res. (2012). doi:10.1007/s00044-012-0089-8
A. Jarrahpour, J. Fathi, M. Mimouni, T. Ben Hadda, J. Sheikh, Z.H. Chohan, Petra, Osiris and Molinspiration (POM) together as a successful support in drug design: antibacterial activity and biopharmaceutical characterization of some azo schiff bases. Med. Chem. Res. 19(7), 1 (2011)
A. Rauf, F. Ahmed, A.M. Qureshi, Aziz-ur-Rehman, A. Khan, M.I. Qadir, M.I. Choudhary, Z.H. Chohan, M.H. Youssoufi, T. Ben Hadda, Synthesis and urease inhibition studies of barbituric and thiobarbituric acid derived sulphonamides. J. Chin. Chem. Soc. 58(4), 1 (2011)
J. Sheikh, A. Parvez, V. Ingle, H. Juneja, R. Dongre, Z.H. Chohan, M.H. Youssoufi, T. Ben Hadda, Synthesis, biopharmaceutical characterization, antimicrobial and antioxidant activities of 1-(4′-O-β-d-glucopyranosyloxy-2′-hydroxyphenyl)-3-aryl-propane-1,3-diones. Eur. J. Med. Chem. 46, 1390 (2011)
A. Parvez, M. Jyotsna, M.H. Youssoufi, T. Ben Hadda, Theoretical calculations and experimental verification of the antibacterial potential of some monocyclic beta-lactames containing two synergetic buried antibacterial pharmacophore sites. Phosphorus, Sulfur, Silicon Relat Elem 7, 1500 (2010)
A. Parvez, J. Meshram, V. Tiwari, J. Sheikh, R. Dongre, M.H. Youssoufi, T.B. Hadda, Pharmacophores modeling in terms of prediction of theoretical physicochemical properties and verification by experimental correlations of novel coumarin derivatives produced via Betti’s protocol. Eur. J. Med. Chem. 45(9), 4370 (2010)
Z.H. Chohan, M.H. Youssoufi, A. Jarrahpour, T.B. Hadda, Identification of antibacterial and antifungal pharmacophore sites for potent bacteria and fungi inhibition: indolenyl sulphonamide derivatives. Eur. J. Med. Chem. 45(3), 1189 (2010)
A. Jarrahpour, M. Motamedifar, M. Zarei, M.H. Youssoufi, M. Mimouni, Z.H. Chohan, T.B. Hadda, Petra, Osiris and Molinspiration together as a guide in drug design: predictions and correlation structure/antibacterial activity relationships of new N-sulfonyl monocyclic β-lactams (Part II). Phosphorus, Sulfur, Silicon Relat Elem 185, 491 (2010)
T. Ben Hadda, M.A. Ali, V. Masand, S. Gharby, T. Fergoug, I. Warad, Tautomeric origin of dual effects of N1-nicotinoyl-3-(4′-hydroxy-3′-methyl phenyl)-5-[(sub)phenyl]-2-pyrazolines on bacterial and viral strains: POM analyses as new efficient bioinformatics’ platform to predict and optimize bioactivity of drugs. Med. Chem. Res. (2012). doi:10.1007/s00044-012-0143-6
J. Tang, K. Maddali, Y. Pommier, Y.Y. Sham, Zhengqiang Wanga, Scaffold rearrangement of dihydroxypyrimidine inhibitors of HIV integrase: docking model revisited. Bioorg. Med. Chem. Lett. 20, 3275 (2010)
K. Majerz-Maniecka, R. Musiol, A. Skórska-Stania, D. Tabak, P. Mazur, B.J. Oleksyn, Jaroslaw Polanski, X-ray and molecular modelling in fragment-based design of three small quinoline scaffolds for HIV integrase inhibitors. Bioorg. Med. Chem. 19, 1606 (2011)
H. Jin, R.Z. Cai, L. Schacherer, S. Jabri, M. Tsiang, M. Fardis, X. Chen, J.M. Chen, C.U. Kim, Design, synthesis, and SAR studies of novel and highly active tri-cyclic HIV integrase inhibitors. Bioorg. Med. Chem. Lett. 16, 3989 (2006)
S. Hare, A.M. Vos, R.F. Clayton, J.W. Thuring, M.D. Cummings, P. Cherepanov, Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc. Natl. Acad. Sci. USA 107(46), 20057 (2010)
A. Hokuldsson, PLS regression methods. J. Chemom. 2, 211 (1988)
P. Ertl, B. Rohde, P. Selzer, Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 43, 3714 (2000)
C. Wei, Z. Liu, D. Zhang, Docking of raltegravir to HIV-1 integrase structure ensemble. J. Theor. Comput. Chem. 9, 1053 (2010)
X.-S. Tai, J. Yin, M.-Y. Hao, Diaquabis(quinoline-2-carboxylato-κ2 N, O) magnesium(II) dihydrate methanol disolvate. Acta Cryst. E63, m1850 (2007)
X.-Y. Liu, L.-H. Liu, Diaqua-bis(5-carb-oxy-2-propyl-1H-imidazole-4-carboxyl-ato-κN,O)magnesium(II) 3.5-hydrate. Acta Crystallogr. Sect. E 66(Pt 3), m305 (2010)
M.A. Sharif, H. Aghabozorg, E. Motyeian, M. Ghadermazi, J. Attar Gharamaleki, Diaquabis(pyridine-2-carboxylato)magnesium(II) 0.15-hydrate. Acta Cryst. E63, m2235–m2236 (2007)
L. Zhu, J. Huang, S.-Y. Han, Z. An, mer-Triaqua(1,10-phenanthroline-κ2 N, N′)(sulfato-κO)magnesium(II). Acta Cryst. E64, m683 (2008)
D. Zhao, F.-F. Li, J. Sha, cis-Bis(1,10-phenanthroline-κ2 N, N′)bis(thiocyanato-κN)magnesium(II). Acta Cryst. E66, m988 (2010)
X.-M. Hao, C.-S. Gu, W.-D. Song, J.-W. Liu, Tetraaqua(1,10-phenanthroline-κ2 N, N′)magnesium(II) bis[(2,4-dichlorophenyl)acetate]. Acta Cryst. E64, m1052 (2008)
K.-M. Park, I. Yoon, J. Seo, Y.H. Lee, S.S. Lee, A magnesium(II) complex of 1,10-phenanthroline-2,9-dicarboxylate. Acta Cryst. E57, m154–m156 (2001)
W. Starosta, J. Leciejewicz, trans-Tetraaquabis(pyridazine-4-carboxylato-O) magnesium(II) dehydrate. Acta Cryst. E67, m316 (2011)
I. Mutikainen, R. Hämäläinen, M. Klinga, O. Orama, U. Turpeinen, Triclinic form of tetraaqua(orotato-N, O)magnesium(II) hydrate at 153 K. Acta Cryst. C52, 2480–2482 (1996). doi:10.1107/S0108270196006567
R. Grubba, W. Wojnowski, K. Baranowska, E. Baum, J. Pikies, Bis(dithiobenzoato-κ2S, S’)bis(tetrahydrofuran-κO)magnesium(II). Acta Cryst. E62, m2080–m2081 (2006)
B.-H. Huang, Y.-L. Peng, C.-C. Lin, Bis[(1Z,3Z)-1,3-bis(4-fluorophenyl)-N, N′-diphenylpropane diiminato] magnesium(II). Acta Cryst. E62, m1977–m1978 (2006). doi:10.1107/S1600536806027954
I.A. Guzei, R.W. McGaff, H.M. Kieler, Why magnesium is five-coordinate in methanol(phthalocyaninato)magnesium(II). Acta Cryst. C61, m472–m475 (2005). doi:10.1107/S0108270105029732
T. Ben Hadda, M. Akkurt, M.F. Baba, M. Daoudi, B. Bennani, A. Kerbal, Z.H. Chohan, Anti-tubercular activity of ruthenium (II) complexes with polypyridines. J. Enzyme Inhib. Med. Chem. 24(2), 457 (2009)
C.-J. Li, Organic reactions in aqueous media with a focus on carbon-carbon bond formations: a decade update. Chem. Rev. 105, 3095 (2005)
S. Friedle, E. Reisner, S.J. Lippard, Current challenges of modeling diiron enzyme active sites for dioxygen activation by biomimetic synthetic complexes. Chem. Soc. Rev. 8, 2768 (2010)
S. Ogo, R. Kabe, K. Uehara, B. Kure, T. Nishimura, S.C. Menon, R. Harada, S. Fukuzumi, Y. Higuchi, T. Ohhara, T. Tamada, R. Kuroki, A dinuclear Ni(mu-H)Ru complex derived from H2. Science 316(5824), 585 (2007)
R. Angamuthu, P. Byers, M. Lutz, A.L. Spek, E. Bouwman, Electrocatalytic CO2 conversion to oxalate by a copper complex. Science 327(5963), 313 (2010)
L.H. Do, T. Hayashi, P. Moenne-Loccoz, S.J. Lippard, Carboxylate as the protonation site in (peroxo)diiron(III) model complexes of soluble methane monooxygenase and related diiron proteins. J. Am. Chem. Soc. 4, 1273 (2010)
C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laurenczy, T.J. Geldbach, G. Sava, P.J. Dyson, In vitro and in vivo evaluation of ruthenium(II)-arene PTA complexes. J. Med. Chem. 48, 4161 (2005)
J.L. Dempsey, A.J. Esswein, D.R. Manke, J. Rosenthal, J.D. Soper, D.G. Nocera, Molecular chemistry of consequence to renewable energy. Inorg. Chem. 44, 6879 (2005)
C. Jin Qin, A. Gavrilova, B. Bosnich, Cooperative bimetallic oxidative addition reactions, Pure Appl. Chem. 73 (2), 221, (2001)
K.H. James, Water oxidation catalyzed by dimeric μ-oxo bridged ruthenium diimine complexes, Coord. Chem. Rev. 249, 313 (2005)
M.A. Ciriano, C. Freund, W.A. Herrmann, F.E. Kühn, C. Limberg, F. Meyer, B.V. Popp, D. Schröder, H. Schwarz, S.S. Stahl, T. Strassner, C. Tejel, K.H. Theopold, J.I. van der Vlugt, Topics in organometallic chemistry (Springer, Berlin, 2007). doi:10.1007/11603818. ISBN 978-3-540-37209-7
Acknowledgments
The authors would like to extend their sincere appreciation to the Deanship of Scientific at King Saud University for its funding of this research through the Research Group Project no RGP-VPP-222.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Lahsasni, S., Ben Hadda, T., Masand, V. et al. POM analyses of Raltegravir derivatives: a new reflection enlightening the mechanism of HIV-integrase inhibition. Res Chem Intermed 41, 5121–5136 (2015). https://doi.org/10.1007/s11164-014-1616-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-014-1616-7