
Abstract
Macroporous microspheres of SAPO-34 (MAMISAPO-34) were fabricated using polystyrene spheres (PS, 2 μm) as hard template. Cubic SAPO-34 (1 μm) synthesized by the conventional hydrothermal method was mixed with kaolin, silica sol, aluminium phosphate sol, template, and deionized water. Spray drying was then performed to prepare 30–50 μm microspheres which were converted to macroporous SAPO-34 by post-hydrothermal and calcination treatments. For comparison, 30–50 μm non-macroporous microspheres (NOMISAPO-34) were also obtained without using PS. XRD, NH3-TPD, and SEM analyses were used to characterize the macroporous and non-macroporous SAPO-34. The results showed that MAMISAPO-34 was more crystalline and had more strong acid sites than NOMISAPO-34. When used in MTO (methanol to olefins) reactions, MAMISAPO-34 had better catalytic performance than NOMISAPO-34, because of its greater crystallinity.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Much effort has been devoted to research on aluminophosphate (AlPO-n) and silicoaluminophosphate (SAPO-n) [1, 2] molecular sieves (where n denotes a particular structure type). These molecular sieves can be used in shape-selective catalysis and phase separation [3, 4]. SAPO-34 is well-known for its excellent performance in methanol to olefins (MTO) reactions [5–8]. However, the lifetime of conventional SAPO-34 is short, because of the easy formation of coke in the microporous system, which often occurs in gas–solid reactions [9, 10]. Mesopores and macropores with pore size above 2 nm can be used to provide good passageways to resolve diffusion limitation problems [11]. Nowadays, much research work has been performed on the synthesis of bimodal pore materials which can increase accessibility to the active sites in micropores [12, 13]. There are two ways of obtaining bimodal porous materials: single-template and double-template methods. The single-template method entails fabricating bimodal pore materials using the EISA (evaporation induced self-assembly) [14] or spontaneous assembly [15] methods. However, samples obtained from the single-template method have low stability because of the loose connections between small crystal particles [16]. The double-template method entails fabricating porous materials using hard templates [17] or soft templates [18] for macroporous or mesoporous structures. For bimodal porous materials, especially for macroporous–microporous composites, a hard template is better than a soft template because of the tunable property of hard templates. It has been reported that macroporous SAPO-34 can be prepared by using a dry gel conversion method. However, the macropores are actually an aggregated structure and could easily be changed by changing the conditions used for synthesis [19]. Polystyrene spheres (PS) as hard templates have attracted much attention [20, 21]. Stein et al. have reported the synthesis of macroporous silica-I zeolites using arrays of monodisperse polystyrene spheres as templates. However, the high hydrothermal temperature required for synthesis of the zeolites (373–473 K) severely obstructed the application of PS, because the glass transition temperature of PS is 353–373 K [22].
Herein we report preparation of bimodal SAPO-34 spheres combining macropores and micropores by use of a spray drying method with PS as hard template, then use of SAPO-34 spheres in MTO reactions. The catalytic performance in MTO reactions was studied and compared with that of non-macroporous SAPO-34 microspheres. To the best of our knowledge, this is the first report of use of PS as hard template for fabricating macroporous SAPO-34 spheres.
Experimental
Materials
All chemicals were analytical grade and used as purchased without further purification. The styrene (99%), triethylamine (TEA, 99%), silica sol, and orthophosphoric acid (H3PO4, 85%) were supplied by Sinopharm Chemical Reagent. The polyvinylpyrrolidone (AR) and azodiisobutyronitrile (AR) were purchased from Aldrich. The pseudo-boehmite, kaolin (a natural material), and aluminium phosphate sol were purchased from Wanlin Chemical and Suzhou Gaolin, and supplied by Shanghai Research Institute of Chemical Industry, respectively. Deionized water was produced by double distillation from quartz.
Preparation
First, polystyrene spheres were synthesized in accordance with the literature [23]. In a typical synthetic method, 105 mL styrene was washed five times with 400 mL 0.1 M NaOH solution and then rinsed five times with 400 mL deionized water. The washed styrene was mixed with 1200 mL deionized water with mechanical agitation. After half an hour, 6 g polyvinylpyrrolidone and 0.2 g azodiisobutyronitrile were added to the mixture, which was then reacted at 343 K under a nitrogen atmosphere with mechanical agitation for 24 h. The latex was then filtered to obtain a white powder, followed by washing five times with both ethanol and deionized water. Finally, the white polystyrene spheres were dried at 333 K overnight in a vacuum oven.
SAPO-34 cubes were synthesized by the conventional method [24]. Typically, 9.0 g pseudo-boehmite was mixed with 33.3 mL deionized water. After stirring for 1 h, 6.1 g silica sol, 7.8 mL H3PO4 (85%), and 22 mL TEA were added successively. The muddy mixture obtained was then sealed in a 100-mL Teflon-lined autoclave, heated, and crystallized at 473 K under autogenic pressure. The product was washed five times with deionized water then dried at 333 K under vacuum, followed by calcination at 823 K for 6 h at a heating rate of 2 K/min. All these procedures were conducted with agitation at a speed of 300 rev/min.
Calcined SAPO-34 (60.0 g) and 30.0 g PS were mixed with 300 mL ethanol. The mixture was heated under reflux at 348 K for 6 h, then the milky mixture was rotary evaporated to furnish white powder. The purpose of this procedure was to make a homogenous mixture of SAPO-34 and PS.
Granulation was completed in a LPG spray dry granulator in Shanghai Research Institute of Chemical Industry. Typically, 117.0 g kaolin, 143.0 g silica sol, 212.0 g deionized water, 85.0 g homogenous mixture of SAPO-34 and PS, and 188.0 g aluminium phosphate sol were vigorously agitated to make a dark brown muddy mixture; the mixture was then spray dried at 383 K to make 30–50 μm microspheres.
For post-hydrothermal treatment 12.0 g of the uncalcined spheres was mixed with 50 mL deionized water and 7 mL H3PO4 (85%) and stirred for 2 h. TEA (14 mL) was then added dropwise, and the mixture was stirred continuously for 2 h until a dark brown muddy mixture was obtained. This mixture was sealed in a 100-mL Teflon-lined autoclave, heated, and crystallized at 473 K under autogenic pressure. The product was washed five times with deionized water, dried at 333 K under vacuum, then calcined at 823 K for 6 h with a heating rate of 2 K/min. Finally, we obtained a dark brown product which can be directly used in fluidized-bed MTO reactions.
For comparison, samples without use of PS (NOMISAPO-34) were prepared following the same procedure as above.
Characterization
To investigate crystallographic properties X-ray diffraction (XRD) measurements were conducted at room temperature with a Rigaku D/Max 2550 VB/PC apparatus (Cu Kα radiation, λ = 1.54056 Å) operated at 40 kV and 100–200 mA. Diffraction patterns were recorded in the angular range 3°–50°. Surface morphology was observed by scanning electron microscopy (Jeol JSM-6360LV). S BET for the samples was determined by nitrogen physical adsorption at 77 K (Micromeritics ASAP 2010). Before measurement, all samples were degassed at 463 K under a pressure of 1 mmHg. The BET surface area was calculated and BJH pore size distribution was estimated using N2 adsorption–desorption isotherms. The macropore size distribution of the calcined microbeads using PS was measured with an Autopore IV 9500 mercury porosimeter (Micromeritics, USA). Temperature-programmed desorption of ammonia (NH3-TPD) was carried out on a Tianjin Xianquan TP-5080 multi-automatic characterization adsorption instrument. The procedure was as follows: 0.05 g sample was heated at 40 K/min to 873 K and kept at this temperature for 1 h. After cooling to 393 K, the gas was switched to NH3 flow. NH3 was absorbed for 1 h at 393 K, then switched to Ar flow under constant temperature for 1 h to sweep out the residual and physical absorbed NH3. Finally, the temperature was raised to 973 K at 10 K/min and the amount of desorbed NH3 was recorded.
Catalytic activity testing
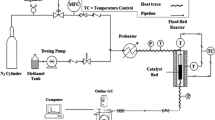
Methanol to olefins reactions were performed in a fluidized bed reactor made of stainless steel tubing (32 mm i.d, 1000 mm length). Catalyst samples (40 g) were activated in flowing nitrogen (100 mL/min) for 120 min at 823 K and cooled to the reaction temperature (723 K) before reaction. The reaction pressure and methanol WHSV (weight hourly space velocity) were 0.1 MP and 0.6 h−1, respectively. Mixtures of H2O and methanol (volume ratio = 1.5:1) were driven through the reactor by nitrogen flow (100 mL/min). The eluent gas was introduced to a Tianmei GC9790 gas chromatograph and analyzed using a TM-PLOT Q column (30 m × 0.53 mm, 40 μm) and an H2 flame ionization detector.
Results and discussion
X-ray diffraction spectroscopy
The wide-angle X-ray diffraction patterns are presented in Fig. 1. MAMISAPO-34 and NOMISAPO-34 both furnish characteristic peaks attributed to the typical CHA structure of SAPO-34, and the intensity of the main peak of (001) for CHA type follows the sequence SAPO-34 cubes > MAMISAPO-34 > NOMISAPO-34.

Wide-angle XRD patterns of various SAPO-34: a SAPO-34 cubes, b MAMISAPO-34, and c NOMISAPO-34
SEM images, N2 adsorption/desorption isotherms, and pore-size distribution
Figure 2a shows an SEM image of uniform PS spheres with a diameter of 2 μm. These particles are used as hard templates in the spray drying process. Figure 2b shows SAPO-34 cubes with an average size of 1 μm. A homogenous mixture of PS and SAPO-34 cubes is shown in the Fig. 2c.

SEM images of polystyrene spheres (a), SAPO-34 cubes (b), and a homogenous mixture of SAPO-34 and PS (c)
Figure 3a, b shows the different morphology of MAMISAPO-34 and NOMISAPO-34 by SEM. There are macropores in MAMISAPO-34, whereas NOMISAPO-34 has no macropores. The surface of NOMISAPO-34 is smooth whereas the surface of MAMISAPO-34 is rough and porous. Figure 3c is a magnification of Fig. 3b, which clearly shows the macropores with a diameter of approximately 2 μm.

SEM images of NOMISAPO-34 (a), MAMISAPO-34 (b) and high magnification of MAMISAPO-34 (c)
Figure 4 shows the N2 physisorption isotherms and BJH pore size distributions for MAMISAPO-34 and NOMISAPO-34. It can be seen that both samples have the representative characteristics of the type IV isotherm and a sharp increment in adsorption near P/P 0 = 0, which is related to the micropores. For both samples, the hysteresis loop at P/P 0 > 0.4 indicates the presence of mesoporous structures. The mesopores may be constructed from the interspace of SAPO-34 crystalline particles. As seen from the pore size distribution curve (Fig. 4, inset), for both SAPO-34s there is a sharp peak at 4 nm, which further confirms the existence of mesopores. The increment near P/P 0 = 1 for MAMISAPO-34 shows the presence of macropores in the MAMISAPO-34 sample; because macropores are absent from NOMISAPO-34 there is only a slight increment near P/P 0 = 1.

N2 physisorption isotherms and BJH pore size distributions for the SAPO-34 samples: (a) MAMISAPO-34; (b) NOMISAPO-34
Figure 5 gives the pore diameter distribution curves of MAMISAPO-34 and NOMISAPO-34 determined by mercury porosimetry measurements. For MAMISAPO-34, there is a sharp peak at 1.9 μm which might be because of the templating effect of PS on the morphology of the microbeads during the process of SAPO-34 zeolitization. For NOMISAPO-34 there is no obvious peak.

Pore diameter distribution of MAMISAPO-34 (a) and NOMISAPO-34 (b) determined by mercury porosimetry measurements
Temperature-programmed desorption
Temperature-programmed desorption of ammonia (NH3-TPD) was used to investigate the acid sites of different samples [25]. Figure 6 shows the desorption curves of MAMISAPO-34 and NOMISAPO-34. Apparently, there are two desorption peaks at 465 and 653 K, which correspond to the weak and strong acid sites [26]. It is reported that weak and strong acid sites originate from surface hydroxyl groups and structure acidity, respectively [27, 28]. As seen from Fig. 6, the desorption curves for MAMISAPO-34 and NOMISAPO-34 are nearly the same, but the desorption peaks at 465 and 653 K for sample MAMISAPO-34 are higher than those of NOMISAPO-34, indicating there are more surface and structure acidity sites for sample MAMISAPO-34. The more surface and structure acidity sites should be attributed to the better crystallinity of sample MAMISAPO-34, as seen from Fig. 1, which ultimately leads to the formation of stronger acidity.

NH3-TPD profiles of MAMISAPO-34 (a) and NOMISAPO-34 (b)
Methanol to olefins (MTO) reactions
For the methanol to olefins (MTO) reactions, conversion of methanol is almost 100%. So we just choose the selectivity and inactivation time to compare the activity of MAMISAPO-34 and NOMISAPO-34. Figure 7 gives the yields of total C2–C4 olefins versus time-on-stream for MTO reactions over MAMISAPO-34 and NOMISAPO-34. The results show that MAMISAPO-34 gives higher yields of C2–C4 olefins than NOMISAPO-34. Moreover, the inactivation time (90 min) of MAMISAPO-34 is longer than that (70 min) of NOMISAPO-34. It is well known that selectivity for C2–C4 olefins has a close relationship with degree of crystallinity [29]. Hence the higher yields of C2–C4 olefins for MAMISAPO-34 might be because of the better crystallization than that of NOMISAPO-34, which is in agreement with the results of XRD. Moreover, the inactivation occurs because of the diffusion limitation, hence the longer inactivation time of MAMISAPO-34 should be attributed to the macroporous structure we can observe from SEM.

Yields of C2–C4 olefins versus time-on-stream for MTO reactions over MAMISAPO-34 and NOMISAPO-34
Figure 8 gives the yields of main products versus time-on-stream over MAMISAPO-34 catalyst. It can be clearly observed from Fig. 8 that the yield of ethylene increases with time, whereas that of propylene decreases because the ethylene molecule is smaller than that of propylene. As the catalytic reaction goes on, the coke first prohibits propene production then ethylene production. Moreover, it is well known that there are two steps in MTO reactions [30]: First, methanol is converted into dimethyl ether (DME) which mainly occurs on the weak acid sites. Second, DME is converted into olefin that mainly occurs on the strong acid sites because of shape selectivity. Coke formation occurs on the strong acid sites in the microporous channels; this prevents the second step of MTO reactions. However, the weak acid sites exist not only in the microporous channels but also on the framework. These are not totally covered by coke and the first step of MTO reactions continues to produce DME. So we observe that the yield of DME increases at the inactivation point time.

Yields of main products versus time-on-stream for MTO reactions over MAMISAPO-34
Mechanism of fabrication of MAMI SAPO-34
A mechanism is proposed for fabrication of MAMISAPO-34. First, the granulated bead containing PS and SAPO-34 crystals is prepared by spray drying. Because the crystallization temperature of 473 K is above the glass transition temperature of PS (353–373 K) but below the melting point of PS (513 K), PS will turn into a glassy state and then diffuse to the outside of the microbead during the crystallization process. Subsequently, the orthophosphoric acid and template molecule will transfer to the position resulting from PS diffusion. Finally, the Si, Al, and P species and templates crystallize near the SAPO-34 crystals under hydrothermal conditions to form macroporous microbeads.
Conclusions
In this work we developed a novel method for fabrication of MAMISAPO-34. Precrystallized SAPO-34 cubes and PS were used as crystal seeds and hard templates, respectively. Spray drying granulated the mixture of SAPO-34 cubes, PS, microporous templates, silica, and aluminium source into 30–50 μm beads and the post-hydrothermal treatment further crystallized the microbeads. The MAMISAPO-34 performed better than NOMISAPO-34 in MTO reactions, because of the improved diffusion in the macropores in MAMISAPO-34. The combination of the hard templating spray-drying method and post-hydrothermal crystallization might be a promising way to obtain new bimodal porous zeolites in the future.
References
S.T. Wilson, B.M. Lok, C.A. Messina, E.R. Cannan, E.M. Flanigen, J. Am. Chem. Soc. 104, 1146 (1982)
M. Hartmann, L. Kevan, Chem. Rev. 99, 639 (1999)
C.M. Wang, Y.D. Wang, Z.K. Xie, Z.P. Liu, J. Phys. Chem. C. 113, 4585 (2009)
J. Van Den Bergh, W.D. Zhu, F. Kapteijn, J.A. Moulijn, K. Yajima, K. Nakayama, T. Tomita, S. Yoshida, Res. Chem. Intermed. 34, 467 (2008)
J. Haggin, Chem. Eng. News. 20, 36 (1983)
W.G. Song, J.F. Haw, J.B. Nicholas, C.S. Heneghan, J. Am. Chem. Soc. 122, 10726 (2000)
B. Arstad, S. Kolboe, J. Am. Chem. Soc. 123, 8137 (2001)
M. Hartmann, L. Kevan, Res. Chem. Intermed. 28, 625 (2002)
D. Chen, H.P. Rebo, A. Grønvold, K. Moljord, A. Holmen, Micropor. Mesopor. Mater. 35–36, 121 (2000)
D. Mores, E. Stavitski, M.H.F. Kox, J. Kornatowski, U. Olsbye, B.M. Weckhuysen, Chem. Eur. J. 14, 11320 (2008)
J. Zhu, Y. Cui, Y. Wang, F. Wei, Chem. Commun. 45, 3282 (2009)
K. Egeblad, C.H. Christensen, M. Kustova, C.H. Christensen, Chem. Mater. 20, 946 (2008)
J.G. Yu, W.G. Wang, B. Cheng, B.B. Huang, X.Y. Zhang, Res. Chem. Intermed. 35, 653 (2009)
C.-T. Hung, H. Bai, Chem. Engin. Sci. 63, 1997 (2008)
L. Chen, S.Y. Zhu, Y.M. Wang, M.-Y. He, New. J. Chem. 34, 2328 (2010)
Y. Yan, H. Wang, Abstracts of Papers, 221st ACS National Meeting, San Diego, CA, United States, 1–5 April 2001
A.I. Carrillo, N. Linares, J.G. Martinez, NSTI Nanotech, Nanotechnology Conference and Trade Show, Technical Proceedings, Boston, MA, United States, 1–5 June 2008, 1, 638–641
M. Choi, H.S. Cho, R. Srivastava, C. Venkatesan, D.-H. Choi, R. Ryoo, Nat. Mater. 5, 718 (2006)
H. Yang, Z. Liu, H. Gao, Z. Xie, J. Mater. Chem. 20, 3227 (2010)
K.H. Rhodes, S.A. Davis, F. Caruso, B. Zhang, S. Mann, Chem. Mater. 12, 2832 (2000)
Y. Qi, P. Dong, S. Chen, X. Wang, Petroleum Sci. 3, 85 (2006)
B.T. Holland, L. Abrams, A. Stein, J. Am. Chem. Soc. 121, 4308 (1999)
T. Sen, G.J.T. Tiddy, J.L. Casci, M.W. Anderson, Chem. Mater. 16, 2044 (2004)
H. Zhou, Y. Wang, F. Wei, D. Wang, Z. Wang, Appl. Catal. A Gen. 341, 112 (2008)
G.V.A. Martins, G. Berlier, C. Bisio, S. Coluccia, H.O. Pastore, L. Marchese, J. Phys. Chem. C. 112, 7193 (2008)
M.J. Van Niekerk, J.C.Q. Fletcher, C.T. O’Connor, Appl. Catal. A Gen. 138, 135 (1996)
H.-J. Chae, Y.-H. Song, K.-E. Jeong, C.-U. Kim, S.-Y. Jeong, J. Phys. Chem. Solid. 71, 600 (2010)
L. Xu, Z.M. Liu, A.P. Du, Y.X. Wei, Z.G. Sun, Stud. Surf. Sci. Catal. 147, 445 (2004)
Y. Hirota, K. Murata, M. Miyamoto, Y. Egashira, N. Nishiyama, Catal. Lett. 140, 22 (2010)
J.W. Park, J.Y. Lee, K.S. Kim, S.B. Hong, G. Seo, Appl. Catal. A Gen. 339, 36 (2008)
Acknowledgments
This work was supported by the National Nature Science Foundation of China (21007016, 20773039, and 20977030), the National Basic Research Program of China (973 Program, 2007CB613301 and 2010CB732306), the Science and Technology Commission of Shanghai Municipality (10520709900), and the Fundamental Research Funds for the Central Universities.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Liu, Y., Wang, L., Zhang, J. et al. Preparation of macroporous SAPO-34 microspheres by a spray drying method using polystyrene spheres as hard template. Res Chem Intermed 37, 949–959 (2011). https://doi.org/10.1007/s11164-011-0302-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-011-0302-2