
Abstract
The incretin hormone, glucagon-like peptide-1 (GLP-1), stimulates insulin secretion and forms the basis of a new drug class for diabetes treatment. GLP-1 has several extra-pancreatic properties which include effects on kidney function. Although renal GLP-1 receptors have been identified, their exact localization and physiological role are incompletely understood. GLP-1 increases natriuresis through inhibition of the sodium-hydrogen ion exchanger isoform 3 in the proximal tubule. This may in part explain why GLP-1 receptor agonists have antihypertensive effects. Glomerular filtration rate is regulated by GLP-1, but the mechanisms are complex and may depend on e.g. glycaemic conditions. Atrial natriuretic peptide or the renin-angiotensin system may be involved in the signalling of GLP-1-mediated renal actions. Several studies in rodents have shown that GLP-1 therapy is renoprotective beyond metabolic improvements in models of diabetic nephropathy and acute kidney injury. Inhibition of renal inflammation and oxidative stress probably mediate this protection. Clinical studies supporting GLP-1-mediated renal protection exist, but they are few and with limitations. However, acute and chronic kidney diseases are major global health concerns and measures improving renal outcome are highly needed. Therefore, the renoprotective potential of GLP-1 therapy need to be thoroughly investigated in humans.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Glucagon-like peptide-1 (GLP-1) is a gut derived hormone primarily recognized for its glucose-dependent stimulation of insulin secretion. GLP-1 is important in the incretin effect, which refers to the observation that orally administered glucose increases plasma insulin levels much more than intravenously administered glucose. Additionally, GLP-1 stimulates insulin synthesis, increases β-cell mass, and inhibits glucagon secretion [1].
The GLP-1 insulinotropic effects are attractive in the treatment of diabetes but native GLP-1 is an inappropriate drug compound due to its short half-life of ~2 min in vivo [2]. Although GLP-1 is cleared by the kidneys, the short half-life is mainly due to degradation by the ubiquitous enzyme dipeptidyl peptidase-4 (DPP-4) [3]. Two different approaches have been applied to overcome this problem: The development of DPP-4 inhibitors (sitagliptin, vildagliptin, saxagliptin, alogliptin, linagliptin) which increase the half-life of endogenous GLP-1 and the development of GLP-1 receptor (GLP-1R) agonists (exendin-4, liraglutide, lixisenatide) which are resistent to DPP-4 degradation.
GLP-1Rs are located in a wide variety of tissues (e.g. pancreas, kidney, heart, brain, lung, and gastrointestinal tract) [4, 5] and native GLP-1 as well as GLP-1 based treatment exert multiple extra-pancreatic effects. Of special clinical interest, type 2 diabetes mellitus (T2DM) patients in long-term treatment with GLP-1R agonists often experience both weight loss [6] and a lowering of blood pressure [7]. Furthermore, GLP-1 therapy inhibits gastrointestinal motility [8], increases cardiac output and heart rate [9], and shows promising properties of both cardioprotection [10] and neuroprotection (preclinical studies) [11].
GLP-1 also has important functions in the kidney. The best described effect is the induction of natriuresis which may in part explain the antihypertensive effect [12, 13]. Rodent studies find GLP-1 therapy to prevent the development of diabetic nephropathy (DN). This remains to be thoroughly explored in humans, but with DN as the leading cause of end-stage renal disease (ESRD) [14] such observation mandates attention.
Our knowledge of the renal effects of GLP-1 is rapidly accumulating, but there is no comprehensive understanding of the underlying mechanisms. This review will focus on the basic physiology as well as the clinical implications of GLP-1 therapy targeting the kidneys.
2 GLP-1 and Kidney Physiology
The effects of GLP-1R agonism on basic kidney physiology have been explored in several rodent studies but only in two dedicated human studies [12, 13]. Generally, the renal effects of GLP-1 have shown to be weaker in humans compared with rodents, but this is most likely explained by the relative differences in the doses used. Thus, apart from species differences it is notable that while interventions increased basal GLP-1 levels ~10-fold in humans [13], the rodents typically had >100-fold increases [15]. Hence, some of the effects demonstrated in animal models might not have clinical relevance.
2.1 Kidney GLP-1 Receptors
The GLP-1R is a G-protein coupled receptor that stimulates adenylate cyclase and protein kinase A (PKA). Studies in rodents [16–19], pigs [20], cattle [21], and humans [20] have identified renal GLP-1Rs at both messenger ribonucleic acid (mRNA) (real-time polymerase chain reaction) and protein level (immunoreactivity). While most studies find GLP-1Rs in the renal vasculature, there is no complete agreement about in which part of the nephron the GLP-1Rs are located. Generally, the receptor is not found in the distal tubules, but some studies find the GLP-1R in the proximal tubules and in the glomerulus at both mRNA and protein level.
In humans, using autoradiography with labelled GLP-1, the GLP-1Rs were found in large and medium sized renal arteries but not in the tubules or glomeruli [5].
The divergent findings may in part be explained by species differences. Recently however, it has been shown that commercially available GLP-1R antisera show a remarkable low specificity and sensitivity [22, 23], and therefore results based on immunoreactivity should be interpreted with caution.
2.2 Electrolyte Excretion and Diuresis
The first study to investigate the renal effects of GLP-1 was published in 2002 and was performed on anesthetized rats [15]. GLP-1 infusions increased sodium excretion and diuresis 12-fold in the innervated kidney and 2-fold in the surgical denervated kidney which had a higher basal sodium excretion [15]. Several subsequent studies in rodents have shown that native GLP-1 [16, 24], exendin-4 [16, 25–28], liraglutide [29, 30] as well as DPP-4 inhibitors [16, 28, 31] all have natriuretic and diuretic effects. The effects are dose dependent and generally seem stronger with GLP-1R agonists than with DPP-4 inhibitors. The increase in natriuresis is accompanied by increased excretion rates of calcium, phosphate, chloride, and in some studies to a lesser extent potassium. Electrolyte excretion rates increase in both healthy and diabetic rodents.
The first data on the renal effects of GLP-1 in humans were published in 2004 and based on a randomised, controlled, cross-over trial [12]. The effect of a 3-h native GLP-1 infusion was investigated during hypertonic saline infusion in healthy subjects and in obese insulin-resistant males. GLP-1 dose-dependently increased the urinary sodium, calcium, chloride, and osmolal excretion rates, whereas potassium excretion remained unchanged. The mean increase in natriuresis was ~60 % in the obese subjects and a little more in the healthy subjects [12, 32]. We have subsequently measured different electrolyte clearance rates in response to a 2-h GLP-1 infusion in healthy males and reached identical conclusions. We found GLP-1 to increase sodium clearance ~40 % whereas the observed increase in diuresis was non-significant [13]. A study designed to explore the systemic hemodynamic effects of a single subcutaneous exenatide injection in healthy males found the sodium:creatinine ratio to double compared with placebo in urine collected over 3 h [9].
2.3 Tubular Reabsorption
There is strong evidence that the increments in electrolyte excretion rates are secondary to decreased tubular proximal reabsorption. Native GLP-1, exendin-4 and to a lesser extent DPP-4 inhibitors markedly decrease the proximal tubular sodium reabsorption in rats [15, 16, 33] estimated with lithium clearance technique [34]. The findings were confirmed with tubular micropuncture technique in rats, where exendin-4 suppressed proximal reabsorption 20-40 % [25]. In healthy humans we found GLP-1 infusion to decrease proximal tubular sodium reabsorption ~10 % (lithium clearance) [13].
The tubular proximal reabsorption in general is predominantly mediated via the Na+/H+ exchanger isoform 3 (NHE3) and to a lesser extent the sodium-glucose linked transporter 2 (SGLT2). Notably, both transporters are up-regulated in diabetes [35].
Several studies have linked GLP-1R activation to down-regulation of NHE3 activity through a PKA-dependent mechanism [16, 18, 31, 33]. This is harmonious with GLP-1 alkalising the urine in rats [16] and humans [12]. NHE3 and DPP-4 physically interact in the brush-boarder epithelium of the proximal tubule which may reflect a functional relationship [31, 36, 37]. Tubular microperfusion studies and functional assays incubated with GLP-1R agonists have shown some signs of direct GLP-1 actions on the tubular reabsorption [16, 20]. However, at present it is not completely established whether GLP-1Rs and DPP-4 enzymes in the tubular brush-border epithelium are relevant for GLP-1 function.
2.4 Renal Perfusion
GLP-1 infusion has shown to increase renal blood flow (RBF) ~22 % in anaesthetized rats measured with a flow probe in the artery of a denervated kidney, while it had no effect on renal medullary blood flow or renal interstitial hydrostatic pressure [15]. Renal plasma flow, estimated as paraaminohippuric acid clearance, increased ~28 % in anaesthetized rats infused with GLP-1 [16]. The exact proportion of preglomerular and postglomerular vasodilatation is unknown. However, to explain the combined effects on RBF, GFR and tubular proximal reabsorption, it seems safe to assume a decrease in preglomerular resistance.
In healthy males we found no effect of GLP-1 infusion on renal plasma flow (95 % CI −3.6 to 8.8 %) estimated as the renal clearance of 123I-hippuran [13]. It is unknown how GLP-1 impacts RBF under diabetic conditions in both rodents and humans.
2.5 Glomerular Filtration Rate
In anaesthetized rats, GLP-1 infusion increased GFR ~39 % in innervated kidneys (estimated with inulin clearance) but had no effect on GFR in denervated kidneys [15]. Exendin-4 increased whole body GFR ~25 % (inulin) and single nephron GFR 33-50 % in anaesthetized hydropenic rats [25]. Others found that exendin-4 increased GFR ~30 % (inulin) in non-diabetic mice but had no acute effect in diabetic db/db mice [28]. DPP-4 inhibition did not significantly increase GFR in non-diabetic mice (inulin) [28] or rats (creatinine clearance (CrCl)) [16]. GLP-1 and exendin-4 both significantly increased GFR (CrCl) in anaesthetized rats [16]. Eight weeks of exendin-4 treatment prevented hyperfiltration (CrCl) in diabetic rodents [19, 38].
A 3-h GLP-1 infusion decreased GFR 6 % (CrCl) in obese insulin-resistant males but did not significantly affect GFR in healthy subjects [12]. We found no effect of GLP-1 infusion on GFR (95 % CI −0.8 to 4.6 %) in healthy males estimated as the renal clearance of 51Cr-EDTA [13]. We and others are currently investigating the effects of GLP-1R agonists on GFR in T2DM patients.
The above indicates that GLP-1 markedly increases GFR in healthy rodents but has limited effect in healthy humans. In contrast, GLP-1 decreases GFR in rodents and humans who are metabolically challenged (with hyperglycaemia and hyperfiltration).
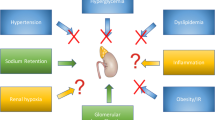
The divergent GFR findings could potentially be explained as depicted in Fig. 1. In rodents GLP-1 increases RBF and therefore GFR, but this effect is minimal in humans receiving pharmacological relevant GLP-1 doses. The GLP-1-mediated decrease in the proximal tubular reabsorption will increase the proximal hydrostatic pressure and therefore decrease GFR due to a decreased glomerular pressure gradient [39]. This effect will likely be most prominent in diabetic subjects who are often in a hyper-reabsorbing state. A potential activation of the tubuloglomerular feedback (TGF) due to GLP-1-mediated increase in sodium delivery to macula densa would also decrease GFR. A micropuncture study, however, suggests that the TGF mechanism is of minor importance when explaining renal GLP-1 effects [25]. Finally, long-term GLP-1 treatment may protect the kidney and preserve GFR, as reviewed later. The precise effect of GLP-1R activation on GFR may therefore depend on species, metabolic state, and type and time-course of the treatment.

A diagram showing potential GLP-1 regulatory pathways of GFR. Depending on the setting, GLP-1 may either increase or decrease GFR
2.6 Blood Pressure
GLP-1 or exendin-4 was administered (between 2 to 12 weeks) to obese db/db mice [26] and Dahl salt-sensitive rats [27, 40] which are prone to salt-induced hypertension. In both models the treatment markedly attenuated hypertension by reducing salt-retention especially during the initial days of treatment. Similarly, sitagliptin attenuated blood pressure rising in young prehypertensive rats [33].
Large clinical studies have shown that GLP-1R agonists often lower blood pressure in T2DM patients [7]. The antihypertensive effects are non-acute in nature [9, 13] but present before substantial weight loss [41]. The late effect probably speaks for a natriuretic mechanism rather than one of vasodilation. However, GLP-1 also acutely increases heart rate and cardiac output which independently tend to increase blood pressure [9, 13]. It is presently not settled to which degree vasodilation and natriuresis contribute to the blood pressure lowering effect.
3 Mechanism of Action
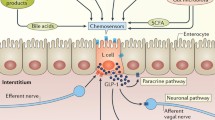
GLP-1Rs are present in the kidneys, but their exact involvement in GLP-1 actions are incompletely understood. Some evidence suggests that GLP-1 affects the kidneys by regulations of atrial natriuretic peptide (ANP) or the renin-angiotensin system (RAS); Fig. 2. The importance of neural signals is unknown, but GLP-1 does impact the denervated kidney [15]. Furthermore, the mechanisms may differ between the various types of GLP-1 therapy.

Possible pathways by which GLP-1 can mediate its renal actions. Direct stimulation of GLP-1Rs located in the kidney. By GLP-1R-mediated modulation of renal RAS activity with down-regulation of ANG2 signaling/concentration. Through GLP-1R-mediated release of ANP from the atria of the heart and secondary ANP receptor stimulation in the kidney. via the central nervous system and efferent autonomic renal nerves
3.1 Physiological Role of GLP-1 Receptors
Although seldom explicitly discussed in the literature, a plausible theory is that renal GLP-1Rs mediate effects to the structure where they are located. Since the exact receptor localization is partly unknown the theory is also partly speculative. However, in vitro experiments do to some degree support direct actions.
Thus, GLP-1Rs in close relation to NHE3 on the apical membrane of the proximal tubular cells potentially mediate the decrease in sodium reabsorption. Alternatively, GLP-1Rs localized basolaterally could signal to the apical side. GLP-1Rs on the vascular walls could mediate afferent or efferent arteriolar vasodilation depending on their position. Finally, GLP-1Rs on the glomerular endothelium, podocytes or mesangial cells could mediate protection of the filtration barrier through inhibition of inflammation and oxidative stress.
One study in rats found hyperglycaemia to decrease the expression of renal GLP-1Rs (protein and mRNA levels) whereas DPP-4 inhibitors increased the expression [42]. This may be important in the aetiology of DN, although the GLP-1R-findings probably should be interpreted with caution.
3.2 GLP-1 – ANP axis
A recent study has challenged the understanding of both GLP-1R-mediated renal and cardiovascular actions [30]. Through several knock-out mouse models it was shown that GLP-1R agonists mediate their antihypertensive effects exclusively via ANP released from atrial cardiomyocytes. Thus, ANP was found to effectuate all natriuretic and vasorelexant effects after its secretion in response to GLP-1R stimulation in the heart atria [30]. The majority of the experiments were performed with liraglutide, but native GLP-1, exendin-4 as well as meal-induced endogenous GLP-1 were capable of releasing ANP. It is notable that liraglutide only reduced blood pressure acutely in hypertensive mouse models (angiotensin II (ANG2)-induced or pressure overload) and predominantly was effective during the evening light-off periods. The observed increase in heart rate was independent of ANP [30]. The above findings define a gut-heart axis in mice and suggest that all acute physiological GLP-1-induced renal actions are secondary to ANP release.
To explore whether a GLP-1 – ANP axis exists in humans, we measured plasma proANP post-hoc in healthy males infused with GLP-1 [43]. Although natriuresis markedly increased during the GLP-1 infusion, there was no effect on proANP (reflects ANP secretion) which questions the existence of a functional important gut-heart axis in humans [43].
3.3 GLP-1 – RAS Interactions
The RAS is a key regulator of blood pressure and water-sodium balance. ANG2, the main effector hormone of the RAS, is very important in both kidney physiology and pathophysiology, e.g. by increasing NHE3 activity and the level of oxidative stress, respectively. These ANG2 effects are opposite to those of GLP-1 and a few studies suggest that this is not a coincidence:
Rodent studies have shown that GLP-1R stimulation very effectively ameliorates ANG2 induced hypertension [26, 30]. In vitro, GLP-1 prevents mesangial cell damage, by complete blocking of ANG2-induced superoxide formation, via PKA [44]. In glomerular endothelium cells, GLP-1R stimulation, via PKA, inhibits ANG2 signalling on cRaf (Ser259) and this provides a biochemical pathway which might explain how GLP-1 inhibits ANG2 actions [45]. In healthy humans infused with GLP-1, we saw a rapid decrease in plasma ANG2 concentration suggesting that circulating RAS components are also affected by GLP-1 [13].
The above findings support the idea that GLP-1 primarily affects kidney physiology through inhibition of RAS activity. Most likely, such mechanism would be mediated through renal GLP-1Rs. Interestingly, GLP-1R-mediated effects in general are compatible with decreased ANG2 signalling. E.g. the effects on insulin secretion [46], β-cell survival [47], gastric motility [48], obesity [49], hypertension, heart protection, and brain protection.
3.4 Neural Signalling
Only limited knowledge exists about GLP-1-mediated neural signalling to the kidneys. In theory, gut-released GLP-1 could affect the central nervous system through either afferent nerves or endocrine signalling. Efferent autonomic nerves to the kidney could subsequently mediate the effects of GLP-1.
In rats, GLP-1 infusion increased RBF and decreased proximal tubular sodium reabsorption in denervated kidneys. GFR did not increase in the denervated kidney which was in contrast to the innervated counterpart [15]. It is difficult to draw definitive conclusions from this one study. However, neural signalling seem to impact GLP-1 actions although the inhibition of NHE3 cannot be mediated exclusively via nerves.
3.5 Differences Between Treatments
Since DPP-4 has many substrates, DPP-4 inhibition may impact the kidneys through pathways not involving GLP-1. Indeed, alogliptin still induced natriuresis in mice lacking the GLP-1R whereas exendin-4 did not [28]. This indicates a mechanistic difference between GLP-1R agonists and DPP-4 inhibitors. However, several other renal studies have shown that DPP-4 inhibitors at least partly function through GLP-1Rs, and find in general that the renal effects of DPP-4 inhibitors are similar although weaker than those of GLP-1R agonists. If renal physiology depend on the activity of DPP-4 in the tubular brush-boarder, it may be of importance whether the specific DPP-4 inhibitor is freely filtered or not.
Similarly, if GLP-1R activation in the tubular lumen is relevant for e.g. inhibition of NHE3, the amount of GLP-1R agonist undergoing glomerular filtration is important. In this light, however, it is very interesting that although exendin-4 and liraglutide both induce natriuresis in rodents, only exendin-4 is freely filtered [50, 51].
4 GLP-1 and Kidney Pathophysiology
Since 2007 multiple studies in rodents have shown that GLP-1 based treatment protects the kidneys in models of DN, hypertensive nephropathy, and acute kidney injury (AKI). Despite these encouraging results, clinical studies are few.
4.1 Diabetic Nephropathy (DN)
The number of patients with diabetes is increasing and today diabetic nephropathy is the most common cause of ESRD [14]. DN is progressive in nature and characterized by persistent albuminuria and a declining GFR. In addition to loss of renal function, patients with DN have a marked increase in cardiovascular morbidity and mortality. Generally, prevention of DN relies on a strict control of glucose and blood pressure, and inhibition of the RAS has proven protective beyond its antihypertensive effects [52]. Despite these treatment strategies, however, there is a massive need for improved renal outcome [53].
Mechanical properties such as increased intra-glomerular pressure and hyperfiltration may partly explain the pathogenesis of DN. In addition, the importance of inflammatory cytokines, reactive oxidative species (ROS), and advanced glycated endproducts (AGE) are increasingly acknowledged. The urinary albumin excretion rate (UAER) is established as a valuable marker of the risk of disease progression.
4.2 GLP-1 and DN: Preclinical Studies
The first study exploring the protective effects of GLP-1R agonism in the kidneys was published in 2007 [38]. Eight weeks of exendin-4 treatment in T2DM db/db mice decreased albuminuria and oxidative stress levels, and diminished histological signs of DN [38]. However, the metabolic status also improved compared with placebo which makes it difficult to draw conclusions on causality.
To show that GLP-1 treatment protects the kidneys beyond metabolic improvements, studies were undertaken in streptozotocin (STZ)-induced type 1 diabetic rodents. STZ destroys the pancreatic beta cells and glycaemic levels are therefore comparable between GLP-1 and placebo treatment arms. In STZ-rodents, the renal effects of 2 to 26 weeks of exendin-4 [19, 45, 54], 4 weeks of liraglutide [55], 24 weeks of vildagliptin [42], 12 weeks of linagliptin [56] and 8 weeks of a long-acting vildagliptin analogue [57] were investigated. In summary, the treatments showed remarkable improvements in UAERs, ROS, inflammatory and fibrogenic cytokines, and renal histopathological changes. The effects generally seemed stronger with the GLP-1R agonists than the DPP-4 inhibitors but were otherwise comparable. One study showed that the treatment was effective on top of ANG2 receptor blocker (ARB) treatment [56] and another that AGEs effects were suppressed [54]. The treatment prevented the initial diabetic hyperfiltration [17, 19] and it preserved GFR in long-term follow-up [42].
Similar improvements were observed during 4 weeks of liraglutide treatment in a mouse model of progressive DN while GLP-1R knock-out mice had more ROS, albuminuria, and mesangial expansion despite comparable glycaemic levels [17].
In some of the above studies there were small but significant GLP-1-mediated improvements in blood pressure or body weight. Since these changes independently will improve renal outcome they represent limitations to the studies.
Several studies have shown that GLP-1 treatment generally improves endothelial cell function, e.g. by reducing ROS formation [58, 59]. In mouse glomerular endothelium, exendin-4 was shown to inhibit ANG2 signalling which may prevent the toxic effects related to increased ANG2 levels in hyperglycaemia [45]. In human cultured mesangial cells, GLP-1R agonists reduced AGE-induced ROS [60], reduced mediators of fibrosis [61], and completely blocked ANG2-induced ROS [44].
4.3 GLP-1 and DN: Clinical Studies
Saxagliptin improved the urinary albumin excretion rate in T2DM patients in a large randomized placebo-controlled trial during a median of 2.1 years follow-up, but also improved glycaemic control [62].
In a parallel study, 31 microalbuminuric T2DM patients were randomly assigned to exendin-4 or glimepiride treatment for 16 weeks on top of their metformin treatment [63]. Exendin-4 significantly reduced albuminuria and other urinary markers of renal injury compared with glimepiride. Glucose levels were comparable between groups but exendin-4 significantly lowered BMI and systolic blood pressure [63].
A non-randomized cross-over study investigated the effect of 4 weeks alogliptin and sitagliptin treatment in 12 microalbuminuric T2DM patients all treated with ARBs [64]. The authors concluded that the stronger DPP-4 inhibitor (alogliptin) decreased albuminuria and markers of oxidative stress compared with the weaker (sitagliptin) despite comparable glycaemic levels [64].
Liraglutide was administered for 12 months to 23 patients with T2DM and overt DN in an uncontrolled study [65]. The usual treatment was continued which for all patients included ARBs. GFR (creatinine) was measured repeatedly from 1 to 3 years before and during the intervention. Liraglutide significantly decreased the GFR decline rate and the UAER. The study has several limitations (no control group, metabolic improvements, and problematical GFR decline rate calculations) but does provide a first suggestion of GLP-1-mediated GFR preservation in humans [65].
4.4 Other models of chronic kidney disease
In a non-diabetic, nitric oxide-depleted rat model of hypertensive nephropathy, 8 weeks of sitagliptin treatment markedly attenuated kidney injury, but also decreased blood pressure making interpretation difficult [66]. Sitagliptin for 8 weeks attenuated renal dysfunction after a surgical 5/6 renal mass reduction in non-diabetic rats [67]. The above could suggest a role for GLP-1 treatment in non-diabetes-related chronic kidney disease.
4.5 GLP-1 and Acute Kidney Injury (AKI)
AKI is a clinical syndrome characterized by a rapid loss of kidney excretory function and accumulation of waste products [68]. The aetiology is multifactorial and includes ischemia-reperfusion injury, nephrotoxic agents, and sepsis. Globally, AKI is a major concern with an increasing incidence and the long-term complications include ESRD and death. Apart from correction of the initiating cause, there are currently few therapeutic options available [69]. The pathogenesis of AKI is complex and incompletely understood, but the generation of ROS, inflammatory reactions and increased apoptosis are believed to play key roles. Thus, it is rational to hypothesize that GLP-1 based treatment can attenuate the severity of AKI and this has currently been tested in two different models of AKI in rodents. AKI induced by surgically induced ischemia-reperfusion and induced by cisplastin, which is a highly nephrotoxic chemotherapeutic agent. Rat models of ischemia-reperfusion injury were pre-treated with exendin-4 [70], vildagliptin [71], or sitagliptin [72] or post-treated with exendin-4 or sitagliptin [73]. All studies found that GLP-1 therapy significantly reduced the severity of AKI as judged by creatinine level, renal histopathological changes, ROS, inflammation, and apoptosis.
Cisplastin-induced renal injury and apoptosis were attenuated by alogliptin and exendin-4 [74]. Furthermore, suppression of the GLP-1R with small interfering RNA partly reversed the beneficial effects indicating that DPP-4 inhibitors act on AKI by increasing GLP-1 levels [74]. Although GLP-1 based treatments look promising in the prevention of AKI in rodents, there are currently no human studies reporting on the topic.
5 Renal Clearance and Safety
When GLP-1 therapy is administered to patients with reduced kidney function, it is important whether the drug is cleared by the kidneys. Although GLP-1R agonists generally have a good renal safety profile, there are case reports of AKI induced by the treatment. The combined effect of GLP-1 therapy and SGLT2 inhibitors in the proximal tubule is perhaps an overlooked issue.
5.1 Renal Clearance and Safety in Renal Impairment
The short half-life of GLP-1 is primarily due to rapid enzymatic degradation [3], but the hormone is also cleared by the kidneys and believed to be freely filtered. The renal extraction fraction of GLP-1 was ~50 % in anaesthetised pigs [51] which clearly exceeds the amount undergoing glomerular filtration. We found no intact GLP-1 in the urine of healthy males infused with GLP-1 [13], which illustrates the effectiveness of the brush-border epithelium.
Exendin-4 is cleared exclusively via the kidneys in a rate equal to glomerular filtration in pigs [51]. The clearance of exendin-4 is markedly affected by the severity of renal impairment and was decreased 84 % in patients with ESRD compared to controls [75]. However, exendin-4 was well tolerated in both mild and moderate renal impairment [75]. Exendin-4 is not recommended if CrCl < 30 mL/min or for the prolonged-release suspension if CrCl < 50 mL/min (European Medicine Agency, EMA).
Liraglutide is not cleared by the kidneys, and is completely degraded within the body [50].
Mild renal impairment has no effect on the safety or efficacy of liraglutide [76, 77]. Liraglutide pharmacokinetics and safety are currently being tested in patients with ESRD [78]. At present, liraglutide is not recommended in patients with CrCl < 60 mL/min due to lack of long-term therapeutic experience (EMA).
Lixisenatide is cleared via glomerular filtration and is not recommended if CrCl < 30 mL/min, and should be used with caution if CrCl < 50 mL/min (EMA).
Sitagliptin, vildagliptin, saxagliptin, and alogliptin are primarily cleared via renal excretion whereas linagliptin primarily is subject to hepatic clearance [79]. Linagliptin is approved for all stages of renal impairment with no dose adjustments required (EMA). Sitagliptin, vildagliptin, and saxagliptin can be used in renal impairment with dosing-adjustments according to guidelines (EMA).
5.2 AKI Induced by GLP-1R Agonists
Case reports have described an acute loss of kidney function associated with exendin-4 [80] and liraglutide treatment [81]. In the great majority of cases the problem seems to be prerenal failure due to volume-depletion. Side-effects of GLP-1R agonists include nausea and vomiting which in severe cases can induce hypovolaemia. Osmotic diuresis during hyperglycaemia and treatment with other diuretic, probably represent additional risk-factors. The topic has recently been reviewed in details [82]. In light of the frequency of GLP-1R agonist prescriptions it does not seem to be a major issue.
5.3 GLP-1 and SGLT2 Inhibitors
A novel therapeutic option in diabetes treatment is SGLT2 inhibitors [83]. Like GLP-1R agonists, these are potent proximal diuretics. In STZ rats, SGLT2 inhibition with dapagliflozin decreased GFR ~20 % acutely and ~15 % chronically (inulin) [84]. A recent study in type 1 diabetes patients showed that 8 weeks of empagliflozin decreased GFR ~19 % (inulin) in a group with baseline hyperfiltration but did not affect those with normal baseline GFR [85]. The mechanism behind the decrease in GFR is probably similar to that of GLP-1 and may involve a decreased glomerular hydrostatic pressure gradient and increased TGF. The combination of SGLT2 inhibitors and GLP-1 therapy has not been thoroughly investigated. Hypothetically, the combined inhibitory effects on SGLT2 and NHE3 in the proximal tubule could act synergistically to decrease proximal reabsorption and GFR. The effect will likely vanish along with a decreasing GFR, but at present, treatment combinations with especially GLP-1R agonists and SGLT2 inhibitors should probably be done with caution.
6 Perspective and Future Direction
From an evolutionary point of view, it may seem surprising that GLP-1 links incretin effects together with natriuresis and anti-oxidative renal effects. The fact that GLP-1 is released in response to food ingestion, however, allows the kidney to differentiate its response to postprandial hyperglycaemia and stress-induced hyperglycaemia. While increased sodium reabsorption, oxidative stress, and inflammation may be useful in stressful situations it is not necessarily healthy subsequent to meal ingestions. Furthermore, GLP-1 might prepare the kidney for the increased sodium load from the meal.
Although many physiological renal effects of GLP-1 are well documented in rodents, the underlying mechanisms are not fully elucidated. Newly elucidated pathways of renal GLP-1-signalling (e.g. via ANP or ANG2) may be applicable to other organs as well. Human studies exploring renal GLP-1 physiology are few, and moreover, there may be differences to the preclinical studies relating to both species and GLP-1-dosing.
Albeit still in the confirmatory state, it is highly interesting that GLP-1 therapy seems to protect the heart [10] and brain [11]. Similarly to the kidneys, the protection seems at least partly mediated through improvements in endothelial function.
At present, several rodent studies have shown that GLP-1 therapy attenuates DN and AKI. A few clinical studies, found GLP-1 therapy to improve surrogate markers of DN and one uncontrolled study additionally found that the therapy preserved GFR. Although the results are encouraging the clinical studies all have limitations which compromises interpretation.
DN and AKI are major global health concerns, and therefore it is crucial that potentially powerful treatment options are thoroughly investigated. The task, however, is not trivial.
In the last 20 years it has taken several large randomized studies to conclude that RAS inhibition is renoprotective beyond antihypertensive effects [52]. Likewise, it has to be validated that GLP-1 therapy is renoprotective beyond the antihypertensive, antidiabetic, and weight-loss effects. Furthermore, a potential treatment has to prove effective either as add on or alternative to RAS inhibition. With an optimally treated control group it will require large studies and several years of follow-up to draw definitive conclusions on changes in GFR decline rates.
Studies exploring the possible benefits of GLP-1 treatment in AKI require shorter follow-up and share some similarities with studies investigating GLP-1 treatment in the prevention of myocardial reperfusion injury [86]. Surprisingly, no clinical studies with GLP-1 therapy and AKI have been reported.
In summary, the incretin hormone, GLP-1, inhibits NHE3 activity in the proximal tubule and thereby induces natriuresis. GLP-1 can affect GFR and RBF, but does not in healthy humans. Renal GLP-1R localization is partly unknown, and alternative signalling pathways may be via ANP, ANG2, or nerves. GLP-1 therapy decreases the level of inflammation and oxidative stress and prevents DN and AKI in rodents. Clinical studies validating these encouraging results are highly warranted.
Abbreviations
- ANG2:
-
angiotensin II
- AKI:
-
acute kidney injury
- ANP:
-
atrial natriuretic peptide
- ARB:
-
angiotensin II receptor blocker
- CrCl:
-
creatinine clearance
- DN:
-
diabetic nephropathy
- DPP-4:
-
dipeptidyl peptidase IV
- EMA:
-
European Medicine Agency
- ESRD:
-
end-stage renal disease
- GLP-1:
-
glucagon-like peptide-1
- GLP-1R:
-
glucagon-like peptide-1 receptor
- GFR:
-
glomerular filtration rate
- mRNA:
-
messenger ribonucleic acid
- NHE3:
-
Na+/H+ exchanger isoform 3
- PKA:
-
protein kinase A
- RAS:
-
renin-angiotensin system
- RBF:
-
renal blood flow
- ROS:
-
reactive oxygen species
- SGLT2:
-
sodium-glucose linked transporter 2
- STZ:
-
streptozotocin
- T2DM:
-
type 2 diabetes mellitus
- TGF:
-
tubuloglomerular feedback
References
Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87(4):1409–39.
Vilsboll T, Agerso H, Krarup T, Holst JJ. Similar elimination rates of glucagon-like peptide-1 in obese type 2 diabetic patients and healthy subjects. J Clin Endocrinol Metab. 2003;88(1):220–4. doi:10.1210/jc.2002-021053.
Meier JJ, Nauck MA, Kranz D, Holst JJ, Deacon CF, Gaeckler D, et al. Secretion, degradation, and elimination of glucagon-like peptide 1 and gastric inhibitory polypeptide in patients with chronic renal insufficiency and healthy control subjects. Diabetes. 2004;53(3):654–62.
Bullock BP, Heller RS, Habener JF. Tissue distribution of messenger ribonucleic acid encoding the rat glucagon-like peptide-1 receptor. Endocrinology. 1996;137(7):2968–78. doi:10.1210/en.137.7.2968.
Körner M, Stöckli M, Waser B, Reubi JC. GLP-1 Receptor Expression in Human Tumors and Human Normal Tissues: Potential for In Vivo Targeting. J Nucl Med. 2007;48(5):736–43. doi:10.2967/jnumed.106.038679.
Vilsboll T, Christensen M, Junker AE, Knop FK, Gluud LL. Effects of glucagon-like peptide-1 receptor agonists on weight loss: systematic review and meta-analyses of randomised controlled trials. Bmj. 2012;344:d7771.
Wang B, Zhong J, Lin H, Zhao Z, Yan Z, He H, et al. Blood pressure-lowering effects of GLP-1 receptor agonists exenatide and liraglutide: a meta-analysis of clinical trials. Diabetes Obes Metab. 2013;15(8):737–49. doi:10.1111/dom.12085.
Nauck MA, Niedereichholz U, Ettler R, Holst JJ, Orskov C, Ritzel R, et al. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. The American journal of physiology. 1997;273(5 Pt 1):E981–8.
Mendis B, Simpson E, MacDonald I, Mansell P. Investigation of the haemodynamic effects of exenatide in healthy male subjects. Br J Clin Pharmacol. 2012;74(3):437–44. doi:10.1111/j.1365-2125.2012.04214.x.
Ussher JR, Drucker DJ. Cardiovascular biology of the incretin system. Endocr Rev. 2012;33(2):187–215. doi:10.1210/er.2011-1052.
Holst JJ, Burcelin R, Nathanson E. Neuroprotective properties of GLP-1: theoretical and practical applications. Current medical research and opinion. 2011;27(3):547–58. doi:10.1185/03007995.2010.549466.
Gutzwiller JP, Tschopp S, Bock A, Zehnder CE, Huber AR, Kreyenbuehl M et al. Glucagon-like peptide 1 induces natriuresis in healthy subjects and in insulin-resistant obese men. J Clin Endocrinol Metab. 2004;89 (6):3055–61. doi:10.1210/jc.2003-03140389/6/3055 [pii]
Skov J, Dejgaard A, Frokiaer J, Holst JJ, Jonassen T, Rittig S, et al. Glucagon-Like Peptide-1 (GLP-1): Effect on Kidney Hemodynamics and Renin-Angiotensin-Aldosterone System in Healthy Men. J Clin Endocrinol Metab. 2013;98(4):E664–71. doi:10.1210/jc.2012-3855.
Ritz E, Rychlik I, Locatelli F, Halimi S. End-stage renal failure in type 2 diabetes: A medical catastrophe of worldwide dimensions. Am J Kidney Dis. 1999;34(5):795–808. doi:10.1016/s0272-6386(99)70035-1.
Moreno C, Mistry M, Roman RJ. Renal effects of glucagon-like peptide in rats. Eur J Pharmacol. 2002;434(3):163–7.
Crajoinas RO, Oricchio FT, Pessoa TD, Pacheco BPM, Lessa LMA, Malnic G, et al. Mechanisms mediating the diuretic and natriuretic actions of the incretin hormone glucagon-like peptide-1. Am J Physiol Ren Physiol. 2011;301(2):F355–F63. doi:10.1152/ajprenal.00729.2010.
Fujita H, Morii T, Fujishima H, Sato T, Shimizu T, Hosoba M et al. The protective roles of GLP-1R signaling in diabetic nephropathy: possible mechanism and therapeutic potential. Kidney Int. 2013. doi:10.1038/ki.2013.427.
Carraro-Lacroix LR, Malnic G, Girardi ACC. Regulation of Na+/H + exchanger NHE3 by glucagon-like peptide 1 receptor agonist exendin-4 in renal proximal tubule cells. Am J Physiol Ren Physiol. 2009;297(6):F1647–F55. doi:10.1152/ajprenal.00082.2009.
Kodera R, Shikata K, Kataoka HU, Takatsuka T, Miyamoto S, Sasaki M, et al. Glucagon-like peptide-1 receptor agonist ameliorates renal injury through its anti-inflammatory action without lowering blood glucose level in a rat model of type 1 diabetes. Diabetologia. 2011;54(4):965–78. doi:10.1007/s00125-010-2028-x.
Schlatter P, Beglinger C, Drewe J, Gutmann H. Glucagon-like peptide 1 receptor expression in primary porcine proximal tubular cells. Regul Pept. 2007;141(1–3):120–8. doi:10.1016/j.regpep.2006.12.016.
Pezeshki A, Muench GP, Chelikani PK. Short communication: expression of peptide YY, proglucagon, neuropeptide Y receptor Y2, and glucagon-like peptide-1 receptor in bovine peripheral tissues. J Dairy Sci. 2012;95(9):5089–94. doi:10.3168/jds.2011-5311.
Pyke C, Knudsen LB. The Glucagon-Like Peptide-1 Receptor—or Not? Endocrinology. 2013;154(1):4–8. doi:10.1210/en.2012-2124.
Panjwani N, Mulvihill EE, Longuet C, Yusta B, Campbell JE, Brown TJ, et al. GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE (−/−) mice. Endocrinology. 2013;154(1):127–39. doi:10.1210/en.2012-1937.
Yu M, Moreno C, Hoagland KM, Dahly A, Ditter K, Mistry M, et al. Antihypertensive effect of glucagon-like peptide 1 in Dahl salt-sensitive rats. J Hypertens. 2003;21(6):1125–35.
Thomson SC, Kashkouli A, Singh P. Glucagon-like peptide-1 receptor stimulation increases GFR and suppresses proximal reabsorption in the rat. Am J Physiol-Renal Physiol. 2013;304(2):F137–F44. doi:10.1152/ajprenal.00064.2012.
Hirata K, Kume S. Araki S-i, Sakaguchi M, Chin-Kanasaki M, Isshiki K et al. Exendin-4 has an anti-hypertensive effect in salt-sensitive mice model. Biochemical and Biophysical Research. Communications. 2009;380(1):44–9.
Liu Q, Adams L, Broyde A, Fernandez R, Baron A, Parkes D. The exenatide analogue AC3174 attenuates hypertension, insulin resistance, and renal dysfunction in Dahl salt-sensitive rats. Cardiovasc Diabetol. 2010;9(1):32.
Rieg T, Gerasimova M, Murray F, Masuda T, Tang T, Rose M, et al. Natriuretic effect by exendin-4, but not the DPP-4 inhibitor alogliptin, is mediated via the GLP-1 receptor and preserved in obese type 2 diabetic mice. Am J Physiol Renal Physiol. 2012;303(7):F963–71. doi:10.1152/ajprenal.00259.2012.
Larsen PJ, Fledelius C, Knudsen LB, Tang-Christensen M. Systemic administration of the long-acting GLP-1 derivative NN2211 induces lasting and reversible weight loss in both normal and obese rats. Diabetes. 2001;50(11):2530–9.
Kim M, Platt MJ, Shibasaki T, Quaggin SE, Backx PH, Seino S, et al. GLP-1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat Med. 2013;19(5):567–75. doi:10.1038/nm.3128.
Girardi AC, Fukuda LE, Rossoni LV, Malnic G, Reboucas NA. Dipeptidyl peptidase IV inhibition downregulates Na + − H + exchanger NHE3 in rat renal proximal tubule. Am J Physiol Renal Physiol. 2008;294(2):F414–22. doi:10.1152/ajprenal.00174.2007.
Gutzwiller JP, Hruz P, Huber AR, Hamel C, Zehnder C, Drewe J, et al. Glucagon-like peptide-1 is involved in sodium and water homeostasis in humans. Digestion. 2006;73(2–3):142–50. doi:10.1159/000094334.
Pacheco BP, Crajoinas RO, Couto GK, Davel AP, Lessa LM, Rossoni LV, et al. Dipeptidyl peptidase IV inhibition attenuates blood pressure rising in young spontaneously hypertensive rats. J Hypertens. 2011;29(3):520–8. doi:10.1097/HJH.0b013e328341939d.
Thomsen K. Lithium Clearance: A New Method for Determining Proximal and Distal Tubular Reabsorption of Sodium and Water. Nephron. 1984;37(4):217–23.
Girardi AC, Di Sole F. Deciphering the mechanisms of the Na+/H + exchanger-3 regulation in organ dysfunction. American journal of physiology Cell physiology. 2012;302(11):C1569–87. doi:10.1152/ajpcell.00017.2012.
Girardi AC, Knauf F, Demuth HU, Aronson PS. Role of dipeptidyl peptidase IV in regulating activity of Na+/H + exchanger isoform NHE3 in proximal tubule cells. American journal of physiology Cell physiology. 2004;287(5):C1238–45. doi:10.1152/ajpcell.00186.2004.
Girardi AC, Degray BC, Nagy T, Biemesderfer D, Aronson PS. Association of Na (+)-H (+) exchanger isoform NHE3 and dipeptidyl peptidase IV in the renal proximal tubule. J Biol Chem. 2001;276(49):46671–7. doi:10.1074/jbc.M106897200.
Park CW, Kim HW, Ko SH, Lim JH, Ryu GR, Chung HW, et al. Long-Term Treatment of Glucagon-Like Peptide-1 Analog Exendin-4 Ameliorates Diabetic Nephropathy through Improving Metabolic Anomalies in db/dB Mice. J Am Soc Nephrol. 2007;18(4):1227–38. doi:10.1681/asn.2006070778.
Persson P, Hansell P, Palm F. Tubular reabsorption and diabetes-induced glomerular hyperfiltration. Acta Physiol (Oxf). 2010;200(1):3–10. doi:10.1111/j.1748-1716.2010.02147.x.
Yu M, Moreno C, Hoagland KM, Dahly A, Ditter K, Mistry M, et al. Antihypertensive effect of glucagon-like peptide 1 in Dahl salt-sensitive rats. J Hypertens. 2003;21(6):1125–35. doi:10.1097/01.hjh.0000059046.65882.49.
Varanasi A, Chaudhuri A, Dhindsa S, Arora A, Lohano T, Vora MR, et al. Durability of effects of exenatide treatment on glycemic control, body weight, systolic blood pressure, C-reactive protein, and triglyceride concentrations. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2011;17(2):192–200. doi:10.4158/ep10199.or.
Liu WJ, Xie SH, Liu YN, Kim W, Jin HY, Park SK, et al. Dipeptidyl Peptidase IV Inhibitor Attenuates Kidney Injury in Streptozotocin-Induced Diabetic Rats. J Pharmacol Exp Ther. 2012;340(2):248–55. doi:10.1124/jpet.111.186866.
Skov J, Holst JJ, Goetze JP, Frokiaer J, Christiansen JS. Glucagon-like peptide-1: effect on pro-atrial natriuretic peptide in healthy males. Endocrine connections. 2013. doi:10.1530/ec-13-0087.
Ishibashi Y, Matsui T, Ojima A, Nishino Y, Nakashima S, Maeda S, et al. Glucagon-like peptide-1 inhibits angiotensin II-induced mesangial cell damage via protein kinase A. Microvasc Res. 2012;84(3):395–8. doi:10.1016/j.mvr.2012.06.008.
Mima A, Hiraoka-Yamomoto J, Li Q, Kitada M, Li C, Geraldes P, et al. Protective effects of GLP-1 on glomerular endothelium and its inhibition by PKCbeta activation in diabetes. Diabetes. 2012;61(11):2967–79. doi:10.2337/db11-1824.
van der Zijl NJ, Moors CC, Goossens GH, Hermans MM, Blaak EE, Diamant M. Valsartan improves {beta}-cell function and insulin sensitivity in subjects with impaired glucose metabolism: a randomized controlled trial. Diabetes Care. 2011;34(4):845–51. doi:10.2337/dc10-2224.
Wang HW, Mizuta M, Saitoh Y, Noma K, Ueno H, Nakazato M. Glucagon-like peptide-1 and candesartan additively improve glucolipotoxicity in pancreatic beta-cells. Metabolism. 2011;60(8):1081–9. doi:10.1016/j.metabol.2010.11.004.
Lu HL, Wang ZY, Huang X, Han YF, Wu YS, Guo X, et al. Excitatory regulation of angiotensin II on gastric motility and its mechanism in guinea pig. Regul Pept. 2011;167(2–3):170–6. doi:10.1016/j.regpep.2011.01.004.
Fogari R, Derosa G, Zoppi A, Rinaldi A, Lazzari P, Fogari E, et al. Comparison of the effects of valsartan and felodipine on plasma leptin and insulin sensitivity in hypertensive obese patients. Hypertension research : official journal of the Japanese Society of Hypertension. 2005;28(3):209–14. doi:10.1291/hypres.28.209.
Malm-Erjefalt M, Bjornsdottir I, Vanggaard J, Helleberg H, Larsen U, Oosterhuis B, et al. Metabolism and excretion of the once-daily human glucagon-like peptide-1 analog liraglutide in healthy male subjects and its in vitro degradation by dipeptidyl peptidase IV and neutral endopeptidase. Drug metabolism and disposition: the biological fate of chemicals. 2010;38(11):1944–53. doi:10.1124/dmd.110.034066.
Simonsen L, Holst JJ, Deacon CF. Exendin-4, but not glucagon-like peptide-1, is cleared exclusively by glomerular filtration in anaesthetised pigs. Diabetologia. 2006;49(4):706–12. doi:10.1007/s00125-005-0128-9.
Vejakama P, Thakkinstian A, Lertrattananon D, Ingsathit A, Ngarmukos C, Attia J. Reno-protective effects of renin-angiotensin system blockade in type 2 diabetic patients: a systematic review and network meta-analysis. Diabetologia. 2012;55(3):566–78.
Rossing P, de Zeeuw D. Need for better diabetes treatment for improved renal outcome. Kidney Int Suppl. 2011;120:S28–32. doi:10.1038/ki.2010.513.
Ojima A, Ishibashi Y, Matsui T, Maeda S, Nishino Y, Takeuchi M et al. Glucagon-Like Peptide-1 Receptor Agonist Inhibits Asymmetric Dimethylarginine Generation in the Kidney of Streptozotocin-Induced Diabetic Rats by Blocking Advanced Glycation End Product–Induced Protein Arginine Methyltranferase-1 Expression. The American journal of pathology. 2013;182 (1):132–41. doi:http://dx.doi.org/10.1016/j.ajpath.2012.09.016.
Hendarto H, Inoguchi T, Maeda Y, Ikeda N, Zheng J, Takei R, et al. GLP-1 analog liraglutide protects against oxidative stress and albuminuria in streptozotocin-induced diabetic rats via protein kinase A-mediated inhibition of renal NAD (P)H oxidases. Metabolism. 2012;61(10):1422–34. doi:10.1016/j.metabol.2012.03.002.
Alter ML, Ott IM, von Websky K, Tsuprykov O, Sharkovska Y, Krause-Relle K, et al. DPP-4 inhibition on top of angiotensin receptor blockade offers a new therapeutic approach for diabetic nephropathy. Kidney & blood pressure research. 2012;36(1):119–30. doi:10.1159/000341487.
Kodera R, Shikata K, Takatsuka T, Oda K, Miyamoto S, Kajitani N et al. Dipeptidyl peptidase-4 inhibitor ameliorates early renal injury through its anti-inflammatory action in a rat model of type 1 diabetes. Biochem Biophys Res Commun. 2013. doi:10.1016/j.bbrc.2013.12.049.
Shiraki A, Oyama J, Komoda H, Asaka M, Komatsu A, Sakuma M, et al. The glucagon-like peptide 1 analog liraglutide reduces TNF-alpha-induced oxidative stress and inflammation in endothelial cells. Atherosclerosis. 2012;221(2):375–82. doi:10.1016/j.atherosclerosis.2011.12.039.
Liu L, Liu J, Wong WT, Tian XY, Lau CW, Wang YX, et al. Dipeptidyl peptidase 4 inhibitor sitagliptin protects endothelial function in hypertension through a glucagon-like peptide 1-dependent mechanism. Hypertension. 2012;60(3):833–41. doi:10.1161/hypertensionaha.112.195115.
Ishibashi Y, Nishino Y, Matsui T, Takeuchi M, Yamagishi S. Glucagon-like peptide-1 suppresses advanced glycation end product-induced monocyte chemoattractant protein-1 expression in mesangial cells by reducing advanced glycation end product receptor level. Metabolism. 2011;60(9):1271–7. doi:10.1016/j.metabol.2011.01.010.
Li W, Cui M, Wei Y, Kong X, Tang L, Xu D. Inhibition of the expression of TGF-beta1 and CTGF in human mesangial cells by exendin-4, a glucagon-like peptide-1 receptor agonist. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2012;30(3):749–57. doi:10.1159/000341454.
Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, et al. Saxagliptin and Cardiovascular Outcomes in Patients with Type 2 Diabetes Mellitus. N Engl J Med. 2013;369(14):1317–26. doi:10.1056/NEJMoa1307684.
Zhang H, Zhang X, Hu C, Lu W. Exenatide reduces urinary transforming growth factor-beta1 and type IV collagen excretion in patients with type 2 diabetes and microalbuminuria. Kidney & blood pressure research. 2012;35(6):483–8. doi:10.1159/000337929.
Fujita H, Taniai H, Murayama H, Ohshiro H, Hayashi H, Sato S et al. DPP-4 inhibition with alogliptin on top of angiotensin II type 1 receptor blockade ameliorates albuminuria via up-regulation of SDF-1alpha in type 2 diabetic patients with incipient nephropathy. Endocrine journal. 2013.
Imamura S, Hirai K, Hirai A. The glucagon-like Peptide-1 receptor agonist, liraglutide, attenuates the progression of overt diabetic nephropathy in type 2 diabetic patients. Tohoku J Exp Med. 2013;231(1):57–61.
Abd El Motteleb DM, Elshazly SM. Renoprotective effect of sitagliptin against hypertensive nephropathy induced by chronic administration of l-NAME in rats: Role of GLP-1 and GLP-1 receptor. Eur J Pharmacol. 2013;720(1–3):158–65. doi:10.1016/j.ejphar.2013.10.033.
Joo KW, Kim S, Ahn SY, Chin HJ, Chae DW, Lee J, et al. Dipeptidyl peptidase IV inhibitor attenuates kidney injury in rat remnant kidney. BMC Nephrol. 2013;14:98. doi:10.1186/1471-2369-14-98.
Bellomo R, Kellum JA, Ronco C. Acute kidney injury. Lancet. 2012;380(9843):756–66. doi:10.1016/s0140-6736(11)61454-2.
Lameire NH, Bagga A, Cruz D, De Maeseneer J, Endre Z, Kellum JA, et al. Acute kidney injury: an increasing global concern. Lancet. 2013;382(9887):170–9. doi:10.1016/s0140-6736(13)60647-9.
Yang H, Li H, Wang Z, Shi Y, Jiang G, Zeng F. Exendin-4 ameliorates renal ischemia-reperfusion injury in the rat. The Journal of surgical research. 2013;185(2):825–32. doi:10.1016/j.jss.2013.06.042.
Glorie LL, Verhulst A, Matheeussen V, Baerts L, Magielse J, Hermans N, et al. DPP4 inhibition improves functional outcome after renal ischemia-reperfusion injury. Am J Physiol Renal Physiol. 2012;303(5):F681–8. doi:10.1152/ajprenal.00075.2012.
Vaghasiya J, Sheth N, Bhalodia Y, Manek R. Sitagliptin protects renal ischemia reperfusion induced renal damage in diabetes. Regul Pept. 2011;166(1–3):48–54. doi:10.1016/j.regpep.2010.08.007.
Chen YT, Tsai TH, Yang CC, Sun CK, Chang LT, Chen HH, et al. Exendin-4 and sitagliptin protect kidney from ischemia-reperfusion injury through suppressing oxidative stress and inflammatory reaction. J Transl Med. 2013;11(1):270. doi:10.1186/1479-5876-11-270.
Katagiri D, Hamasaki Y, Doi K, Okamoto K, Negishi K, Nangaku M et al. Protection of Glucagon-Like Peptide-1 in Cisplatin-Induced Renal Injury Elucidates Gut-Kidney Connection. J Am Soc Nephrol. 2013. doi:10.1681/asn.2013020134.
Linnebjerg H, Kothare PA, Park S, Mace K, Reddy S, Mitchell M, et al. Effect of renal impairment on the pharmacokinetics of exenatide. Br J Clin Pharmacol. 2007;64(3):317–27. doi:10.1111/j.1365-2125.2007.02890.x.
Davidson JA, Brett J, Falahati A, Scott D. Mild renal impairment and the efficacy and safety of liraglutide. Endocrine practice : official journal of the American College of Endocrinology and the American Association of Clinical Endocrinologists. 2011;17(3):345–55. doi:10.4158/ep10215.ra.
Jacobsen LV, Hindsberger C, Robson R, Zdravkovic M. Effect of renal impairment on the pharmacokinetics of the GLP-1 analogue liraglutide. Br J Clin Pharmacol. 2009;68(6):898–905. doi:10.1111/j.1365-2125.2009.03536.x.
Idorn T, Knop FK, Jorgensen M, Jensen T, Resuli M, Hansen PM et al. Safety and efficacy of liraglutide in patients with type 2 diabetes and end-stage renal disease: protocol for an investigator-initiated prospective, randomised, placebo-controlled, double-blinded, parallel intervention study. BMJ Open. 2013;3 (4). doi:10.1136/bmjopen-2013-002764.
Scheen AJ. Pharmacokinetics of dipeptidylpeptidase-4 inhibitors. Diabetes Obes Metab. 2010;12(8):648–58. doi:10.1111/j.1463-1326.2010.01212.x.
Weise WJ, Sivanandy MS, Block CA, Comi RJ. Exenatide-associated ischemic renal failure. Diabetes Care. 2009;32(2):e22–3. doi:10.2337/dc08-1309.
Narayana SK, Talab SK, Elrishi MA. Liraglutide-induced acute kidney injury. Practical Diabetes. 2012;29(9):380–2. doi:10.1002/pdi.1727.
Filippatos TD, Elisaf MS. Effects of glucagon-like peptide-1 receptor agonists on renal function. World journal of diabetes. 2013;4(5):190–201. doi:10.4239/wjd.v4.i5.190.
Abdul-Ghani MA, Norton L, Defronzo RA. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr Rev. 2011;32(4):515–31. doi:10.1210/er.2010-0029.
Thomson SC, Rieg T, Miracle C, Mansoury H, Whaley J, Vallon V, et al. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol. 2012;302(1):R75–83. doi:10.1152/ajpregu.00357.2011.
Cherney DZ, Perkins BA, Soleymanlou N, Maione M, Lai V, Lee A et al. The Renal Hemodynamic Effect of SGLT2 Inhibition in Patients with Type 1 Diabetes. Circulation. 2013. doi:10.1161/circulationaha.113.005081.
Lonborg J, Vejlstrup N, Kelbaek H, Botker HE, Kim WY, Mathiasen AB, et al. Exenatide reduces reperfusion injury in patients with ST-segment elevation myocardial infarction. Eur Heart J. 2012;33(12):1491–9. doi:10.1093/eurheartj/ehr309.
Conflict of interest
J.S. has a PhD fellowship in a joint collaboration between Aarhus University Hospital and Novo Nordisk.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Skov, J. Effects of GLP-1 in the Kidney. Rev Endocr Metab Disord 15, 197–207 (2014). https://doi.org/10.1007/s11154-014-9287-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-014-9287-7