
Abstract
The early events in the triplet 4-carboxybenzophenone (CB)-induced oxidation of N-acetyl-methionine methyl ester (N-Ac-Met-OCH3) are investigated in aqueous solution. Upon electron transfer from the methionine residue of N-Ac-Met-OCH3 to 3CB*, the resulting sulfur radical cation undergoes further reactions: (1) back electron transfer, (2) escape of the radical ions from the solvent cage, or (3) proton transfer and escape of the radicals. The yields and paths of these reactions are shown to depend strongly on the pH of the solution, and, similar to the previously reported results for dipeptides (Met-Gly and Gly-Met), on the structural nature of the methionine substituents. In the experiments performed in this work, low quencher concentrations were used to avoid formation of intermolecular transients (e.g., dimeric sulfur-centered radical cation (S∴S)+). Under these experimental conditions, the one-electron oxidized sulfur does not seem to become stabilized in an (S∴N)+ three-electron bonded intramolecular complex. The proposed mechanism is further supported by the stable products analysis. A detailed mechanism involving characterization of the transients is discussed and compared to that of methionine and methionine-containing dipeptides (Met-Gly and Gly-Met). Moreover, a newly installed transient absorption laser system is described in details.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Oxidative damage to biologically relevant material (i.e. aminoacids, peptides, proteins) has been the subject of many investigations due to its role in medicine, biology and basic chemistry [1, 2]. One of the sites likely attacked by oxidative agents is a sulfur atom in methionine residues [3–5]. The methionine residue in some biological systems can be attacked by oxidative species causing oxidative stress and biological aging [1–5]. There are numerous studies on the possible stabilization of the sulfide radical cation (>S●+) through intramolecular complexation with nucleophilic sites present in the neighboring groups, and donating electron lone pairs necessary to form a two-centered three-electron 2σ/1σ* bond [3–5]. Direct one-electron oxidation of methionine-containing compounds is possible, e.g., by using 4-carboxybenzophenone (CB) [6] or benzophenone (BP) [7–9] as the triplet sensitizers in aqueous or organic solvents, respectively. The use of laser flash photolysis technique (despite some difficulties like spectral overlap of the sensitizer and the intermediates) creates conditions where the one-electron oxidized species are formed directly via photosensitized electron transfer from the Met-containing compound onto CB [6]. In this work, the triplet CB is used to accept an electron from the sulfur moiety of the amino acid yielding a sulfur-centered radical cation in the initial step of the photoreaction. The progress of the process is monitored by resolving the transient spectra after selected delay times, and the concentration profiles for individual components are extracted from the multi-component spectra [8, 9].
Materials and methods
Benzophenone (BP), 4-carboxybenzophenone (CB), and N-Ac-Methionine ester (N-Ac-Met-OCH3) where obtained from Aldrich. The deionized water for the experiments was purified using a commercial system from Millipore, model Simplicity (Billerica, MA, USA).
Steady-state irradiations were performed on a standard optical bench system equipped with a high-pressure mercury lamp (HBO 200; Narva, Berlin, Germany) together with a water filter, cut-off glass filter (<300 nm), and an interference filter (313 nm). The sample solution was placed in a standard, rectangular, quartz cell (1 cm pathlength) and purged with high-purity argon for at least 20 min prior to irradiation. Absorption changes were monitored by taking the absorption spectra on a Cary 300 Bio spectrophotometer from Varian. The decomposition of the substrates of the photosensitized reaction and the photoproducts were monitored using an HPLC 600E system from Waters equipped with a diode array UV–Vis detector (Waters 996). Analyses were carried out on an XTerra RP-18 reverse phase column (4.6 mm × 250 mm, 5 μm particle size; Waters). For the HPLC-MS analyses, a similar system, equipped with a ZQ electrospray mass detector from Waters & Micromas, was used (Scheme 1).

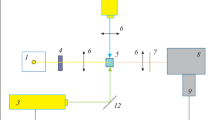
The experimental set-up for nanosecond laser flash photolysis (see text for details)
The newly installed nanosecond laser flash photolysis set-up was used in this work to investigate the photosensitized oxidation reactions. This kind of equipment (presented schematically above) is commonly used to investigate fast photochemical reactions, being an example of so-called pump-probe techniques. The set-up consists of: (1) a Nd:YAG laser as a pump (Spectra-Physics); (2) a pulsed xenon lamp to probe the excited sample; (3) a monochromator and a photomultiplier tube to detect and convert the acquired signals; and (4) a variety of electronics (digital scope, delay generator, shutters, photodiode) to synchronize the computer-controlled system. The system works in a so-called “kinetic-mode” and allows a sensitivity better than 0.001 OD units. In more detail, the set-up consists of the following elements:
-
a Nd:YAG laser from Spectra Physics (Mountain View, CA, USA), model INDI 40-10, equipped with a harmonic generation module to achieve 266, 355 and 532 nm excitation wavelengths at 10 Hz repetition rate, pulse duration 6–8 ns, max. pulse energy 450 mJ at 1,064 nm (155 mJ at 355 nm);
-
temperature controlled sample flow system from Quantum Northwest (Liberty Lake, WA, USA) with Peltier cooling;
-
150 W pulsed Xe lamp system with the lamp pulser from Applied Photophysics (Surrey, UK);
-
Monochromator from Acton (MA, USA), model Spectra Pro SP-2155 (2 grating turret);
-
Photomultiplier from Hamamatsu (Japan), model R955 (working on 5 dynodes amplification), powered from Stanford Research System (Sunnyvale, CA, USA) PS-310 Power Supply;
-
real-time digital oscilloscope from LeCroy, model Wave Runner 6100A (1 GHz, 10 GS/s) with GPIB port for digitized data transfer;
-
computer equipped with DAQ and timer PCI cards from National Instruments, software based on LabView 8.0;
-
optics and mechanics from Standa (Vilnius, Lithuania).
The monitoring Xe lamp light path was limited using a pair of regulated iris diaphragms (Newport, CA, USA), and the system was aligned in such a way that only the volume of the sample that was excited by the laser was monitored.
The set-up and the procedure of resolving the transient spectra allows us to obtain the concentration profiles for all of the transient species [short-lived excited triplet states, radicals, radical ions, other intramolecular and intermolecular transient species such as (S∴N)+ or (S∴S)+ intermediates] and to follow the reaction kinetics.
All flash photolysis experiments were carried out in rectangular quartz fluorescence cells (1 cm monitoring light pathlength). Relative actinometry was used to calculate the transients’ concentrations, and the triplet–triplet absorption maximum of the BP triplet state in acetonitrile solution was measured (ε520 = 6,500 M−1 cm−1) according to the procedure described in [10].
The new set-up and the procedure of resolving the transient spectra allowed us to obtain the concentration profiles for all of the transient species [CB triplet, ketyl radical, ketyl radical anion, intramolecular (S∴N)+, intermolecular (S∴S)+] and to determine appropriate primary quantum yields.
Results and discussion
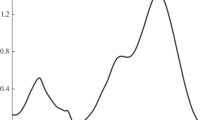
The rate constants for the quenching of CB/BP excited triplet states by N-Ac-Met-OCH3 were measured by fitting the decay traces of the 3CB* (or 3BP*) monitored at 520 nm, and acquired for different quencher concentrations (Fig. 1). The decay traces followed first order kinetics, and the k q values were computed using the Stern–Volmer equation shown below:
where τ is the observed CB/BP triplet lifetime, τ 0 is the natural triplet lifetime, and k q is the quenching rate constant. The rate constants evaluated from this procedure were 3.3 × 109 and 1.4 × 109 M−1 s−1 for CB and BP triplets, respectively. The order of magnitude (among other considerations, see below) suggests that the quenching occurs via electron transfer.

Stern–Volmer plot for the quenching of CB (2 mM) triplet state by N-Ac-Met-OCH3 at pH = 7
For the electron transfer reactions, one would expect that radical intermediates should be present in the transient absorption spectra. The absorption spectra of the benzophenone (and 4-carboxybenzophenone) triplet state and their radical ions and ketyl radicals have previously been reported [11], and used as reference spectra in the spectral-resolution procedure described in detail elsewhere [12]. In brief, the transient spectra at a given delay time after the laser pulse, were resolved into the component spectra associated with the individual components (e.g., triplet, radical anion, ketyl) using a multiple linear regression method [13]. Using this procedure, it was straightforward to produce the concentration profiles of the individual transients with time, as shown in Figs. 2, 3, 4, and 5.

Spectral resolution of the transient spectrum taken 600 ns after the 355 nm laser flash in an aqueous solution of CB (2 mM) and N-Ac-Met-OCH3 (~1 mM) at pH = 7

Spectral resolution of the transient spectrum taken 600 ns after the 355 nm laser flash in an aqueous solution of CB (2 mM) and N-Ac-Met-OCH3 (~1 mM) at pH = 10

Concentration profiles of transients (CB triplet, CB radical anion, and CBH neutral radical) in the flash photolysis of an aqueous solution of CB (2 mM) and N-Ac-Met-OCH3 (~1 mM) at pH = 7 (a) and pH = 10 (b). Concentrations of isolated spectral components calculated from the least squares regression analysis in the transient absorption spectra at various time intervals

Spectral resolution of the transient spectrum taken 1 μs after the 355 nm laser flash in acetonitrile:water (1:1) solution of BP (2 mM) and N-Ac-Met-OCH3 (~1 mM)
After the 355 nm laser pulse, the CB excited triplet is reduced while N-Ac-Met-OCH3 undergoes one-electron oxidation. The species expected to be formed upon electron transfer are: the radical anion of CB (CB●−), its protonated form CBH●, the sulfur-centered radical cation (>S●+), and the C-centered radical derived from the quencher (-S-C●H-). The reference spectra of the transients produced from CB are well known [6]; however, they were acquired again on the new flash photolysis set-up. Using the reference spectra together with the spectral-resolution procedure, it was possible to follow the concentration changes of the transients produced from the quenching of 3CB* by N-Ac-Met-OCH3 in aqueous solution. As can be seen in Figs. 2 and 4a, three intermediates are formed: the CB triplet immediately and, after its decay, CB●− and CBH● (see Scheme 2 for details). At pH = 7, the dominant product is CBH●, however, CB●− is also formed with a smaller yield. In basic solution at pH 10 (Figs. 3, 4b) the radical anion is formed with a higher yield, while the initially formed ketyl radical is being converted to its deprotonated form, and the equilibrium between the two forms is reached after a few microseconds.

The formation of the intermediate: the CB triplet together with CB●− and CBH●
It is worthwhile to point out that, in basic aqueous solution, proton transfer (k H) occurs in a solvent cage as a primary process, as can be seen in Fig. 4 (concentration profiles of CB●− and CBH● at pH = 10). After the charge separation, this process is then followed by equilibration. This does not favor CBH● and, consequently most of CBH● radicals are converted into CB●− anions. This equilibrium is reached on the microsecond timescale, and can be clearly seen in Fig. 4, and in Scheme 3.

The acid-base equilibrium between CB●− and CBH●
It should be noted that, in the case of N-Ac-Met-OCH3 photooxidation, no stabilization through (S∴N)+ three-electron bond formation was observed (see Figs. 2, 3, and 5 for spectral resolutions), although the lone pair on the nitrogen seems to be available for the S-centered radical cation to form an energetically-favored five-membered ring. Similarly, for Gly-Met dipeptide, no stabilization of the radical through the (S∴N)+ bond formation was observed, while substantial amounts of such stabilized radicals were seen upon Met-Gly photosensitized oxidation (see Table 1).
All the observations described above suggest that the accessibility of the nucleophilic site (namely, the N-atom of either the amino or peptide group) may be hampered by steric hindrance. Moreover, in the case of the Gly-Met and N-Ac-Met-OCH3, the lone pair on the nitrogen atom might be delocalized, making it inaccessible for the sulfur-p-electrons in the initially formed sulfur-centered radical cation. This last conclusion may though be in contradiction to some recently published results [14], where a similar two-centered three-electron (S∴N)+ intermediate was formed between an oxidized sulfur atom and a nitrogen atom from a peptide bond.
In an acetonitrile:water (1:1) solution, only one main transient is observed after the BP triplet decay. This species (possibly accompanied with some traces of radical anion, see Fig. 5) exhibits a characteristic absorption with a maximum at 540 nm, and it was attributed to the BP ketyl radical (BPH●). This lack of BP●− formation is likely due to the fast proton transfer within a solvent cage, yielding the BP radical. This proton is presumably donated by the quencher molecule, yielding the relatively stable C-centered radical [18], which could not be observed in the flash photolysis due to its inconvenient absorption overlap with the BP ground state.
Despite of those difficulties, the supporting evidence for the proposed reaction mechanism came from the steady-state irradiation experiments, where the product of recombination of the BP ketyl radical and a C-centered radical of N-Ac-Met-OCH3 (formed upon deprotonation of the initially formed S-centered radical cation) was detected and identified by means of HPLC-MS analysis.
Conclusions
The high values of the quenching rate constants together with a lack of accessible energy levels to make triplet transfer possible, suggests electron transfer as a quenching mechanism for all the reaction conditions (quenching of triplet CB and BP in aqueous and acetonitrile solutions, respectively). This conclusion is further supported by the direct observation of intermediate species (being the primary products of electron transfer) observed in laser flash photolysis experiments as well as by the identified stable product as a result of the radical recombination reactions. There are three main routes by which the resulting radical ion pair can react: (1) back electron transfer to regenerate the reactants in their ground states; (2) proton transfer within the ion pair; and (3) charge separation yielding appropriate radical ions.
Moreover, it is suggested that the lack of intramolecular three-electron (S∴N)+ bond formation is due to the delocalization of the nitrogen lone electron pair within the N-acetyl group, making the lone electron pair inaccessible for an interaction with the p-orbital of sulfur in the radical cation.
References
E.R. Stadtman, in Free Radicals, Oxidative Stress, and Antioxidants, vol. 296, ed. by T. Ozben (Plenum Press, New York, 1998), pp. 51–64
M. Davies, The oxidative environment and protein damage. Biochim. Biophys. Acta 1703, 93–109 (2005)
R.S. Glass, in Sulfur-Centered Reactive Intermediates in Chemistry and Biology, vol. 97, ed. by C. Chatgilialoglu, K.-D. Asmus (Plenum Press, New York, 1990), pp. 213–226
K.-D. Asmus, M. Göbl, K.-O. Hiller, S. Mahling, J. Möning, S∴N and S∴O three-electron-bonded radicals and radical cations in aqueous solutions. J. Chem. Soc. Perkin Trans. 2, 641–646 (1985)
D. Pogocki, K. Serdiuk, C. Schöneich, Computational characterization of sulfur-oxygen three-electron-bonded radicals in methionine and methionine-containing peptides: important intermediates in one-electron oxidation processes. J. Phys. Chem. A 107, 7032–7042 (2003)
K. Bobrowski, B. Marciniak, G.L. Hug, 4-Carboxybenzophenone-sensitized photooxidation of sulfur-containing amino acids. Nanosecond laser flash photolysis and pulse radiolysis studies. J. Am. Chem. Soc. 114, 10279–10288 (1992)
K. Bobrowski, B. Marciniak, G.L. Hug, A reinvestigation of the mechanism of photoreduction of benzophenones by alkyl sulfides. J. Photochem. Photobiol. A 81, 159–168 (1994)
P. Filipiak, J. Bartoszewicz, G.L. Hug, H. Kozubek, J. Paczkowski, B. Marciniak, Modification of photochemical pathways of sensitized oxidation of phenylthioacetic acid. Effects of solvent and tetrabutylammonium salt. J. Photochem. Photobiol. A 19, 167–175 (2007)
K. Bobrowski, G.L. Hug, D. Pogocki, B. Marciniak, C. Schöneich, Stabilization of sulfide radical cations through complexation with the peptide bond: mechanism relevant to oxidation of proteins containing multiple methionine residues. J. Phys. Chem. B 111, 9608–9620 (2007)
I. Carmichael, G.L. Hug, Triplet–triplet absorption spectra of organic molecules in condensed phases. J. Phys. Chem. Ref. Data 15, 1 (1986)
S. Baral-Tosh, S.K. Chattopadhyay, P.K. Das, A laser flash photolysis study of paraquat reduction by photogenerated aromatic ketyl radicals and carbonyl triplets. J. Phys. Chem. 88, 1404–1408 (1984)
B. Marciniak, K. Bobrowski, G.L. Hug, Quenching of triplet states of aromatic ketones by sulfur-containing amino acids in solution. Evidence for electron transfer. J. Phys. Chem. 97, 11937–11943 (1993)
P.R. Bevington, Data Reduction and Error Analysis for the Physical Sciences (McGraw-Hill, New York, 1969)
G.L. Hug, K. Bobrowski, D. Pogocki, G. Hörner, B. Marciniak, Conformational influence on the type of stabilization of sulfur radical cations in cyclic peptides. ChemPhysChem 8, 2202–2210 (2007)
G.L. Hug, K. Bobrowski, H. Kozubek, B. Marciniak, Photooxidation of methionine derivatives by the 4-carboxybenzophenone triplet state in aqueous solution. Intracomplex proton transfer involving the amino group. Photochem. Photobiol. 68, 785–796 (1998)
G.L. Hug, B. Marciniak, Comparison of electron-transfer/diffusion models as applied to fluorescence quenching data. J. Phys. Chem. 99, 1478–1483 (1995)
G.L. Hug, B. Marciniak, K. Bobrowski, Sensitized photooxidation of sulfur-containing amino acids and peptides in aqueous solution. Photoinduced electron transfer between sulfur-containing simple peptides and the 4-carboxy-benzophenone triplet state in aqueous solution. J. Photochem. Photobiol. A 95, 81–88 (1996)
M. Bonifacic, H. Mockel, D. Bahnemann, K.D. Asmus, Formation of positive-ions and other primary species in oxidation of sulfides by hydroxyl radicals. J. Chem. Soc. Perkin Trans. 2, 675–685 (1975)
Acknowledgments
This work was performed under the COST Action CM 0603. The authors would like to thank Dr. G. Burdzinski and Dr. J. Karolczak from the Center for Ultrafast Laser Spectroscopy, AMU, Poznan, Poland for their help in building the laser flash photolysis system. We gratefully acknowledge fruitful discussions with Dr. Gordon L. Hug from the Notre Dame Radiation Laboratory, IN, USA.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pedzinski, T., Markiewicz, A. & Marciniak, B. Photosensitized oxidation of methionine derivatives. Laser flash photolysis studies. Res Chem Intermed 35, 497–506 (2009). https://doi.org/10.1007/s11164-009-0046-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-009-0046-4