
Abstract
The pharaoh cuttle Sepia pharaonis Ehrenberg, 1831 (Mollusca: Cephalopoda: Sepiida) is a broadly distributed species of substantial fisheries importance found from east Africa to southern Japan. Little is known about S. pharaonis phylogeography, but evidence from morphology and reproductive biology suggests that Sepia pharaonis is actually a complex of at least three species. To evaluate this possibility, we collected tissue samples from Sepia pharaonis from throughout its range. Phylogenetic analyses of partial mitochondrial 16S sequences from these samples reveal five distinct clades: a Gulf of Aden/Red Sea clade, a northern Australia clade, a Persian Gulf/Arabian Sea clade, a western Pacific clade (Gulf of Thailand and Taiwan) and an India/Andaman Sea clade. Phylogenetic analyses including several Sepia species show that S. pharaonis sensu lato may not be monophyletic. We suggest that “S. pharaonis” may consist of up to five species, but additional data will be required to fully clarify relationships within the S. pharaonis complex.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The pharaoh cuttle Sepia pharaonis Ehrenberg 1831 is a broadly distributed species found from east Africa to southern Japan (Nesis 1987; Roper et al. 1984). The species is a significant component of cephalopod catches in several Middle Eastern, south Asian and southeast Asian fisheries (including Vietnam, Thailand, Yemen and India). For example, in Yemen alone, over 10,000 tonnes of cephalopod were landed in 2003; Sepia pharaonis constituted about 80% of this total (FAO 2005). Estimated cuttle production throughout the Indian Ocean has risen steadily since the late 1980’s; nearly 80,000 tonnes were taken in 2003 (FAO 2000), with Sepia pharaonis probably constituting 40–50% of this total. Even this value is probably an underestimate, as approximately 30,000 tonnes of Sepia pharaonis exported from India is not included in this total. Nesis (1987) noted that “This species [Sepia pharaonis] is the most important object of the cuttlefish fishery in the northern part of the Indian Ocean and southeastern Asia”.
Despite the clear economic importance of this species, relatively little is known about S. pharaonis phylogeography and population genetics. However, there is some evidence that S. pharaonis may actually be a complex of closely related species. Norman (2000) suggested that Sepia pharaonis consists of three forms: Sepia pharaonis (sensu stricto) (found in the western Indian Ocean from the Red Sea to the Arabian Gulf; the eastern limit of the range of this form is unknown); Sepia “pharaonis” II (Japan to the Gulf of Thailand, Philippines and north Australia) and Sepia “pharaonis” III (Maldives to Andaman Sea coast of Thailand). The three forms seem to differ in both morphology and reproductive patterns. While mating, S. pharaonis s. s. males show zebra lines on the third arm pair, while S. “pharaonis” II males have broken lines and S. “pharaonis” III males have spots (Norman 2000). In addition, S. pharaonis s. s. spawn between August and October, while S. “pharaonis” II (in Hong Kong) spawn from March–May and S. pharaonis in India spawn year round (Norman 2000).
To study S. pharaonis phylogeography, we collected partial mitochondrial 16S DNA sequence data from Sepia pharaonis samples from the coasts of Yemen, Oman, Iran, India, Thailand, Taiwan and Australia with the help of an international network of collaborators. Phylogenetic analyses were used to assess phylogeographic patterns within S. pharaonis sensu lato.
Materials and methods
Tissue specimen collection, DNA extraction and sequencing
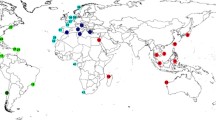
Tissue samples were collected from Sepia pharaonis individuals from Australia, India, Iran, Oman, Taiwan, Thailand and Yemen (Fig. 1; Table 1) and shipped to the first author (FEA) in 80–100% EtOH. Total DNA was extracted using a DNEasy kit (QIAGEN). A ∼500-bp fragment of the mitochondrial large subunit (16S) RNA was amplified via PCR using a Perkin-Elmer 9700 thermal cycler, oligonucleotide primers described elsewhere (Anderson 2000; Geller et al. 1997), HotStar Master Mix (QIAGEN) following manufacturer’s protocols (half-reactions) and a thermal cycling regime as follows: 94° (1 minute)—42° (1 minute)—72° (1:30), repeated for 35 cycles, with a 7-minute terminal extension step at 72°. PCR products were gel-purified using a MinElute kit (QIAGEN) and directly sequenced using BigDye Terminator mix (Applied Biosystems). Sequences were run out on an ABI 377 automated sequencer and edited using Sequencher 4.1 (GeneCodes). Redundant haplotypes were recorded and removed prior to phylogenetic analysis.

Map showing the type localities for Sepia pharaonis (*) and sampling localities for this study: 1 = Red Sea (RS), 2 = Gulf of Aden (GofA), 3 = Persian Gulf (PG), 4 = Arabian Sea (AS), 5 = Gulf of Oman (GofO), 6 = Kochi (CFH), 7 = Veraval (VRL), 8 = Vishakapatanam (VSK), 9 = Phuket (PH), 10 = Prachuap (PR), 11 = Chumphon (CHU), 12 = Taiwan (TAI), 13 = Gulf of Carpenteria (GofC), 14 = Northeast Queensland (NEQ)
Sequence alignment and phylogenetic analyses
The 16S sequences generated here were analyzed alone (“pharaonis only”) or along with 16S sequences for several other Sepia taxa obtained from Genbank (“pharaonis with outgroups”; Table 2). For both sets of analyses, sequences were aligned using CLUSTAL X (Thompson et al. 1997) with default settings and edited manually in Se-Al v. 2.0a11 (Rambaut 1996). A few regions could not be aligned with confidence across all taxa in the “pharaonis with outgroups” data set; these regions were retained in the Sepia pharaonis sequences but coded as “?” (missing data) in all other sequences (data sets and alignments are available from FEA upon request). Maximum parsimony (MP), maximum parsimony bootstrap and Bayesian analyses were performed for each data set in PAUP* (Swofford 2002) and MrBayes 3.1.1 (Ronquist and Huelsenbeck 2003). Useful reviews of Bayesian phylogenetic methodology can be found in Lewis (2001) and Holder and Lewis (2003). For MP analyses, branch-and-bound searches were performed for the “pharaonis only” data set and heuristic searches (1,000 random addition sequence replicates, holding 10 trees at each step) were used for the larger “pharaonis with outgroups” data set. Two sets of MP analyses were performed for each data set: one with gaps treated as missing data, one with gaps treated as a “fifth base”. All inferred indels were one base pair in length except for one that was two bases long; positions for this indel were weighted 0.5 for all analyses where gaps were treated as a fifth base. For Bayesian analyses, best-fitting DNA substitution models were chosen by first estimating a neighbor-joining tree using LogDet distances in PAUP*. The LogDet transformation (Lockhart et al. 1994) was used for distance correction because it is robust to changing base compositions across the tree (which can cause systematic error for analyses based on uncorrected distances) (Swofford et al. 1996). Likelihoods of the 16S data under all standard nucleotide substitution models available in MrBayes 3.1.1 were calculated using PAUP*. These likelihood scores were used to select a best-fitting substitution model using “MrDT-ModSel”, a modification of DT-ModSel (Minin et al. 2003) developed by FEA to compare only substitution models that are available in MrBayes 3.1.1. Four Bayesian analyses, each consisting of one cold and three heated Metropolis-coupled Markov chains, were run in MrBayes 3.1.1, with random starting trees and trees sampled every 500 generations. A topological similarity criterion—the average standard deviation in partition frequency values across independent runs—was used to automatically assess convergence of the runs (when this value reached 0.005, the runs were stopped). Upon topological convergence, the first 25% of trees from each run were removed as burn-in. The post burn-in trees from all four runs were assumed to be independent samples from the posterior probability distribution, and thus were combined to produce a phylogram and a 50% majority-rule consensus tree.
Results
MP analyses of the “pharaonis only” data set resulted in 44 trees (treelength = 61.5) for the analysis where gaps were treated as a “fifth base” and 22 trees (treelength = 53) for the “gaps = missing” analysis (Fig. 2). The consensus phylogram of trees resulting from Bayesian analyses (HKY85 + I model; consensus of 21,948 trees from four combined runs, run length of 3,657,500 generations) of the “pharaonis only” data set is shown in Fig. 3.

Strict consensus cladogram of 44 trees (treelength = 61.5) for “gaps = fifth base” analysis and 22 trees (treelength = 53) for “gaps = missing” parsimony analysis of the Sepia pharaonis haplotype data set (consensus trees for both analyses are identical). Numbers above the branches are bootstrap support values with gaps treated as missing data; numbers below the branches are bootstrap support values with gaps treated as a fifth state. Numbers in parentheses after haplotype codes denote the number of sampled individuals that possessed that haplotype. Haplotype codes are listed in Table 1; large letters denote clades described in the text

Bayesian phylogram (branch lengths equal to the estimated number of substitutions per site averaged across all post-burn-in trees) depicting relationships among Sepia pharaonis sequences (HKY85 + I model). Numbers on branches are clade posterior probability estimates; other notations as in Fig. 2
MP analyses of the “pharaonis with outgroups” data set resulted in 33,707 trees (treelength = 211.5) for the “gaps = fifth base” analysis and 132 trees (treelength = 203) for the “gaps = missing” analysis. The strict consensus tree for the “gaps = missing” analysis is shown in Fig. 4 (the strict consensus tree for the “gaps = fifth base” analysis is somewhat less resolved). During analysis of several MP bootstrap pseudoreplicates of this data set, the maxtrees limit (the number of trees retained in memory by PAUP*, in this case 100,000) was reached, limiting the effectiveness of the bootstrap analysis. The consensus phylogram of trees resulting from Bayesian analyses (GTR + Γ + I model, consensus of 21,088 trees from four combined runs, run length of 3,514,500 generations) of this data set is shown in Fig. 5.

Strict consensus cladogram of 132 trees (treelength = 203) resulting from parsimony analysis of all available Sepia sequences, with gaps treated as missing data. The cladogram is arbitrarily rooted with Sepia officinalis. Nodes not seen in the strict consensus tree resulting from the “gaps = fifth base” analysis of this data set are marked with asterisks (*). Numbers above the branches are bootstrap support values with gaps treated as missing data; numbers below the branches are bootstrap support values with gaps treated as a fifth state. All other notations are as in Fig. 2

Bayesian phylogram (branch lengths equal to the estimated number of substitutions per site averaged across all post-burn-in trees) depicting position of Sepia pharaonis haplotypes within Sepia (GTR + Γ + I model), arbitrarily rooted with Sepia officinalis. Numbers on branches are clade posterior probability estimates; all other notations are as in Fig. 2. Only posterior probabilities >0.9 are shown
All trees reveal strong support for two groups: an Australia clade (denoted clade E) and a Red Sea/Gulf of Aden clade (clade A). A clade consisting of all samples from the coast of India and the Andaman Sea coast of Thailand (clade C) was strongly supported in all analyses but one (the Bayesian analysis of the “pharaonis only” data set; Fig. 3). A fourth clade consisting of all samples from the Gulf of Thailand and Taiwan (clade D) was weakly supported but consistently recovered (except in Fig. 5, where the position of one sequence from clade D is unresolved). All four sequences obtained thus far from the Persian Gulf, Gulf of Oman and Arabian Sea were identical (clade B). There is some support for a clade including clades B, C and D. This clade is recovered in all MP analyses of both data sets—bootstrap support values = 81 (“gaps = missing”) and 95 (“gaps = fifth state”) for the “pharaonis only” data set and 51 (“gaps = missing”) and 53 (“gaps = fifth state”) for the “pharaonis with outgroups” data set—but this clade has a low posterior probability in all Bayesian analyses (0.253 for the “pharaonis only” data set; 0.384 for the “pharaonis with outgroups” data set).
The trees based on analyses of the “pharaonis with outgroups” data set (Figs. 4 and 5) suggest that the 16S gene region used here is insufficient for resolving either the phylogenetic position of Sepia pharaonis within Sepia or the relationships among S. pharaonis subclades. However, there is still strong (>75%) parsimony bootstrap support and high posterior probabilities (>0.9) for clades A, C and E. By contrast, support for a monophyletic S. pharaonis was very low (parsimony bootstrap support values <10%, posterior probability = 0.0134).
Conclusion
Norman (2000) suggested that Sepia pharaonis sensu lato consists of three forms: Sepia pharaonis sensu stricto, Sepia “pharaonis” II and Sepia “pharaonis” III. Although we were unable to sample cuttles from the type localities for S. pharaonis (Fig. 1), it seems likely that our clade A (southern Red Sea and Gulf of Aden) represents S. pharaonis s. s. (see further discussion below). Our clade D (Gulf of Thailand and Taiwan) may correspond with S. “pharaonis” II, but our Australian samples constitute a separate, rather distantly related group (clade E). In this preliminary study, we have thus far been unable to obtain samples from Indonesia or the Philippines; samples from these regions, as well as from northwest Australia, would clarify the status of clade D with respect to Norman’s S. “pharaonis” II. Clade C (India and Andaman Sea) roughly corresponds to S. “pharaonis” III. Intriguingly, cuttles sampled from the west coast of India (i.e., Kochi and Veraval) are genetically very similar (or even identical; i.e., haplotype “PH 3/CFH 1/VRL 1/VSK 2”) to samples from the Andaman coast of Thailand. If clade C represents S. “pharaonis” III, the range of this form may extend westward well past the Maldives, at least to the northwestern corner of the Indian subcontinent. Our clade B (consisting of all samples from the Persian Gulf, Gulf of Oman and the Arabian Sea) may be closely related to clades C and D. Although the posterior probability of a B/C/D clade is quite low, there is some MP bootstrap support (as high as 95% in one analysis) for such a clade.
MP and Bayesian analyses of the “pharaonis with outgroups” data set produce rather unresolved consensus phylogenies. Bootstrap support values and posterior probabilities of a monophyletic Sepia pharaonis are minimal, but support for any particular groupings of S. pharaonis subclades with other Sepia taxa is also low; this is likely due to the relatively small amount of sequence data surveyed here. Additional data from other genes should help resolve the relationships among the subclades of S. pharaonis.
Our results show that Sepia “pharaonis” is a complex of three to five clades, perhaps corresponding to species, and suggest that a thorough taxonomic revision of this species complex—incorporating additional molecular and morphological data—is warranted. The appropriate application of the binomen Sepia pharaonis will also need to be re-evaluated in light of this research. Norman (2000) noted that the type locality of S. pharaonis sensu lato is the Gulf of Suez (it is sometimes listed as “Massawa, Gulf of Suez”), but there is some confusion on this point. Ehrenberg (1831) described the type locality as “Prope Tor Arabiae sinaiticae s. petraeae et prope insulam Massauam ad Habessiniae littus huius speciei formas frequentes observavimus”. A rough translation of this is “Near El-Tor in the South Sinai and near the island of Massawa on the shore of Abyssinia (Eritrea), we have frequently observed this species”. Two locations are being described here: one in the Gulf of Suez, and one in modern-day Eritrea (Fig. 1). Although we were unable to collect specimens from either El-Tor or Massawa, we believe that our samples from the Yemeni coast of the Red Sea are likely to be very similar to those from El-Tor and Massawa (which are both in the Red Sea). If our S. pharaonis subclades are found to represent several distinct species, the binomen Sepia pharaonis should probably be restricted to the Red Sea/Gulf of Aden lineage (clade A) found in this study.
Our work on the Sepia pharaonis complex is ongoing, and will include acquisition of tissue samples from additional regions (especially Indonesia, the Philippines and the type localities), investigation of additional gene regions (both mitochondrial and nuclear) and morphological comparisons among the members of the clades recovered here.
References
Anderson FE (2000) Phylogeny and historical biogeography of the loliginid squids (Mollusca: Cephalopoda) based on mitochondrial DNA sequence data. Mol Phylogenet Evol 15:191–214
Ehrenberg CG (1831) Symbolae physicae seu icones et descriptiones animalium evertebratorum sepositis insectis quae ex itinere per Africam Borealem et Asiam Occidentalem - novae aut illustratae redierunt. Decas 1 Mollusca
FAO (Food and Agriculture Organization) (2000) FISHSTAT Plus—Universal software for fishery statistical time series, version 2.3. Publisher, Rome
FAO (Food and Agriculture Organization) (2005) FAO Fisheries Global Information System. Web site address: http://www.fao.org/figis/servlet/static?dom=root&xml=index.xml (accessed February 28, 2006)
Geller JB, Grosholz E, Ruiz G (1997) Cryptic invasions of the crab Carcinus detected by molecular phylogeography. Mol Ecol 6:256–262
Holder M, Lewis PO (2003) Phylogeny estimation: traditional and Bayesian approaches. Nat Rev Gen 4:275–284
Lewis PO (2001) Phylogenetic systematics turns over a new leaf. Trends Ecol Evol 16:30–37
Lockhart PJ, Steel MA, Hendy MD, Penny D (1994) Recovering evolutionary trees under a more realistic model of sequence evolution. Mol Biol Evol 11:605–612
Minin V, Abdo Z, Joyce P, Sullivan J (2003) Performance-based selection of likelihood models for phylogeny estimation. Syst Biol 52:674–683
Nesis KN (1987) Cephalopods of the world. TFH Publications, Neptune City, NJ
Norman MD (2000) Cephalopods, a world guide: Pacific Ocean, Indian Ocean, Red Sea, Atlantic Ocean, Caribbean, Arctic, Antarctic. ConchBooks, Hackenheim, Germany, 320 pp
Rambaut A (1996) Se-Al: Sequence Alignment Editor. Available at http://evolve.zoo.ox.ac.uk/
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Roper CFE, Sweeney MJ, Nauen CE (1984) Cephalopods of the world: an annotated and illustrated catalogue of species of interest to fisheries. United Nations Development Programme; Food and Agricultural Organization of the United Nations, New York, NY, 277 pp
Swofford DL (2002) PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods). Sinauer Associates, Sunderland, MA
Swofford DL, Olsen GJ, Waddell PJ, Hillis DM (1996) Phylogenetic inference. In: Hillis DM, Moritz C, Mable BK (eds) Molecular Systematics. Sinauer Associates, Sunderland, MA, pp 407–514
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 24:4876–4882
Acknowledgements
We would like to thank the organizers of the Cephalopod International Advisory Council 2003 meeting in Phuket, Thailand, that brought several of the authors together and sparked this project, and the SIUC Department of Zoology for providing travel funds to FEA for that meeting. The research was supported by NSF Grant DEB-0235794 to FEA. We also thank Alonso Córdoba, Stephanie Clutts, Mike Venarsky and Adria Pilsits for assistance with sequencing. Special thanks to Maggie Helwig for help with the translation of the type locality of Sepia pharaonis, and to Martina A. C. Roeleveld for confirming the identity of one of the specimens from which tissue was taken and depositing it as a voucher specimen at the South African Museum.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Anderson, F.E., Valinassab, T., Ho, CW. et al. Phylogeography of the pharaoh cuttle Sepia pharaonis based on partial mitochondrial 16S sequence data. Rev Fish Biol Fisheries 17, 345–352 (2007). https://doi.org/10.1007/s11160-007-9042-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11160-007-9042-1