
Abstract
Obesity and its associated metabolic disorders are growing health concerns in the US and worldwide. In the US alone, more than two-thirds of the adult population is classified as either overweight or obese [1], highlighting the need to develop new, effective treatments for these conditions. Whereas the hormone oxytocin is well known for its peripheral effects on uterine contraction during parturition and milk ejection during lactation, release of oxytocin from somatodendrites and axonal terminals within the central nervous system (CNS) is implicated in both the formation of prosocial behaviors and in the control of energy balance. Recent findings demonstrate that chronic administration of oxytocin reduces food intake and body weight in diet-induced obese (DIO) and genetically obese rodents with impaired or defective leptin signaling. Importantly, chronic systemic administration of oxytocin out to 6 weeks recapitulates the effects of central administration on body weight loss in DIO rodents at doses that do not result in the development of tolerance. Furthermore, these effects are coupled with induction of Fos (a marker of neuronal activation) in hindbrain areas (e.g. dorsal vagal complex (DVC)) linked to the control of meal size and forebrain areas (e.g. hypothalamus, amygdala) linked to the regulation of food intake and body weight. This review assesses the potential central and peripheral targets by which oxytocin may inhibit body weight gain, its regulation by anorexigenic and orexigenic signals, and its potential use as a therapy that can circumvent leptin resistance and reverse the behavioral and metabolic abnormalities associated with DIO and genetically obese models.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
While the nonapeptide oxytocin is historically recognized for its role in parturition [2], lactation [3], and osmoregulation [4], it has gained more recent attention for its effects on prosocial behavior [5, 6] and therapeutic potential in the treatment of autism spectrum disorder (ASD) [5, 6], schizophrenia [5, 7] and obesity [8–14]. In fact, 225 completed, ongoing or future investigations in humans list oxytocin in studies on caloric intake, gastric emptying, or obesity (ClinicalTrials.gov registry, National Institutes of Health). In light of the growing obesity epidemic, which impacted over 78 million adults and 12.5 million children and adolescents in the U.S. in 2009–2010 [15], combined with the relative ineffectiveness of existing weight loss strategies, this review will focus on timely findings that assess oxytocin’s ability to reduce food intake and body weight in diet-induced obese (DIO) [8, 10, 12–14] and genetically obese rodent models [9, 11, 12], highlight potential downstream CNS and peripheral mechanisms that could potentially elicit these effects, and discuss its regulation by anorexigenic and orexigenic signals.
1.1 Oxytocin production and release
Oxytocin is produced primarily from parvocellular paraventricular nucleus (pPVN) and magnocellular neurons in both the PVN and the supraoptic nucleus (SON) whereas more limited amounts are produced in the anterior hypothalamus, bed nucleus of the stria terminalis (BNST), medial preoptic area, medial amygdala and in the periphery [16–18]. Its release occurs locally in the SON and PVN via somatodendrites and distally via terminals that originate from magnocellular PVN and SON projections to the posterior pituitary and pPVN projections to sites that include the ventral tegmental area (VTA) [19], nucleus of the solitary tract (NTS) [20, 21] and spinal cord [21]. Several studies suggest that the major source of circulating oxytocin derives from the posterior pituitary, as oxytocin concentrations in the extracellular space of the SON are nearly 100-fold higher than circulating levels [22], oxytocin levels in the rat hypothalamus are 3-fold higher compared to heart and uterus [16], and oxytocin levels in pituitary extracts are over 250-fold higher than in heart perfusate [16]. Whereas recent findings point to the nodose ganglion [23] and gastrointestinal (GI) tract [24] as sites of oxytocin receptor (OXTR) expression and the GI tract as a site of synthesis [25], the extent to which OXTR signaling within the GI tract or vagal sensory afferent nerves contributes to energy homeostasis is unknown.
1.2 OXTR signaling and distribution
To date only one subtype of the OXTR has been identified. The majority of research has focused on the association between OXTR variants or polymorphisms and ASD and schizophrenia, but recent developments point to a link between rare OXTR variants and severe early-onset obesity in humans [26]. The extent to which these OXTR variants are causally related to variation in energy regulation in humans awaits further investigation. Nonetheless, the importance of oxytocin signaling to energy balance is clearly reflected in the wide distribution of OXTRs in relevant areas of the CNS such as the basal ganglia (nucleus accumbens (NAc), central amygdala), hypothalamus (PVN, SON, ventromedial hypothalamus (VMH)), and hindbrain (NTS, area postrema (AP)) [27–30], as well as in several peripheral tissues, including adipocytes [31–33] and GI tract [25].
The second messenger systems coupled to OXTRs in the CNS have only been recently identified [29, 34]. The classical uterine OXTR is a G protein-coupled receptor (GPCR) that signals through two G proteins, Gq/11 and Gi/0. It signals primarily through phospholipase C-β via Gq-binding proteins, stimulating production of inositol trisphosphate and 1,2-diacylglycerol that leads to the release of intracellular Ca2+ and phosphorylation of target proteins [28]. In contrast to GPCR signaling in uterine OXTRs, neuronal Gq is inhibitory and Gi stimulates an inward rectifying current [35] thereby eliciting changes in neuronal activation.
2 Effects of oxytocin on reductions in food intake and body weight
2.1 Role of oxytocin as an anorexigenic agent
Oxytocin was first reported to inhibit food intake following systemic administration in rodents by Arletti and colleagues in 1989 [36, 37]. Subsequent studies showed these effects could be reproduced following administration of much lower doses when given directly into the CNS [8, 9, 12–14, 38–40] and these effects were completely blocked by pretreatment of an oxytocin antagonist [36, 37, 39]. This ability to reduce food intake appears to be through a specific effect of oxytocin to reduce meal size [18, 38, 41] and increase latency to the first meal [37]. Meal-related stimuli are associated with activation of PVN and SON oxytocin neurons, release of oxytocin into the circulation, and activation of hindbrain neurons that regulate meal size. These stimuli include food intake [41–45], refeeding following a 24-[18] or 48-h fast [43, 45], the satiety signal cholecystokinin (CCK-8) [46, 47], gastric distension [47–49], activation of gastric vagal afferents [50, 51] and changes in osmolality following food intake [43]. Although circulating levels of oxytocin were not measured in these studies, the dietary signals associated with activation of PVN oxytocin neurons include branched-chain amino acids (e.g. leucine [41]), the dietary fat-derived signal, oleolyethanolamide (OEA) [52], and sucrose [44]. Peak circulating levels in the dark cycle generally correspond to typical patterns of food intake in mice [14]. Not surprisingly, energy deficits induced by prolonged fasting are associated with reductions in oxytocin mRNA when samples are restricted to the pPVN [53] or include the entire PVN [9, 54], and this effect is restored by refeeding [9]. Together, these studies suggest that nutrient excess is linked with increased hypothalamic oxytocin signaling while energy deficits are coupled with reductions in hypothalamic oxytocin signaling.
2.2 Physiological relevance of oxytocin in the control of food intake
Several lines of evidence have established a link between oxytocin signaling and food intake. For example, oxytocin antagonists stimulate intake of chow [9, 13, 14, 39, 41, 55], glucose [38], and sucrose [44] through a specific increase in meal size [38, 41]. Recent findings in OXTR null mice show increases in meal size during the dark cycle [18] despite no change in daily food intake [18, 56]. However, increases in daily food intake are observed in mice with reductions in mature hypothalamic oxytocin levels [57] (MAGED1 deficient mice). Interestingly, MAGED1 is a member of MAGE gene family associated in patients with Prader-Willi Syndrome, characterized by hyperphagia and reductions in number and size of PVN oxytocin neurons [58]. Likewise, rodent mutations of the single-minded 1 gene, SIM1 (e.g. Sim1 haploinsufficient mice) are also associated with hyperphagia and reductions in PVN oxytocin expression. These deficits in food intake in Sim1 haploinsufficient mice can be restored with oxytocin treatment [9]. Consistent with these findings, lentiviral knockdown of PVN oxytocin mRNA expression in adult mice results in increased intake of both low and high fat diets (HFD) [13]. In contrast, a recent finding showed that ablation of nearly 95 % of PVN oxytocin neurons via Cre-mediated diphtheria toxin treatment in adult oxytocin-Ires-Cre mice has no effect on chow or HFD intake [59]. However, caution must be used in interpreting these latter results as they may be due, in part, to ablation of other neuropeptides or neurotransmitters expressed in PVN oxytocin neurons. Further studies will also need to take into account the potential impact of gliosis in projection sites [60], background strain [44] as well as site specific developmental changes in OXTR expression [28, 61] that may be related to age [28, 61] or diet exposure in these mice at the time the loss in PVN oxytocin signaling occurred to determine the extent to which these factors may play a role in these differential effects.
2.3 Does oxytocin inhibit food intake by reducing gastric emptying and gastrointestinal transit?
Historical studies have shown that injections of small quantities of oxytocin into the DVC inhibit gastric motility in rats [62] and that oxytocin excites both NTS and dorsal motor nucleus of the vagus (DMV) cells that are activated in response to gastric distension [63]. Verbalis and colleagues speculated that oxytocin elicited suppression of gastric motility results from the effects of oxytocin to excite NTS neurons which, in turn, inhibit DMN neurons that stimulate gastric motility, resulting in an inhibition of gastric motility [4]. While previous findings show that systemic administration of oxytocin has either no effect on gastric emptying rate in humans [64] and rats [65] or a stimulatory effect on gastric motility in rabbits ([66]; attributed to species differences), recent findings show that systemic administration of oxytocin reduces gastric emptying in rats [67, 68] consistent with the presence of oxytocin receptors along the GI tract [24]. These effects are blocked by an oxytocin antagonist [67, 68]. They also appear to be dependent on CCK release and CCK1R signaling as these effects are blocked by the CCK1R antagonists, devazepide [67, 68] and lorglumide [67, 68], but not by the CCK2R antagonist, L-365,260 [67, 68]. These findings raise the question as to whether the effects of systemic oxytocin to inhibit gastric emptying require activation of hindbrain NTS OXTRs and whether the ability of peripheral oxytocin to reduce food intake also requires reductions in gastric emptying. Testing the effects of both central and systemic administration of oxytocin to reduce food intake in rats with gastric cannulas that remain open (sham feeding; absence of gastric distension) or closed (real feeding) would be helpful in determining the extent to which reductions in gastric emptying contribute to the anorexigenic response to oxytocin.
2.4 Does oxytocin reduce food intake by suppressing feeding reward circuitry?
Existing studies implicate an important role of endogenous oxytocin to reduce intake of highly palatable foods, such as sucrose [44, 69, 70] and HFD [8, 10, 12–14]. While exogenous administration of oxytocin reduces intake of HFD [8, 10, 12–14] and sucrose [71], it appears that endogenous oxytocin may preferentially inhibit intake of carbohydrates. Although PVN oxytocin neurons are activated towards the end of a meal consisting of HFD or sucrose, sucrose appears to activate a greater proportion of PVN oxytocin neurons relative to intake of fat (intralipid) [44]. Moreover, systemic administration of an oxytocin antagonist that readily crosses the blood brain barrier (L-368,799) [72] stimulates intake of sucrose, but not chow or intralipid [44]. Similarly, oxytocin knockout mice exhibit increased intake of sucrose [69], but not chow [69]. Work from Olszewski and colleagues provide a mechanism whereby opioids inhibit oxytocin signaling when a diet high in sugar is administered [73], and this may explain why the ability of sucrose to activate PVN and SON oxytocin neurons is impaired following long-term exposure to sucrose [74]. Furthermore, the ability of oxytocin to stimulate CCK-8 release in the SON is inhibited by morphine [75] although it is not clear the extent to which morphine also impacts the ability of oxytocin to enhance the hindbrain satiety and neuronal response to CCK-8. Together, these findings suggest that oxytocin signaling is sufficient to limit intake of rewarding foods high in sucrose, but chronic exposure to sucrose leads to impaired regulation and overconsumption.
The CNS pathways that potentially contribute to the effects of oxytocin to suppress intake of palatable food are not fully understood. Recent findings suggest that oxytocin may suppress food intake, in part, through a mechanism that involves inhibition of feeding reward circuitry in the NAc. Systemic oxytocin reduces drug reward behavior such as methamphetamine (METH) elicited self-administration [76] and METH-seeking behavior, which coincides with a suppression of METH-elicited neuronal activation in the NAc [76, 77]. These findings are consistent with studies that show central administration of oxytocin reduces METH-elicited hyperactivity, dopamine release in the striatum and NAc [78] and conditioned place preference [78]. Whether these effects require activation of OXTRs in the NAc or NTS with subsequent activation of ascending NTS-NAc projections remains to be determined.
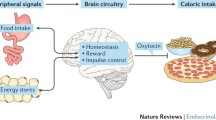
As reviewed by Paul Kenny [27] signaling via glucagon-like peptide-1 (GLP-1) and noradrenergic (NA) neuronal projections as well as NTS proopiomelanocortin (POMC) neurons may represent shared components that underlie feeding and drug reward circuitry within the NTS (see 6.1; Fig. 1). DIO is associated with both a reduction in the hindbrain satiety [79] and NTS neuronal response to CCK-8 [80]. These impairments may be driven by reductions in hindbrain oxytocin signaling in DIO animals, leading to an inability of CCK-8 to activate NTS neurons that express GLP-1 [81], NA (including prolactin releasing peptide (PrRP)) [82–85] and POMC [86, 87] and thus elicit satiety [88–91]. NTS GLP-1 and NA neurons project to feeding reward areas in the limbic system such as the VTA [92, 93] and NAc [92, 94–96]. GLP-1 also inhibits food intake following administration into the VTA [92] and NAc [92, 97], the latter of which occurs at doses that do not elicit a conditioned taste aversion (CTA). Furthermore, GLP-1 administration into the NAc core appears to preferentially inhibit intake of sucrose and HFD relative to chow [92]. Reductions in CNS GLP-1 signaling in adult rats also result in a predisposition to DIO [98]. Opiate administration into the NTS inhibits POMC neurons [86] whose overexpression in the NTS prevents DIO [99] and reduces adult-onset obesity [100]. Although the outgoing NTS POMC neuronal circuits that contribute to these effects are unknown (Fig. 1), fourth ventricular (4 V) administration of MTII inhibits sucrose intake, and supports a potential role of melanocortin 4 receptors (MC4R) in the NTS in the suppression of highly palatable food [101]. Although current data support potential roles of NTS GLP-1 and POMC neurons in suppressing intake of palatable food, the role of NTS NA neurons is less clear. While there is reduced sensitivity of NTS noradrenergic neurons to lipids in DIO or genetically obese mice [102, 103], the existing data favor an interpretation that the drug reward is enhanced by increased NA signaling in the NTS. For example, the reward associated with morphine is eliminated in dopamine-B-hydroxylase (DBH; enzymatic precursor to noradrenaline) null mice but restored following viral reexpression of DBH into the NTS [104]. Together, these findings indicate that, while NA neurons in the NTS may not be essential to drug reward, the nature of GLP-1, NA, and POMC neuronal signaling in the NTS in feeding reward has not been fully elucidated [27].

A schematic of proposed CNS circuitry involved in oxytocin regulation of energy balance. Leptin activates pPVN oxytocin neurons through a MC4R-dependent mechanism resulting in the release of oxytocin in the hindbrain. Oxytocin is proposed to activate ascending cNTS NA and GLP-1 neuronal projections to the PVN and NAc (denoted by purple arrows to PVN and NAc) as well as POMC neurons. These combined effects are proposed to reduce body weight by reducing food intake and increasing energy expenditure. Abbreviations: α-1R Alpha-1 adrenoceptor, ARC Arcuate nucleus, CART Cocaine-amphetamine-regulated transcript, CCK Cholecystokinin, GLP-1 Glucagon-like peptide-1, GLP-1R Glucagon-like peptide-1 receptor, LepRbLong form of the Leptin receptor, MC4R Melanocortin 4 receptor, NA Noradrenergic, NAc Nucleus accumbens, NTS Nucleus of the solitary tract, OXY Oxytocin, OXTR Oxytocin receptor, PrRP Prolactin releasing peptide, PVN Paraventricular nucleus, POMC Proopiomelanocortin
3 Physiological relevance of oxytocin in the control of body weight
Although both central [8, 12–14] and peripheral [10, 12, 14] administration of oxytocin reduces body weight the best evidence for the role of endogenous oxytocin signaling in the control of body weight stems from pharmacological and genetic loss of function studies. Oxytocin antagonists increase body weight gain [13, 14] and mice with global loss in oxytocin and OXTRs develop adult-onset obesity [56, 105]. Moreover, anatomically selective loss of oxytocin in the PVN in adult mice is associated with increased body weight gain on both low and HFD [13]. Similarly, recent reports show that ablation of PVN oxytocin neurons following diphtheria toxin treatment results in increased body weight gain on a HFD [59]. Mice with reductions in MAGED1 and reductions in mature hypothalamic oxytocin are also associated with adult-onset obesity and reduced activity. Reductions in the number and size of PVN oxytocin neurons are observed in patients with Prader-Willi syndrome that are characterized by hyperphagia and obesity [58]. Likewise, humans with mutations of SIM1 are associated with severe obesity in humans [106, 107] and Sim1 haploinsufficient mice are characterized by hyperphagia, obesity and reductions in PVN oxytocin expression. These deficits in relation to the body weight gain observed in Sim1 haploinsufficient mice can be restored with oxytocin treatment [9]. DIO mice are also associated with impairments in oxytocin release within the PVN and reductions in serum oxytocin [13]. These findings in DIO mice are attributed to increased PVN expression of synaptotagmin-4 (SYT4), a negative regulator of oxytocin release both in the PVN and in periphery [13]. Consistent with a link between increased oxytocin signaling and reductions in body weight is the finding that SYT4 null mice are lean and resistant to the development of DIO [13].
3.1 Weight loss associated with oxytocin treatment is not fully explained by reductions in food intake: Effects on energy expenditure
The effects of oxytocin to increase energy expenditure appear to be a principal driving force behind its ability to reduce body weight. Oxytocin produces reductions in body weight at doses that are ineffective at reducing food intake when administered chronically [8]. In cases where chronic oxytocin treatment inhibits both food intake and body weight gain its ability to reduce body weight is maintained even after treatment has ended [10] or food intake has returned to control values [10]. These findings are consistent with data which show that body weight loss attributed to oxytocin exceeds that from pair-fed control animals [8, 12]. Additionally, oxytocin and OXTR null mice develop adult-onset obesity, yet have no impairments in daily food intake [18, 56, 69, 105]. OXTR null mice also exhibit impairments in cold induced thermogenesis [56] and oxytocin null mice have reduced urinary adrenaline consistent with impairments in the sympathetic nervous system [105]. Oxytocin activates sympathetic preganglionic neurons [108], including the stellate ganglia [109]. With well characterized polysynaptic projections to brown adipose tissue [110], stellate ganglia [111], and spinal cord [21], these findings suggest that oxytocin plays an important role in regulating sympathetic nervous system activity. Consistent with this are findings that oxytocin increases heart rate [30, 112], body temperature [30] and oxygen consumption in mice [13, 14] and prevents the decrease in energy expenditure associated with reductions in body weight [12]. In addition, transgenic mouse models with reductions in PVN oxytocin signaling, such as Sim1 haploinsufficient mice [113] and SYT4 null mice [13] are characterized, in part, by reduced energy expenditure. Recent reports show that ablation of PVN oxytocin neurons following diphtheria toxin treatment results in increased body weight gain and reductions in energy expenditure in the absence of any changes in food intake in animals maintained on a HFD [59] confirming a role for PVN oxytocin neurons in the regulation of energy expenditure. In contrast, there are no changes in food intake, obesity and energy expenditure in lean mice [59]. These findings implicate an enhanced sensitivity to changes in oxytocin signaling in DIO relative to lean animals. While additional studies need to confirm the extent to which the metabolic phenotype following complete ablation of PVN oxytocin neurons could be attributed to nonspecific effects independent of a mechanism mediated by oxytocin signaling, these findings are consistent with the enhanced sensitivity to changes in oxytocin signaling in DIO [10, 12, 13] and genetically obese rodent models [9]. In contrast to the work from Wu and colleagues [59], other studies indicate that selective lentiviral knockdown of PVN oxytocin mRNA in adult mice increases food intake and body weight gain in animals fed either a low or HFD [13]. Together, these studies raise awareness for the need to examine energy expenditure following selective ablation of OXTRs in distinct forebrain and hindbrain sites that are linked to the regulation of energy expenditure, including those that receive projections from the pPVN (e.g. NTS and spinal cord) that may be impacted by gliosis and astrocytic infiltration that accompany ablation of neurons in response to diphtheria toxin administration [60]. It would also help extend the field by using the recently described OXTR-Venus knock-in mice (OXTR Venus ∆ Neo/+) [30] to assess downstream targets that could potentially underlie these effects on energy expenditure. One such downstream target which could contribute to the effects of oxytocin on energy expenditure is GLP-1 [114], and evidence in support of GLP-1 being a downstream target of oxytocin action is discussed later in this review.
3.2 Weight loss associated with oxytocin treatment is not fully explained by reductions in food intake: Effects on lipolysis
Oxytocin may also reduce body weight due, in part, to its ability to increase lipolysis through a direct effect on adipocytes [31–33, 115] or an indirect mechanism involving polysynaptic projections from the PVN to white adipose tissue [116, 117]. OXTRs are expressed on primary and cultured adipocytes [31–33] and oxytocin elicits direct effects on these cells [115]. In vitro data from 3 T3-L1 adipocytes show that oxytocin increases enzymes associated with lipolysis [8] and results in increased glycerol release [8]. In addition, chronic oxytocin treatment in vivo results in reductions in fat mass [8, 10, 14], particularly adipocyte area from both mesenteric and epididymal fat [10]. Consistent with these findings, in vivo data from animals with global loss in oxytocin signaling show increases in abdominal fat [56, 105] and increases in perirenal, mesenteric, and epididymal fat pad weights relative to littermate controls [56]. Similarly, selective ablation of oxytocin neurons in the PVN and SON [59] and postnatal ablation of PVN Sim1 neurons (resulting in a 50 % drop in hypothalamic oxytocin mRNA expression) [113] are associated with increases in body fat. These findings corroborate those from pair-feeding studies [8, 12], studies that report oxytocin to be effective at reducing body weight at doses ineffective at reducing food intake [8], and studies that show reductions in body weight persist well beyond the normalization of food intake and cessation of treatment [10]. Together, these recent studies unveil potential mechanisms whereby oxytocin reduces body fat through separate or combined effects to promote lipolysis and increase energy expenditure.
3.3 Evidence to support role of descending oxytocin pPVN-NTS projections in regulation of body weight
Several lines of evidence support the hypothesis that a neural pathway from the pPVN to the NTS (Fig. 1) is a critical component of CNS circuits that regulate the long-term control of body adiposity [55, 89, 118–120]. Decerebrate animals (all connections between hypothalamus and hindbrain severed) are unable to mount a normal compensatory response to energy deficits by increasing food intake, despite intact feeding responses to many short-term, meal-related stimuli [121]. The NTS not only integrates descending information originating from the hypothalamus, but it receives ascending information from the GI tract through its extensive innervation by vagal afferent projections that originate from the gut [122]. Substances may also access the NTS through the nearby AP, a region that lacks a blood brain barrier and is thus capable of directly responding to nutrient and blood-borne factors released during the course of a meal. It is well established that the meal-related satiety signals, CCK-8 and gastric distension, activate neurons in the AP and NTS [82, 123]. Lesions of both the AP and NTS eliminate the ability of CCK-8 to inhibit food intake [124]. Together, these studies reveal the importance of these areas in integrating information pertaining to meal-related satiety signals (CCK-8, gastric distension) to control feeding [122, 123, 125, 126].
Further understanding of the role of caudal brainstem neurons in the regulation of regulation of food intake has been facilitated by identifying the neuronal circuits involved in this pPVN-NTS pathway. Oxytocin fibers comprise 11–16 % of pPVN projections to the medulla and spinal cord [127] of which nearly 6 % project directly to the DVC [46], specifically the NTS and DMV. Previous data show that oxytocin fibers in the NTS arise solely from the pPVN [20] and are in close anatomical proximity to areas in the NTS that respond to CCK-8 [89]. PVN lesions are also associated with an attenuated satiety response to CCK [128] and destruction of either PVN neurons [129] or this hindbrain projection results in hyperphagia and obesity [130].
3.4 Role of descending pPVN-NTS oxytocin projections in contributing to leptin signaling
Descending pPVN-NTS oxytocin projections are hypothesized to contribute to the ability of leptin to enhance the hindbrain neuronal and satiety response to CCK-8 [55, 90]. Leptin activates pPVN oxytocin neurons [55, 131, 132], some of which project to the caudal NTS [55] [132], and prevents the drop in PVN oxytocin mRNA expression associated with long-term fasting [54, 132]. Blockade of OXTR signaling attenuates leptin’s ability to inhibit food intake [55, 59] and body weight [132]. Moreover, blockade of endogenous oxytocin signaling attenuates the ability of leptin to enhance the hindbrain neuronal response to CCK-8 [55], and reduces the effectiveness of CCK-8 to inhibit food intake [88, 89]. Together, these findings suggest that endogenous oxytocin contributes to the ability of leptin to reduce food intake through a mechanism that involves enhancing the hindbrain response to satiety signals such as CCK.
Evidence to date suggests that leptin activates pPVN oxytocin neurons through both a direct and an indirect melanocortin-dependent mechanism. One recent study in rats provides evidence in support of a direct effect by showing that leptin elicits phosphorylation of signal transducer and activator of transcription-3 (pSTAT3) signaling in pPVN oxytocin neurons that project to the NTS [132]. However, the majority of evidence in both rats and mice supports an indirect mechanism of leptin to activate pPVN oxytocin neurons. For example, leptin elicits little pSTAT3 signaling in pPVN [133] consistent with low levels of expression of leptin receptors [134]. In addition, the ability of leptin to induce Fos in PVN neurons and inhibit food intake is completely blocked by pretreatment with the melanocortin 3/4 receptor (MC3/MC4R) antagonist, SHU9119 [135]. Furthermore, CNS administration of the MC3/MC4R agonist, alpha-melanocyte-stimulating hormone or a selective MC4R agonist (cyclo(β-Ala-His-D-Phe-Arg-Trp-Glu)-NH2), activates PVN oxytocin neurons [9, 136], which express MC4R [137]. pPVN neurons that project to the NTS also express MC4R [138] and PVN administration of alpha-MSH induces Fos in both PVN and NTS neurons [45]. Importantly, blockade of endogenous oxytocin also blocks the effects of MTII to reduce food intake [139]. Together, these findings support both a direct action as well as an indirect effect of leptin to activate downstream pathways that secrete alpha-MSH from POMC neurons onto pPVN MC4R/oxytocin neurons that project to the NTS, resulting in activation of CCK-sensing neurons in the hindbrain and an enhanced satiety response to CCK.
3.5 Proposed impact of leptin resistance on activation of downstream descending oxytocin pPVN-NTS projections
The use of leptin as therapeutic agent to treat obesity was initially an exciting concept, but has since been disappointing due to the development of “leptin resistance”, characterized by an impaired ability of leptin to activate both intracellular signals in the ARC and downstream signaling pathways in rodents and in all likelihood humans [140]. These impairments result in the failure of leptin to reduce food intake [141] and body weight [142], despite elevated levels of leptin in the circulation [141]. Leptin resistance is selective in the ARC [133], and impairments in leptin signaling occur within one week of exposure to a HFD in rodents [133]. Although the causes of leptin resistance are not well understood, many factors are thought to contribute to leptin resistance, including defective transport of leptin across the blood brain barrier [141, 143], hyperleptinemia [144], defects in leptin intracellular signaling in the ARC through upregulation of suppressor of cytokine signaling 3 (SOCS-3) [145], leptin promoter DNA methylation [146], hypothalamic inflammation [147], and increased levels of hypothalamic protein-tyrosine phosphatase 1B [148, 149]. One predicted outcome of leptin resistance is a failure of leptin to activate downstream pathways that secrete alpha-MSH from POMC neurons onto pPVN MC4R/oxytocin neurons that project to the NTS, resulting in a defect in both the activation of CCK-sensing neurons in the hindbrain and the satiety response to CCK, both of which occur in animals fed a HFD [79, 80]. Recent studies illustrate that mice deficient in SOCS-3 in the mediobasal hypothalamus (MBH) have restored leptin sensitivity, reductions in food intake and body weight gain, and increased hindbrain responsiveness to satiety signals [90]. In addition, blockade of endogenous oxytocin attenuates the response to satiety signaling in these animals [90]. Moreover, leptin resistant animals have reductions in oxytocin content in the DVC relative to leptin sensitive MBH SOCS-3 null mice. Together, these findings suggest that leptin resistance is associated with downstream impairments in oxytocin release within the NTS [90]. The extent to which this impairment is important in causing or maintaining the metabolic and behavioral abnormalities in DIO remains to be determined.
3.6 What is the potential role of forebrain OXTRs in the regulation of body weight?
Few studies have examined the impact of neuroanatomically specific knockdown or overexpression of OXTRs on the control of food intake and body weight. Surprisingly, one study found a lean phenotype is associated with postnatal loss of OXTRs in the lateral septum, hippocampus and ventral pallidum [150]. However, the extent to which OXTRs were ablated in other forebrain areas linked to the regulation of energy expenditure (e.g. PVN, VMH) was not discussed nor was the possibility that developmental upregulation of OXTRs in extrahypothalamic areas or in the periphery could contribute to these effects. As indicated earlier, lentiviral reduction in PVN oxytocin mRNA expression is associated with a clear phenotype on food intake and body weight [13], but it is unclear whether this phenotype resulted from loss in PVN oxytocin autoreceptors and/or loss in outgoing oxytocin projections to NTS, spinal cord, sympathetic ganglia or brown adipose tissue. Although much work has been done to elucidate the role of PVN oxytocin neurons, examining the impact of selective loss in OXTRs in the PVN and VMH would better help delineate the role of forebrain OXTR signaling in the regulation of energy balance.
3.7 Does systemic administration of oxytocin inhibit body weight through a mechanism that requires OXTRs in the CNS?
While there is a lot of excitement regarding the therapeutic potential of systemic oxytocin to reduce food intake and body weight following systemic administration, it is not clear the extent to which this route of administration reduces food intake through receptors in the brain vs. periphery. Systemic oxytocin increases Fos induction in PVN oxytocin neurons [77, 151] and increases the release of oxytocin within the PVN [14]. Oxytocin may, in turn, activate PVN oxytocin neurons via autoreceptors making it hard to differentiate a central from peripheral mechanism. The evidence supporting the notion that circulating oxytocin enters the brain is mixed as studies report that only 0.01 to 0.002 % enters the brain following intravenous administration [152, 153] but does so within only 10 min postinjection [152]. Furthermore, recent findings in rats and mice show increases in oxytocin levels in both the hippocampus and amygdala within 30-min of an intraperitoneal injection [154]. In addition, peripheral administration of oxytocin recapitulates the effects of CNS administration on food intake [12, 14], body weight [12, 14], and Fos induction in hindbrain areas [10, 12, 14, 40, 123, 151, 155] that express OXTRs [28–30]. Access to OXTRs in the AP and NTS could be achieved through leaks in the blood brain barrier [156], tanycytes [156] or other transporters [157, 158]. Since such small doses of oxytocin are required to inhibit food intake following CNS administration little would actually need to be transported into the brain in order to elicit an effect which may explain why large acute systemic doses are required to inhibit food intake [10, 12, 14, 36, 37]. Consistent with oxytocin being able to cross the blood brain barrier [152, 153] recent findings show that the anxiolytic activity following systemic administration of oxytocin is blocked by intracerebroventricular (ICV) administration of a nonpenetrant oxytocin antagonist [159]. Future efforts should examine the effectiveness of systemic administration of oxytocin to reduce food intake and body weight in animals with timed and localized ablation of OXTR in the hindbrain.
4 Oxytocin is a common downstream effector of anorexigenic signals
4.1 Stimulation of oxytocin neurons by anorexigenic signals
Cocaine-amphetamine-regulated transcript (CART)
Earlier we discussed reports that show PVN oxytocin neurons are activated by both leptin and MC3/MC4R ligands and appear to be critical components of both leptin and melanocortin signaling. The extent to which oxytocin contributes to the anorexigenic response to CART, a downstream target of leptin and expressed on ARC POMC neurons, is unknown. CART activates magnocellular and pPVN oxytocin neurons and increases serum levels of oxytocin [160]. Future studies could address the extent to which CART [161] inhibits food intake in animals pretreated with selective oxytocin antagonists or in animals with global or site specific ablation in either OXTRs or oxytocin.
Fat mass and obesity associated gene (FTO)
Recent data show that FTO, a gene linked to the regulation of food intake and body weight, has a high degree of colocalization with magnocellular and pPVN oxytocin neurons [162], and increases the expression of oxytocin in vitro [163]. The exact role of FTO remains unclear as overexpression leads to increased food intake and obesity [164] yet global loss [165] and mutations in FTO [166] lead to increased food intake, reduced body weight and increased energy expenditure. It remains to be determined if overexpression or absence of FTO produces the predicted changes in oxytocin signaling in vivo.
Leucine
The branched chain amino acid, leucine, inhibits meal size, in part, by activating ARC POMC neurons, PVN oxytocin neurons, and OXTRs in the NTS [41]. Administration of an oxytocin antagonist into the 4 V blocks the effects of MBH application of leucine to inhibit food intake, suggesting that endogenous oxytocin in the NTS contributes to the ability of MBH leucine to inhibit food intake [41]. Recent studies show that this is not the only mechanism by which leucine inhibits food intake as sites within the caudal hindbrain also contribute to the anorexigenic response to leucine [167].
Nesfatin-1
Nesfatin-1, a peptide derived from the precursor nucleobindin-2 (NUCB2), is expressed in the CNS, including the hypothalamus and hindbrain, and colocalizes with PVN oxytocin neurons [11]. It activates magnocellular and pPVN oxytocin neurons [11], increases the release of PVN oxytocin [11] and inhibits food intake through an oxytocin dependent mechanism [139]. Recent studies show that, similar to oxytocin, it inhibits food intake through a central [11, 139] and peripheral route of administration [168]. It remains to be determined if nesfatin-1, like oxytocin, is also capable of activating PVN oxytocin neurons when given by a peripheral route of administration.
OEA
OEA, which is produced in small-intestinal enterocytes following dietary fat absorption, inhibits feeding via peroxisome proliferator-activated receptor-alpha (PPAR-alpha) and the release of oxytocin [52]. OEA increases Fos mRNA in the PVN, increases oxytocin mRNA in both PVN and SON, and increases plasma oxytocin [52]. While these studies suggest that oxytocin is downstream of PPAR-alpha, recent studies indicate that a single dose of oxytocin fails to inhibit food intake in PPAR-alpha KO mice [8]. Together, these studies suggest that PPAR-alpha and oxytocin may reciprocally regulate one another, but additional dose–response studies would be helpful to confirm these findings.
PPAR-gamma coactivator (PGC)-1 alpha
Mice that lack the transcriptional coactivator, PGC-1 alpha, develop adult-onset obesity [169], similar to what is observed in OXTR [56], oxytocin null mice [105], and MAGED1 deficient mice [57]. PGC-1 alpha is coexpressed with oxytocin in the zebrafish hypothalamus [170] and forms a complex that contains PGC1-alpha and the oxytocin promoter [170] in fed, but not fasted zebrafish [170]. Moreover, PGC-1alpha overexpression increases the expression of oxytocin in muscles and neurons [170] and reductions in PGC-1 alpha also decrease oxytocin mRNA [170] suggesting that PGC1-alpha contributes to oxytocin synthesis.
5 Oxytocin as a downstream target of orexigenic signals
5.1 Inhibition of oxytocin neurons by orexigenic signals
Agouti-related protein (AGRP)
Recent advances in optogenetics and electrophysiology have determined that AGRP fibers appear to contact PVN oxytocin neurons [171] and that AGRP stimulates food intake by inhibiting these neurons [171]. This can be reversed with bilateral co-photostimulation of PVN oxytocin neurons and ARC-PVN AGRP axonal projections [171].
Opioids
Previous studies have shown that opioid receptors are expressed on PVN and SON oxytocin neurons [172] and that opioid agonists can inhibit further activation of PVN oxytocin towards the end of a meal [73]. The ability of oxytocin to stimulate CCK-8 release in the SON is inhibited by morphine [75]. The effectiveness of LiCl to induce Fos in PVN and SON oxytocin neurons is also inhibited by opioids [173]. Olszewski and colleagues also show that opioid-induced inhibition of oxytocin signaling is particularly evident when a diet high in sugar is administered [73]. Interestingly, others have also shown this to occur during periods of hyperphagia in mid to late pregnancy [174, 175], but sex steroids (e.g. progesterone) may play a more predominant role in the inhibition of oxytocin neurons during pregnancy [22].
SYT4
Work from Zhang and colleagues demonstrate that lentiviral expression of SYT4 into the PVN of SYT4 null mice results in impaired release of oxytocin within the PVN, increased HFD intake and body weight gain in DIO animals [13]. While these findings reveal another important negative regulator of oxytocin regulation in the control of food intake and body weight, remaining issues to address are whether SYT4 is expressed on pPVN neurons that project to the NTS and the extent to which SYT4 is regulated by leptin and MTII. In addition to alterations in oxytocin signaling being an important downstream effector of both anorexigenic and orexigenic signals, recent studies have identified potential downstream intracellular signals and CNS targets that underlie the effects of oxytocin.
6 Potential downstream NTS targets that contribute to the effects of oxytocin on food intake and body weight
It is well appreciated that endogenous oxytocin acts, in part, through OXTRs in the NTS to inhibit food intake through a reduction in meal size [8, 10, 11, 40, 41, 55, 88, 89, 130, 176]. Some of the outstanding questions in the field are as follows: What are the potential signals and downstream targets that contribute to these effects? To what extent does activation of OXTRs in forebrain areas contribute to the inhibition of food intake? To what extent does systemic administration of oxytocin inhibit food intake through a mechanism that requires OXTRs in the CNS?
6.1 Potential OXTR signaling mechanism (s) in NTS
NTS receptors
Recent electrophysiological data show oxytocin excites NTS neurons through both a postsynaptic [177] as well as a presynaptic mechanism involving release of glutamate and subsequent activation of NTS neurons through 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid receptors (AMPA) [177]. These findings extend immunocytochemical data that show central [40, 123, 155] and peripheral [10, 12, 14, 77, 151] administration of oxytocin induces Fos largely in the caudal NTS (cNTS) where OXTRs are expressed [28–30].
Potential intracellular signals in NTS
Stimulation of PVN OXTRs leads to transactivation of the epidermal growth factor receptor and activation of a MEK-extracellular signal-regulated kinase (ERK) mitogen-activated protein kinase (MAP kinase) pathway [34]. Ingestion of food increases Erk1/2 phosphorylation in PVN oxytocin neurons [41]. Although it remains to be determined if ERK signaling in the NTS mediates the effects of oxytocin to reduce food intake, ERK signaling in this site contributes to the ability of CCK-8 to inhibit food intake [178]. Further studies are required to determine if ERK signaling also contributes to the ability of oxytocin to enhance the satiety response to CCK [88, 89].
6.2 cNTS A2 NA neurons
cNTS NA neurons are activated by visceral afferents [82], gastric distension [126], PYY(3–36) [179], nicotine [180], high protein meals [181], CCK-8 [82, 182], and oxytocin [183–185]. Oxytocin fibers that originate from the pPVN are found in anatomical proximity to NTS NA neurons [186]. Previous findings indicate that oxytocin contributes to the satiety response to CCK-8 [89, 91] and to the ability of leptin to enhance the hindbrain neuronal response to CCK-8 [55]. Previous data that show a) CCK-8 activates NTS NA neurons [82, 182] via a presynaptic mechanism involving release of glutamate [82]; b) the satiety response to CCK-8 is attenuated in rats with cNTS NA lesions [83]; c) cNTS NA neurons are potential downstream targets of leptin and CCK-8 [187], and d) CCK-8 increases ERK1/2 expression in cNTS NA neurons [188]. Together, these findings raise an intriguing possibility that cNTS NA neuronal signaling through ERK1/2 contributes to the ability of oxytocin to inhibit food intake. Interestingly, in mice, cNTS NA neurons do not appear to be a direct target of leptin action [189], which may represent a species difference with respect to downstream targets of leptin action or that NTS NA neurons are indirect targets of leptin action in both species. Regardless, these findings raise the possibility that NTS NA neurons contribute to the ability of oxytocin to inhibit food intake by enhancing the satiety response to CCK-8.
6.3 Do NA projections from the cNTS to PVN contribute to oxytocin-elicited anorexia?
Both CCK-8 and oxytocin induce Fos in PVN oxytocin neurons. CCK-8-elicited activation of PVN oxytocin neurons appears to occur through ascending NA innervation of the PVN from the A2 region of the cNTS [95, 127] (Fig. 1) as lesions of cNTS NA neurons blunt the ability of CCK-8 to activate PVN oxytocin neurons [83]. Furthermore alpha-1 adrenoceptor activation in the PVN is associated with reductions in food intake [190, 191] and alpha-1 adrenoceptor 1D mRNA is expressed on PVN oxytocin and corticotrophin-releasing hormone (CRH) neurons [192]. Further studies need to address whether oxytocin activates PVN neurons that express alpha-1 adrenoceptor 1D subunits and determine the extent to which reductions in alpha-1 adrenoceptor signaling in the PVN contribute to the effects of oxytocin to inhibit food intake.
6.4 cNTS PrRP
The majority of cNTS NA neurons express PrRP [193] (reviewed in [85]), a peptide found to suppress food intake [84, 194]. Recent findings show that PrRP receptors are expressed on both magnocellular and pPVN oxytocin neurons [18]. In addition, the ability of refeeding (following a 24-h fast) and CCK-8 to activate PVN oxytocin neurons and increase circulating oxytocin is suppressed in PrRP-deficient mice [18]. Furthermore, the effects of leptin [195] and CCK-8 [84] to reduce food intake are impaired in PrRp-deficient mice, which likely contributes to PrRp-deficient mice having increased meal size [195]. Similar to OXTR [56], oxytocin null mice [105], and MAGED1 deficient mice [57] both PrRp [195] and PrRP receptor deficient mice [196, 197] develop late onset-obesity. Further studies need to determine the extent to which reductions in PrRP receptor signaling in the PVN impair the ability of oxytocin to inhibit food intake.
6.5 cNTS GLP-1 neurons
CNS GLP-1 derives predominantly from posttranslational processing of preproglucagon (PPG) in the NTS. NTS GLP-1 neurons, which are distinct from NA (and PrRP) neurons [198–200], are activated by vagal afferents [201], leptin [201], gastric distension [202], CCK-8, noradrenaline, adrenaline [81], and oxytocin [40]. NTS oxytocin fibers are found in anatomical proximity to NTS GLP-1 neurons [40], and central administration of oxytocin induces Fos in NTS GLP-1 neurons [40]. Furthermore, ICV administration of the GLP-1 receptor (GLP-1R) antagonist (des His1 Glu9-exendin 4) attenuates the ability of oxytocin to reduce food intake [40]- findings which raise the possibility that local release of GLP-1 in the NTS or via ascending projections to the forebrain may contribute to these effects.
6.6 Which GLP-1 neuronal projections potentially contribute to oxytocin-elicited anorexia?
NTS GLP-1 neurons provide a source of GLP-1 within the cNTS [201, 203], but they also project to numerous sites in the forebrain, all of which express OXTRs, including the ARC [203], dorsomedial hypothalamus (DMH) [203], PVN [203] (Fig. 1), VTA [92] and NAc [92, 94] (Fig. 1; see 2.4). Of these possibilities it appears that activation of GLP-1Rs in the PVN or NTS most likely to contribute to oxytocin elicited anorexia. For instance, GLP-1 inhibits food intake following administration into the PVN [204], where GLP-1Rs are expressed on PVN oxytocin neurons [205]. Furthermore, GLP-1 fibers are also in anatomical proximity to PVN oxytocin neurons [198] and CNS administration of GLP-1 [206] recapitulates the effects of oxytocin to induce Fos in PVN oxytocin neurons [77, 151] although this effect may also occur through a direct action of oxytocin on autoreceptors on PVN oxytocin neurons [207]. Within the NTS, GLP-1R agonists and antagonists inhibit and stimulate food intake, respectively [97]. As discussed earlier, ICV administration of oxytocin elicits Fos in NTS GLP-1 neurons [40]. Although the ARC receives GLP-1 projections [203] and expresses Fos in response to oxytocin [10], GLP-1Rs in the ARC appear to be principally involved with regulation of glucose homeostasis [208], as GLP-1R agonists fail to reduce food intake following administration in this area [208]. GLP-1 also inhibits food intake following administration into the DMH [209], which receives GLP-1 projections [203], but oxytocin fails to elicit Fos in this area [10]. Further studies need to address whether a) oxytocin activates GLP-1R in the PVN; b) reductions in GLP-1R signaling in the PVN and NTS impair the ability of oxytocin to reduce food intake, and c) oxytocin inhibits food intake in transgenic mice with specific ablation of OXTRs in NTS GLP-1 neurons [210].
6.7 cNTS POMC neurons
NTS oxytocin terminals are in anatomical proximity to NTS POMC neurons, oxytocin evokes the release of intracellular Ca2+ from NTS POMC neurons, and third ventricular (3 V) administration of SHU9119 blocks the effects of oxytocin to inhibit feeding [11]. Both hindbrain melanocortin [87] and oxytocin signaling [88, 89] contributes to the ability of CCK-8 to reduce food intake. Furthermore, CCK-8 increases ERK1/2 expression in NTS POMC neurons [188], and this effect is abolished following hindbrain MC3/MC4R blockade [211]. While it still remains to be determined whether activation of ERK1/2 signaling through NTS POMC neurons is required to mediate the effects of OXTR signaling in the NTS, ERK is a downstream intracellular signal induced by ingestion of food in PVN oxytocin neurons [41]. Together, these findings suggest that activation of ERK1/2 in NTS POMC neurons may contribute to the effects of oxytocin to suppress food intake.
7 Oxytocin as a potential therapeutic target?
Given the uncertainties to the extent to which systemic oxytocin targets OXTRs in the CNS, one of the benefits to the use of oxytocin as a potential therapeutic agent is that oxytocin is one of the few hormones that can stimulate its own [77, 151, 212, 213] release via autoreceptors in the magnocellular SON [212, 213] and PVN [207] following a central or peripheral route of administration. CNS administration of oxytocin upregulates PVN oxytocin mRNA expression [8] and systemic administration of oxytocin activates PVN oxytocin neurons [77, 151] and stimulates release of oxytocin in the PVN [14]. Long-term changes in function [214] may be linked to this mechanism of stimulation [215, 216]. During parturition, oxytocin concentrations in the CSF are as high as levels in the periphery and likely remain high as a result of the reduced clearance in brain (T1/2 = 19 min; [152]) vs. periphery (T1/2 = 6 min; [217]). The prolonged bioavailability of CNS oxytocin in combination with its self-stimulatory properties are thought to contribute, in part, to its positive effects on prosocial behavior [207] and may contribute to its prolonged effects on body weight loss following cessation of treatment [10] .
The fact that oxytocin inhibits food intake in DIO rats with impaired leptin signaling [8, 10, 12–14] and in animal models with defective leptin signaling, such as the obese Zucker [11] and Koletsky rats [12], as well as obese animals with Sim1 haploinsufficiency [9] make it an attractive therapeutic target in both DIO and genetic models of obesity. To date only one paper reports tolerance in the effectiveness of ICV administration of a potent oxytocin agonist [e-L- β-MePhe2]oxytocin to reduce food intake over 3 days [39]. Importantly, these effects do not appear to interfere with its ability to reduce body weight as chronic administration of oxytocin over 14–17 days produces sustained reductions in body weight [8, 10] raising the question as to how long this weight loss could be maintained. Recent findings in DIO mice show that chronic administration of oxytocin produced a sustained loss in body weight loss over a 6-week period [14]. This is particularly exciting given that other substances, such as exendin-4, typically fail to maintain body weight loss after 17 days [218]. Of obvious clinical significance are findings that indicate oxytocin reduces anxiety [219], blood pressure [220], the size of cardiac infarcts [221], and is not causally related to the development of a CTA [222] in rats. Moreover, CNS [13] administration of oxytocin fails to elicit a CTA at doses that suppress food intake and body weight in rodents [12, 14, 36].
While it remains to be determined whether chronic systemic administration of oxytocin reduces body weight in the absence of unwanted peripheral side effects in women (e.g. uterine cramping), the intranasal route of administration appears to effectively allow oxytocin to access the CNS in mice, rats, nonhuman primates and humans within 30–35 min after administration [154, 223, 224] without causing unwanted side effects [225]. In humans, intranasal administration of vasopressin (structurally similar to oxytocin) reaches the CSF within 10 min and levels are maintained for at least 80 min following administration [226]. One uncertainty is the extent to which these levels reflect exogenous oxytocin and/or endogenous oxytocin release following stimulation of hypothalamic oxytocin neurons that project within the CNS. Another uncertainty is whether intranasal delivery restricts oxytocin to the CNS as the increase in plasma oxytocin levels could result from either direct entry of intranasal oxytocin into the circulation or oxytocin-elicited autostimulation of hypothalamic oxytocin neurons that project to the posterior pituitary. One means to circumvent direct entry of drugs into the circulation without altering concentrations in the CSF is to include a vasoconstrictor such as phenylephrine in the intranasal treatment [227]. Regardless, this route of administration has yielded positive prosocial effects in nonhuman primates [223], male and female schizophrenics [228, 229], and in men and women who experience social rejection [230]. The added benefit of potentially ameliorating obesity and its related complications only adds to the excitement surrounding oxytocin as a potential therapeutic strategy in humans. While rodent and limited nonhuman primate and human studies are encouraging thus far, OXTRs have the capacity to interact with and modulate other GPCRs such as the β2 adrenergic receptor [231], and multi-drug therapies may need to be considered to maximize the benefits of targeting the oxytocin signaling pathway [232]. Caution should be used prior to administration in children and young adults as some studies suggest that behavioral deficits occur in response to chronic administration in developing prairie voles [233]. Additional studies should continue to address the long-term neuroendocrine and behavioral effects following multiple doses in developing animals [233, 234].
8 Conclusions
It is clear that disruptions in oxytocin signaling contribute to obese phenotypes through what appear to be impairments in both food intake and energy expenditure. It remains to be determined whether such disruptions in oxytocin signaling are critical to causing and/or maintaining the obese phenotype in DIO. While there is tremendous excitement over the potential use of oxytocin as a therapeutic agent, we await the results of ongoing human trials for further confirmation of its effectiveness as a weight loss strategy in the absence of unwanted side effects. It is clear that there are several means by which oxytocin, given centrally or peripherally, can influence body weight, namely through reductions in food intake, increases in energy expenditure, and lipolysis. In light of the finding that oxytocin is synthesized in the GI tract, and the observation that peripheral administration can stimulate the release of CNS oxytocin, additional studies are required to address the relevance of peripheral stores of oxytocin on energy balance and whether activation of CNS oxytocin circuitry is, in fact, required to maximize its therapeutic potential when given systemically.
References
Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. [Comparative Study]. JAMA. 2012;307(5):491–7. doi:10.1001/jama.2012.39.
den Hertog CE, de Groot AN, van Dongen PW. History and use of oxytocics. [Historical Article Review]. Eur J Obstet Gynecol Reprod Biol. 2001;94(1):8–12.
Braude R, Mitchell KG. Observations on the relationship between oxytocin and adrenaline in milk ejection in the sow. J Endocrinol. 1952;8(3):238–41.
Verbalis JG, Blackburn RE, Hoffman GE, Stricker EM. Establishing behavioral and physiological functions of central oxytocin: insights from studies of oxytocin and ingestive behaviors. [Research Support, U.S. Gov’t, P.H.S. Review]. Adv Exp Med Biol. 1995;395:209–25.
Striepens N, Kendrick KM, Maier W, Hurlemann R. Prosocial effects of oxytocin and clinical evidence for its therapeutic potential. [Research Support, Non-U.S. Gov’t Review]. Front Neuroendocrinol. 2011;32(4):426–50. doi:10.1016/j.yfrne.2011.07.001.
Yamasue H, Yee JR, Hurlemann R, Rilling JK, Chen FS, Meyer-Lindenberg A, et al. Integrative approaches utilizing oxytocin to enhance prosocial behavior: from animal and human social behavior to autistic social dysfunction. [Review]. J Neurosci. 2012;32(41):14109–17. doi:10.1523/JNEUROSCI.3327-12.2012.
Montag C, Brockmann EM, Bayerl M, Rujescu D, Muller DJ, Gallinat J. Oxytocin and oxytocin receptor gene polymorphisms and risk for schizophrenia: A case–control study. World J Biol Psychiatry. 2012. doi:10.3109/15622975.2012.677547.
Deblon N, Veyrat-Durebex C, Bourgoin L, Caillon A, Bussier AL, Petrosino S, et al. Mechanisms of the anti-obesity effects of oxytocin in diet-induced obese rats. [Research Support, Non-U.S. Gov’t]. PLoS One. 2011;6(9):e25565. doi:10.1371/journal.pone.0025565.
Kublaoui BM, Gemelli T, Tolson KP, Wang Y, Zinn AR. Oxytocin deficiency mediates hyperphagic obesity of Sim1 haploinsufficient mice. Mol Endocrinol. 2008;22(7):1723–34. doi:10.1210/me.2008-0067.
Maejima Y, Iwasaki Y, Yamahara Y, Kodaira M, Sedbazar U, Yada T. Peripheral oxytocin treatment ameliorates obesity by reducing food intake and visceral fat mass. [Research Support, Non-U.S. Gov’t]. Aging (Albany NY). 2011;3(12):1169–77.
Maejima Y, Sedbazar U, Suyama S, Kohno D, Onaka T, Takano E, et al. Nesfatin-1-regulated oxytocinergic signaling in the paraventricular nucleus causes anorexia through a leptin-independent melanocortin pathway. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Cell Metab. 2009;10(5):355–65. doi:10.1016/j.cmet.2009.09.002.
Morton GJ, Thatcher BS, Reidelberger RD, Ogimoto K, Wolden-Hanson T, Baskin DG, et al. Peripheral oxytocin suppresses food intake and causes weight loss in diet-induced obese rats. [Comparative Study Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, Non-P.H.S.]. Am J Physiol Endocrinol Metab. 2012;302(1):E134–144. doi:10.1152/ajpendo.00296.2011.
Zhang G, Bai H, Zhang H, Dean C, Wu Q, Li J, et al. Neuropeptide exocytosis involving synaptotagmin-4 and oxytocin in hypothalamic programming of body weight and energy balance. [Comparative Study Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Neuron. 2011;69(3):523–35. doi:10.1016/j.neuron.2010.12.036.
Zhang G, Cai D. Circadian intervention of obesity development via resting-stage feeding manipulation or oxytocin treatment. [Evaluation Studies Research Support, N.I.H., Extramural]. Am J Physiol Endocrinol Metab. 2011;301(5):E1004–1012. doi:10.1152/ajpendo.00196.2011.
Ogden CL, Carroll MD, Kit BK, Flegal, KM (2012) Prevalence of obesity in the United States, 2009–2010. NCHS Data Brief.
Jankowski M, Hajjar F, Kawas SA, Mukaddam-Daher S, Hoffman G, McCann SM, et al. Rat heart: a site of oxytocin production and action. [In Vitro Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Proc Natl Acad Sci U S A. 1998;95(24):14558–63.
Rosen GJ, de Vries GJ, Goldman SL, Goldman BD, Forger NG. Distribution of oxytocin in the brain of a eusocial rodent. [Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, Non-P.H.S.]. Neuroscience. 2008;155(3):809–17. doi:10.1016/j.neuroscience.2008.05.039.
Yamashita M, Takayanagi Y, Yoshida M, Nishimori K, Kusama M, Onaka T. Involvement of prolactin releasing peptide in activation of oxytocin neurones in response to food intake. J Neuroendocrinol. 2013. doi:10.1111/jne.12019.
Shahrokh DK, Zhang TY, Diorio J, Gratton A, Meaney MJ. Oxytocin-dopamine interactions mediate variations in maternal behavior in the rat. Endocrinology. 2010;151(5):2276–86. doi:10.1210/en.2009-1271.
Rinaman L. Oxytocinergic inputs to the nucleus of the solitary tract and dorsal motor nucleus of the vagus in neonatal rats. J Comp Neurol. 1998;399(1):101–9.
Sawchenko PE, Swanson LW. Immunohistochemical identification of neurons in the paraventricular nucleus of the hypothalamus that project to the medulla or to the spinal cord in the rat. [Research Support, U.S. Gov’t, P.H.S.]. J Comp Neurol. 1982;205(3):260–72. doi:10.1002/cne.902050306.
Douglas AJ, Johnstone LE, Leng G. Neuroendocrine mechanisms of change in food intake during pregnancy: a potential role for brain oxytocin. [Research Support, Non-U.S. Gov’t Review]. Physiol Behav. 2007;91(4):352–65. doi:10.1016/j.physbeh.2007.04.012.
Welch MG, Tamir H, Gross KJ, Chen J, Anwar M, Gershon MD. Expression and developmental regulation of oxytocin (OT) and oxytocin receptors (OTR) in the enteric nervous system (ENS) and intestinal epithelium. J Comp Neurol. 2009;512(2):256–70. doi:10.1002/cne.21872.
Qin J, Feng M, Wang C, Ye Y, Wang PS, Liu C. Oxytocin receptor expressed on the smooth muscle mediates the excitatory effect of oxytocin on gastric motility in rats. [Research Support, Non-U.S. Gov’t]. Neurogastroenterol Motil. 2009;21(4):430–8. doi:10.1111/j.1365-2982.2009.01282.x.
Ohlsson B, Truedsson M, Djerf P, Sundler F. Oxytocin is expressed throughout the human gastrointestinal tract. [Research Support, Non-U.S. Gov’t]. Regul Pept. 2006;135(1–2):7–11. doi:10.1016/j.regpep.2006.03.008.
Wheeler E, Huang N, Bochukova EG, Keogh JM, Lindsay S, Garg S, et al. Genome-wide SNP and CNV analysis identifies common and low-frequency variants associated with severe early-onset obesity. Nat Genet. 2013. doi:10.1038/ng.2607.
Kenny PJ. Common cellular and molecular mechanisms in obesity and drug addiction. [Research Support, N.I.H., Extramural Review]. Nat Rev Neurosci. 2011;12(11):638–51. doi:10.1038/nrn3105.
Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiol Rev. 2001;81(2):629–83.
Verbalis JG. The brain oxytocin receptor(s)? [Review]. Front Neuroendocrinol. 1999;20(2):146–56. doi:10.1006/frne.1999.0178.
Yoshida M, Takayanagi Y, Inoue K, Kimura T, Young LJ, Onaka T, et al. Evidence that oxytocin exerts anxiolytic effects via oxytocin receptor expressed in serotonergic neurons in mice. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.]. J Neurosci. 2009;29(7):2259–71. doi:10.1523/JNEUROSCI.5593-08.2009.
Gould BR, Zingg HH. Mapping oxytocin receptor gene expression in the mouse brain and mammary gland using an oxytocin receptor-LacZ reporter mouse. [Research Support, Non-U.S. Gov’t]. Neuroscience. 2003;122(1):155–67.
Schaffler A, Binart N, Scholmerich J, Buchler C. Hypothesis paper Brain talks with fat–evidence for a hypothalamic-pituitary-adipose axis? [Review]. Neuropeptides. 2005;39(4):363–7. doi:10.1016/j.npep.2005.06.003.
Tsuda T, Ueno Y, Yoshikawa T, Kojo H, Osawa T. Microarray profiling of gene expression in human adipocytes in response to anthocyanins. [Research Support, Non-U.S. Gov’t]. Biochem Pharmacol. 2006;71(8):1184–97. doi:10.1016/j.bcp.2005.12.042.
van den Burg EH, Neumann ID. Bridging the gap between GPCR activation and behaviour: oxytocin and prolactin signalling in the hypothalamus. [Review]. J Mol Neurosci. 2011;43(2):200–8. doi:10.1007/s12031-010-9452-8.
Gravati M, Busnelli M, Bulgheroni E, Reversi A, Spaiardi P, Parenti M, et al. Dual modulation of inward rectifier potassium currents in olfactory neuronal cells by promiscuous G protein coupling of the oxytocin receptor. [Comparative Study Research Support, Non-U.S. Gov’t]. J Neurochem. 2010;114(5):1424–35. doi:10.1111/j.1471-4159.2010.06861.x.
Arletti R, Benelli A, Bertolini A. Influence of oxytocin on feeding behavior in the rat. Peptides. 1989;10(1):89–93.
Arletti R, Benelli A, Bertolini A. Oxytocin inhibits food and fluid intake in rats. Physiol Behav. 1990;48(6):825–30.
Lokrantz CM, Uvnas-Moberg K, Kaplan JM. Effects of central oxytocin administration on intraoral intake of glucose in deprived and nondeprived rats. [Research Support, Non-U.S. Gov’t]. Physiol Behav. 1997;62(2):347–52.
Olson BR, Drutarosky MD, Chow MS, Hruby VJ, Stricker EM, Verbalis JG. Oxytocin and an oxytocin agonist administered centrally decrease food intake in rats. Peptides. 1991;1991:113–8.
Rinaman L, Rothe EE. GLP-1 receptor signaling contributes to anorexigenic effect of centrally administered oxytocin in rats. Am J Physiol Regul Integr Comp Physiol. 2002;283(1):R99–106.
Blouet C, Jo YH, Li X, Schwartz GJ. Mediobasal hypothalamic leucine sensing regulates food intake through activation of a hypothalamus-brainstem circuit. J Neurosci. 2009;29(26):8302–11. doi:10.1523/JNEUROSCI.1668-09.2009.
Johnstone LE, Fong TM, Leng G. Neuronal activation in the hypothalamus and brainstem during feeding in rats. [Comparative Study Research Support, Non-U.S. Gov’t]. Cell Metab. 2006;4(4):313–21. doi:10.1016/j.cmet.2006.08.003.
Lucio-Oliveira F, Franci CR. Effect of the interaction between food state and the action of estrogen on oxytocinergic system activity. [Research Support, Non-U.S. Gov’t]. J Endocrinol. 2012;212(2):129–38. doi:10.1530/JOE-11-0272.
Olszewski PK, Klockars A, Olszewska AM, Fredriksson R, Schioth HB, Levine AS. Molecular, immunohistochemical, and pharmacological evidence of oxytocin’s role as inhibitor of carbohydrate but not fat intake. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Endocrinology. 2010;151(10):4736–44. doi:10.1210/en.2010-0151.
Singru PS, Wittmann G, Farkas E, Zseli G, Fekete C, Lechan RM. Refeeding-activated glutamatergic neurons in the hypothalamic paraventricular nucleus (PVN) mediate effects of melanocortin signaling in the nucleus tractus solitarius (NTS). [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Endocrinology. 2012;153(8):3804–14. doi:10.1210/en.2012-1235.
Olson BR, Hoffman GE, Sved AF, Stricker EM, Verbalis JG. Cholecystokinin induces c-fos expression in hypothalamic oxytocinergic neurons projecting to the dorsal vagal complex. Brain Res. 1992;569(2):238–48.
Renaud LP, Tang M, McCann MJ, Stricker EM, Verbalis JG. Cholecystokinin and gastric distension activate oxytocinergic cells in rat hypothalamus. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Am J Physiol. 1987;253(4 Pt 2):R661–665.
Nelson EE, Alberts JR, Tian Y, Verbalis JG. Oxytocin is elevated in plasma of 10-day-old rats following gastric distension. Brain Res Dev Brain Res. 1998;111(2):301–3.
Zhang J, Liu S, Tang M, Chen JD. Optimal locations and parameters of gastric electrical stimulation in altering ghrelin and oxytocin in the hypothalamus of rats. [Research Support, Non-U.S. Gov’t]. Neurosci Res. 2008;62(4):262–9. doi:10.1016/j.neures.2008.09.004.
Tang M, Zhang J, Xu L, Chen JD. Implantable gastric stimulation alters expression of oxytocin- and orexin-containing neurons in the hypothalamus of rats. [Research Support, Non-U.S. Gov’t]. Obes Surg. 2006;16(6):762–9. doi:10.1381/096089206777346745.
Ueta Y, Kannan H, Higuchi T, Negoro H, Yamaguchi K, Yamashita H. Activation of gastric afferents increases noradrenaline release in the paraventricular nucleus and plasma oxytocin level. [Comparative Study Research Support, Non-U.S. Gov’t]. J Auton Nerv Syst. 2000;78(2–3):69–76.
Gaetani S, Fu J, Cassano T, Dipasquale P, Romano A, Righetti L, et al. The fat-induced satiety factor oleoylethanolamide suppresses feeding through central release of oxytocin. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. J Neurosci. 2010;30(24):8096–101. doi:10.1523/JNEUROSCI.0036-10.2010.
Flak JN, Jankord R, Solomon MB, Krause EG, Herman JP. Opposing effects of chronic stress and weight restriction on cardiovascular, neuroendocrine and metabolic function. [Research Support, N.I.H., Extramural]. Physiol Behav. 2011;104(2):228–34. doi:10.1016/j.physbeh.2011.03.002.
Tung YC, Ma M, Piper S, Coll A, O’Rahilly S, Yeo GS. Novel leptin-regulated genes revealed by transcriptional profiling of the hypothalamic paraventricular nucleus. [Research Support, Non-U.S. Gov’t]. J Neurosci. 2008;28(47):12419–26. doi:10.1523/JNEUROSCI.3412-08.2008.
Blevins JE, Schwartz MW, Baskin DG. Evidence that paraventricular nucleus oxytocin neurons link hypothalamic leptin action to caudal brain stem nuclei controlling meal size. Am J Physiol Regul Integr Comp Physiol. 2004;287(1):R87–96. doi:10.1152/ajpregu.00604.2003.
Takayanagi Y, Kasahara Y, Onaka T, Takahashi N, Kawada T, Nishimori K. Oxytocin receptor-deficient mice developed late-onset obesity. [Research Support, Non-U.S. Gov’t]. Neuroreport. 2008;19(9):951–5. doi:10.1097/WNR.0b013e3283021ca9.
Dombret C, Nguyen T, Schakman O, Michaud JL, Hardin-Pouzet H, Bertrand MJ, et al. Loss of Maged1 results in obesity, deficits of social interactions, impaired sexual behavior and severe alteration of mature oxytocin production in the hypothalamus. [Research Support, Non-U.S. Gov’t]. Hum Mol Genet. 2012;21(21):4703–17. doi:10.1093/hmg/dds310.
Swaab DF, Purba JS, Hofman MA. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: a study of five cases. [Case Reports Research Support, Non-U.S. Gov’t]. J Clin Endocrinol Metab. 1995;80(2):573–9.
Wu Z, Xu Y, Zhu Y, Sutton AK, Zhao R, Lowell BB, et al. An obligate role of oxytocin neurons in diet induced energy expenditure. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. PLoS One. 2012;7(9):e45167. doi:10.1371/journal.pone.0045167.
Wu Q, Howell MP, Palmiter RD. Ablation of neurons expressing agouti-related protein activates fos and gliosis in postsynaptic target regions. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. J Neurosci. 2008;28(37):9218–26. doi:10.1523/JNEUROSCI.2449-08.2008.
Lukas M, Bredewold R, Neumann ID, Veenema AH. Maternal separation interferes with developmental changes in brain vasopressin and oxytocin receptor binding in male rats. Neuropharmacology. 2010;58(1):78–87. doi:10.1016/j.neuropharm.2009.06.020.
Rogers RC, Hermann GE. Oxytocin, oxytocin antagonist, TRH, and hypothalamic paraventricular nucleus stimulation effects on gastric motility. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Peptides. 1987;8(3):505–13.
McCann MJ, Rogers RC. Oxytocin excites gastric-related neurones in rat dorsal vagal complex. [Research Support, U.S. Gov’t, P.H.S.]. J Physiol. 1990;428:95–108.
Borg J, Simren M, Ohlsson B. Oxytocin reduces satiety scores without affecting the volume of nutrient intake or gastric emptying rate in healthy subjects. [Research Support, Non-U.S. Gov’t]. Neurogastroenterol Motil. 2011;23(1):56–61. doi:10.1111/j.1365-2982.2010.01599.x. e55.
McCann MJ, Verbalis JG, Stricker EM. LiCl and CCK inhibit gastric emptying and feeding and stimulate OT secretion in rats. [Research Support, U.S. Gov’t, P.H.S.]. Am J Physiol. 1989;256(2 Pt 2):R463–468.
Li L, Kong X, Liu H, Liu C. Systemic oxytocin and vasopressin excite gastrointestinal motility through oxytocin receptor in rabbits. [Research Support, Non-U.S. Gov’t]. Neurogastroenterol Motil. 2007;19(10):839–44. doi:10.1111/j.1365-2982.2007.00953.x.
Wu CL, Doong ML, Wang PS. Involvement of cholecystokinin receptor in the inhibition of gastrointestinal motility by oxytocin in ovariectomized rats. [Research Support, Non-U.S. Gov’t]. Eur J Pharmacol. 2008;580(3):407–15. doi:10.1016/j.ejphar.2007.11.024.
Wu CL, Hung CR, Chang FY, Pau KY, Wang PS. Pharmacological effects of oxytocin on gastric emptying and intestinal transit of a non-nutritive liquid meal in female rats. [Comparative Study Research Support, Non-U.S. Gov’t]. Naunyn Schmiedebergs Arch Pharmacol. 2003;367(4):406–13. doi:10.1007/s00210-003-0690-y.
Amico JA, Vollmer RR, Cai HM, Miedlar JA, Rinaman L. Enhanced initial and sustained intake of sucrose solution in mice with an oxytocin gene deletion. [Research Support, N.I.H., Extramural]. Am J Physiol Regul Integr Comp Physiol. 2005;289(6):R1798–1806. doi:10.1152/ajpregu.00558.2005.
Sclafani A, Rinaman L, Vollmer RR, Amico JA. Oxytocin knockout mice demonstrate enhanced intake of sweet and nonsweet carbohydrate solutions. [Research Support, N.I.H., Extramural]. Am J Physiol Regul Integr Comp Physiol. 2007;292(5):R1828–1833. doi:10.1152/ajpregu.00826.2006.
Mullis K, Kay K, Williams DL. Oxytocin action in the ventral tegmental area affects sucrose intake. Brain Res. 2013. doi:10.1016/j.brainres.2013.03.026.
Boccia ML, Goursaud AP, Bachevalier J, Anderson KD, Pedersen CA. Peripherally administered non-peptide oxytocin antagonist, L368,899, accumulates in limbic brain areas: a new pharmacological tool for the study of social motivation in non-human primates. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Horm Behav. 2007;52(3):344–51. doi:10.1016/j.yhbeh.2007.05.009.
Olszewski PK, Levine AS. Central opioids and consumption of sweet tastants: when reward outweighs homeostasis. [Review]. Physiol Behav. 2007;91(5):506–12. doi:10.1016/j.physbeh.2007.01.011.
Mitra A, Gosnell BA, Schioth HB, Grace MK, Klockars A, Olszewski PK, et al. Chronic sugar intake dampens feeding-related activity of neurons synthesizing a satiety mediator, oxytocin. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Peptides. 2010;31(7):1346–52. doi:10.1016/j.peptides.2010.04.005.
Leng G, Brown CH, Murphy NP, Onaka T, Russell JA. Opioid-noradrenergic interactions in the control of oxytocin cells. [Research Support, Non-U.S. Gov’t]. Adv Exp Med Biol. 1995;395:95–104.
Carson DS, Cornish JL, Guastella AJ, Hunt GE, McGregor IS. Oxytocin decreases methamphetamine self-administration, methamphetamine hyperactivity, and relapse to methamphetamine-seeking behaviour in rats. [Research Support, Non-U.S. Gov’t]. Neuropharmacology. 2010;58(1):38–43. doi:10.1016/j.neuropharm.2009.06.018.
Carson DS, Hunt GE, Guastella AJ, Barber L, Cornish JL, Arnold JC, et al. Systemically administered oxytocin decreases methamphetamine activation of the subthalamic nucleus and accumbens core and stimulates oxytocinergic neurons in the hypothalamus. [Research Support, Non-U.S. Gov’t]. Addict Biol. 2010;15(4):448–63. doi:10.1111/j.1369-1600.2010.00247.x.
Qi J, Yang JY, Song M, Li Y, Wang F, Wu CF. Inhibition by oxytocin of methamphetamine-induced hyperactivity related to dopamine turnover in the mesolimbic region in mice. [Research Support, Non-U.S. Gov’t]. Naunyn Schmiedebergs Arch Pharmacol. 2008;376(6):441–8. doi:10.1007/s00210-007-0245-8.
Covasa M, Ritter RC. Rats maintained on high-fat diets exhibit reduced satiety in response to CCK and bombesin. Peptides. 1998;19(8):1407–15.
Covasa M, Grahn J, Ritter RC. High fat maintenance diet attenuates hindbrain neuronal response to CCK. Regul Pept. 2000;86(1–3):83–8.
Hisadome K, Reimann F, Gribble FM, Trapp S. CCK stimulation of GLP-1 neurons involves alpha1-adrenoceptor-mediated increase in glutamatergic synaptic inputs. [In Vitro Research Support, Non-U.S. Gov’t]. Diabetes. 2011;60(11):2701–9. doi:10.2337/db11-0489.
Appleyard SM, Marks D, Kobayashi K, Okano H, Low MJ, Andresen MC. Visceral afferents directly activate catecholamine neurons in the solitary tract nucleus. [In Vitro Research Support, N.I.H., Extramural]. J Neurosci. 2007;27(48):13292–302. doi:10.1523/JNEUROSCI.3502-07.2007.
Rinaman L. Hindbrain noradrenergic lesions attenuate anorexia and alter central cFos expression in rats after gastric viscerosensory stimulation. [Research Support, U.S. Gov’t, P.H.S.]. J Neurosci. 2003;23(31):10084–92.
Bechtold DA, Luckman SM. Prolactin-releasing Peptide mediates cholecystokinin-induced satiety in mice. [Research Support, Non-U.S. Gov’t]. Endocrinology. 2006;147(10):4723–9. doi:10.1210/en.2006-0753.
Maniscalco JW, Kreisler AD, Rinaman L. Satiation and stress-induced hypophagia: examining the role of hindbrain neurons expressing prolactin-releasing Peptide or glucagon-like Peptide 1. Front Neurosci. 2012;6:199. doi:10.3389/fnins.2012.00199.
Appleyard SM, Bailey TW, Doyle MW, Jin YH, Smart JL, Low MJ, et al. Proopiomelanocortin neurons in nucleus tractus solitarius are activated by visceral afferents: regulation by cholecystokinin and opioids. [Comparative Study In Vitro Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, P.H.S.]. J Neurosci. 2005;25(14):3578–85. doi:10.1523/JNEUROSCI.4177-04.2005.
Fan W, Ellacott KL, Halatchev IG, Takahashi K, Yu P, Cone RD. Cholecystokinin-mediated suppression of feeding involves the brainstem melanocortin system. [Comparative Study Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Nat Neurosci. 2004;7(4):335–6. doi:10.1038/nn1214.
Baskin DG, Kim F, Gelling RW, Russell BJ, Schwartz MW, Morton GJ, et al. A new oxytocin-saporin cytotoxin for lesioning oxytocin-receptive neurons in the rat hindbrain. [Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, Non-P.H.S.]. Endocrinology. 2010;151(9):4207–13. doi:10.1210/en.2010-0295.
Blevins JE, Eakin TJ, Murphy JA, Schwartz MW, Baskin DG. Oxytocin innervation of caudal brainstem nuclei activated by cholecystokinin. Brain Res. 2003;993(1–2):30–41.
Matarazzo V, Schaller F, Nedelec E, Benani A, Penicaud L, Muscatelli F, et al. Inactivation of Socs3 in the hypothalamus enhances the hindbrain response to endogenous satiety signals via oxytocin signaling. [Research Support, Non-U.S. Gov’t]. J Neurosci. 2012;32(48):17097–107. doi:10.1523/JNEUROSCI.1669-12.2012.
Olson BR, Drutarosky MD, Stricker EM, Verbalis JG. Brain oxytocin receptor antagonism blunts the effects of anorexigenic treatments in rats: evidence for central oxytocin inhibition of food intake. [Research Support, U.S. Gov’t, P.H.S.]. Endocrinology. 1991;129(2):785–91.
Alhadeff AL, Rupprecht LE, Hayes MR. GLP-1 neurons in the nucleus of the solitary tract project directly to the ventral tegmental area and nucleus accumbens to control for food intake. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Endocrinology. 2012;153(2):647–58. doi:10.1210/en.2011-1443.
Mejias-Aponte CA, Drouin C, Aston-Jones G. Adrenergic and noradrenergic innervation of the midbrain ventral tegmental area and retrorubral field: prominent inputs from medullary homeostatic centers. [Comparative Study Research Support, U.S. Gov’t, P.H.S.]. J Neurosci. 2009;29(11):3613–26. doi:10.1523/JNEUROSCI.4632-08.2009.
Dossat AM, Lilly N, Kay K, Williams DL. Glucagon-like peptide 1 receptors in nucleus accumbens affect food intake. [Research Support, N.I.H., Extramural]. J Neurosci. 2011;31(41):14453–7. doi:10.1523/JNEUROSCI.3262-11.2011.
Delfs JM, Zhu Y, Druhan JP, Aston-Jones GS. Origin of noradrenergic afferents to the shell subregion of the nucleus accumbens: anterograde and retrograde tract-tracing studies in the rat. [Research Support, U.S. Gov’t, P.H.S.]. Brain Res. 1998;806(2):127–40.
Wang ZJ, Rao ZR, Shi JW. Tyrosine hydroxylase-, neurotensin-, or cholecystokinin-containing neurons in the nucleus tractus solitarii send projection fibers to the nucleus accumbens in the rat. [Research Support, Non-U.S. Gov’t]. Brain Res. 1992;578(1–2):347–50.
Hayes MR, Bradley L, Grill HJ. Endogenous hindbrain glucagon-like peptide-1 receptor activation contributes to the control of food intake by mediating gastric satiation signaling. [Research Support, N.I.H., Extramural]. Endocrinology. 2009;150(6):2654–9. doi:10.1210/en.2008-1479.
Barrera JG, Jones KR, Herman JP, D’Alessio DA, Woods SC, Seeley RJ. Hyperphagia and increased fat accumulation in two models of chronic CNS glucagon-like peptide-1 loss of function. [Research Support, N.I.H., Extramural]. J Neurosci. 2011;31(10):3904–13. doi:10.1523/JNEUROSCI.2212-10.2011.
Zhang Y, Rodrigues E, Gao YX, King M, Cheng KY, Erdos B, et al. Pro-opiomelanocortin gene transfer to the nucleus of the solitary track but not arcuate nucleus ameliorates chronic diet-induced obesity. [Research Support, N.I.H., Extramural]. Neuroscience. 2010;169(4):1662–71. doi:10.1016/j.neuroscience.2010.06.001.
Li G, Zhang Y, Rodrigues E, Zheng D, Matheny M, Cheng KY, et al. Melanocortin activation of nucleus of the solitary tract avoids anorectic tachyphylaxis and induces prolonged weight loss. [Evaluation Studies Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, Non-P.H.S.]. Am J Physiol Endocrinol Metab. 2007;293(1):E252–258. doi:10.1152/ajpendo.00451.2006.
Baird JP, Palacios M, LaRiviere M, Grigg LA, Lim C, Matute E, et al. Anatomical dissociation of melanocortin receptor agonist effects on taste- and gut-sensitive feeding processes. [Comparative Study Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Am J Physiol Regul Integr Comp Physiol. 2011;301(4):R1044–1056. doi:10.1152/ajpregu.00577.2010.
Covasa M, Ritter RC. Reduced sensitivity to the satiation effect of intestinal oleate in rats adapted to high-fat diet. [Research Support, U.S. Gov’t, P.H.S.]. Am J Physiol. 1999;277(1 Pt 2):R279–285.
Donovan MJ, Paulino G, Raybould HE. Activation of hindbrain neurons in response to gastrointestinal lipid is attenuated by high fat, high energy diets in mice prone to diet-induced obesity. [Research Support, N.I.H., Extramural]. Brain Res. 2009;1248:136–40. doi:10.1016/j.brainres.2008.10.042.
Olson VG, Heusner CL, Bland RJ, During MJ, Weinshenker D, Palmiter RD. Role of noradrenergic signaling by the nucleus tractus solitarius in mediating opiate reward. [Research Support, N.I.H., Extramural]. Science. 2006;311(5763):1017–20. doi:10.1126/science.1119311.
Camerino C. Low sympathetic tone and obese phenotype in oxytocin-deficient mice. Obesity (Silver Spring). 2009;17(5):980–4. doi:10.1038/oby.2009.12.
Holder Jr JL, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. [Case Reports Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.]. Hum Mol Genet. 2000;9(1):101–8.
Swarbrick MM, Evans DS, Valle MI, Favre H, Wu SH, Njajou OT, et al. Replication and extension of association between common genetic variants in SIM1 and human adiposity. [Research Support, N.I.H., Extramural Research Support, N.I.H., Intramural Research Support, Non-U.S. Gov’t]. Obesity (Silver Spring). 2011;19(12):2394–403. doi:10.1038/oby.2011.79.
Backman SB, Henry JL. Effects of oxytocin and vasopressin on thoracic sympathetic preganglionic neurones in the cat. [Comparative Study Research Support, Non-U.S. Gov’t]. Brain Res Bull. 1984;13(5):679–84.
Armour JA, Klassen GA. Oxytocin modulation of intrathoracic sympathetic ganglionic neurons regulating the canine heart. [Research Support, Non-U.S. Gov’t]. Peptides. 1990;11(3):533–7.
Oldfield BJ, Giles ME, Watson A, Anderson C, Colvill LM, McKinley MJ. The neurochemical characterisation of hypothalamic pathways projecting polysynaptically to brown adipose tissue in the rat. [Research Support, Non-U.S. Gov’t]. Neuroscience. 2002;110(3):515–26.
Jansen AS, Wessendorf MW, Loewy AD. Transneuronal labeling of CNS neuropeptide and monoamine neurons after pseudorabies virus injections into the stellate ganglion. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Brain Res. 1995;683(1):1–24.
Higa KT, Mori E, Viana FF, Morris M, Michelini LC. Baroreflex control of heart rate by oxytocin in the solitary-vagal complex. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Am J Physiol Regul Integr Comp Physiol. 2002;282(2):R537–545. doi:10.1152/ajpregu.00806.2000.
Xi D, Gandhi N, Lai M, Kublaoui BM. Ablation of Sim1 neurons causes obesity through hyperphagia and reduced energy expenditure. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. PLoS One. 2012;7(4):e36453. doi:10.1371/journal.pone.0036453.
Lockie SH, Heppner KM, Chaudhary N, Chabenne JR, Morgan DA, Veyrat-Durebex C, et al. Direct control of brown adipose tissue thermogenesis by central nervous system glucagon-like peptide-1 receptor signaling. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Diabetes. 2012;61(11):2753–62. doi:10.2337/db11-1556.
Muchmore DB, Little SA, de Haen C. A dual mechanism of action of ocytocin in rat epididymal fat cells. [Comparative Study In Vitro Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. J Biol Chem. 1981;256(1):365–72.
Shi H, Bartness TJ. Neurochemical phenotype of sympathetic nervous system outflow from brain to white fat. [Research Support, U.S. Gov’t, P.H.S.]. Brain Res Bull. 2001;54(4):375–85.
Stanley S, Pinto S, Segal J, Perez CA, Viale A, DeFalco J, et al. Identification of neuronal subpopulations that project from hypothalamus to both liver and adipose tissue polysynaptically. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Proc Natl Acad Sci U S A. 2010;107(15):7024–9. doi:10.1073/pnas.1002790107.
Ho JM, Blevins JE. Coming full circle: contributions of central and peripheral oxytocin actions to energy balance. Endocrinology. 2013;154(2):589–96. doi:10.1210/en.2012-1751.
Schwartz MW, Woods SC, Porte Jr D, Seeley RJ, Baskin DG. Central nervous system control of food intake. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S. Research Support, U.S. Gov’t, P.H.S. Review]. Nature. 2000;404(6778):661–71. doi:10.1038/35007534.
Woods SC, Schwartz MW, Baskin DG, Seeley RJ. Food intake and the regulation of body weight. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S. Research Support, U.S. Gov’t, P.H.S. Review]. Annu Rev Psychol. 2000;51:255–77. doi:10.1146/annurev.psych.51.1.255.
Kaplan JM, Seeley RJ, Grill HJ. Daily caloric intake in intact and chronic decerebrate rats. Behav Neurosci. 1993;107:876–81.
Shapiro RE, Miselis RR. The central organization of the vagus nerve innervating the stomach of the rat. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. J Comp Neurol. 1985;238(4):473–88. doi:10.1002/cne.902380411.
Olson BR, Freilino M, Hoffman GE, Stricker EM, Sved AF, Verbalis JG. c-Fos expression in rat brain and brainstem nuclei in response to treatments that alter food intake and gastric motility. Mol Cell Neurosci. 1993;4:93–106.
Edwards GL, Ladenheim EE, Ritter RC. Dorsomedial hindbrain participation in cholecystokinin-induced satiety. [Research Support, U.S. Gov’t, P.H.S.]. Am J Physiol. 1986;251(5 Pt 2):R971–977.
Fraser KA, Raizada E, Davison JS. Oral-pharyngeal-esophageal and gastric cues contribute to meal-induced c-fos expression. Am J Physiol. 1995;268(1 Pt 2):R223–230.
Willing AE, Berthoud HR. Gastric distension-induced c-fos expression in catecholaminergic neurons of rat dorsal vagal complex. [Research Support, U.S. Gov’t, P.H.S.]. Am J Physiol. 1997;272(1 Pt 2):R59–67.
Sawchenko PE, Swanson LW. The organization of noradrenergic pathways from the brainstem to the paraventricular and supraoptic nuclei in the rat. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Brain Res. 1982;257(3):275–325.
Crawley JN, Kiss JZ. Paraventricular nucleus lesions abolish the inhibition of feeding induced by systemic cholecystokinin. Peptides. 1985;6(5):927–35.
Leibowitz SF, Hammer NJ, Chang K. Hypothalamic paraventricular nucleus lesions produce overeating and obesity in the rat. Physiol Behav. 1981;27(6):1031–40.
Kirchgessner AL, Sclafani A. PVN-hindbrain pathway involved in the hypothalamic hyperphagia-obesity syndrome. Physiol Behav. 1988;42:517–28.
Qi Y, Henry BA, Oldfield BJ, Clarke IJ. The action of leptin on appetite-regulating cells in the ovine hypothalamus: demonstration of direct action in the absence of the arcuate nucleus. Endocrinology. 2010;151(5):2106–16. doi:10.1210/en.2009-1283.
Perello M, Raingo J. Leptin activates oxytocin neurons of the hypothalamic paraventricular nucleus in both control and diet-induced obese rodents. PLoS One. 2013;8(3):e59625. doi:10.1371/journal.pone.0059625.
Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Endocrinology. 2004;145(11):4880–9. doi:10.1210/en.2004-0726.
Baskin DG, Schwartz MW, Seeley RJ, Woods SC, Porte Jr D, Breininger JF, et al. Leptin receptor long-form splice-variant protein expression in neuron cell bodies of the brain and co-localization with neuropeptide Y mRNA in the arcuate nucleus. [Research Support, U.S. Gov’t, Non-P.H.S. Research Support, U.S. Gov’t, P.H.S.]. J Histochem Cytochem. 1999;47(3):353–62.
Seeley RJ, Yagaloff KA, Fisher SL, Burn P, Thiele TE, van Dijk G, et al. Melanocortin receptors in leptin effects. [Letter]. Nature. 1997;390(6658):349. doi:10.1038/37016.
Olszewski PK, Wirth MM, Shaw TJ, Grace MK, Billington CJ, Giraudo SQ, et al. Role of alpha-MSH in the regulation of consummatory behavior: immunohistochemical evidence. [Research Support, U.S. Gov’t, Non-P.H.S. Research Support, U.S. Gov’t, P.H.S.]. Am J Physiol Regul Integr Comp Physiol. 2001;281(2):R673–680.
Liu H, Kishi T, Roseberry AG, Cai X, Lee CE, Montez JM, et al. Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoter. [In Vitro Research Support, U.S. Gov’t, P.H.S.]. J Neurosci. 2003;23(18):7143–54.
Blevins JE, Morton GJ, Williams DL, Caldwell DW, Bastian LS, Wisse BE, et al. Forebrain melanocortin signaling enhances the hindbrain satiety response to CCK-8. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.]. Am J Physiol Regul Integr Comp Physiol. 2009;296(3):R476–484. doi:10.1152/ajpregu.90544.2008.
Yosten GL, Samson WK. The anorexigenic and hypertensive effects of nesfatin-1 are reversed by pretreatment with an oxytocin receptor antagonist. [Research Support, N.I.H., Extramural]. Am J Physiol Regul Integr Comp Physiol. 2010;298(6):R1642–1647. doi:10.1152/ajpregu.00804.2009.
Myers Jr MG, Leibel RL, Seeley RJ, Schwartz MW. Obesity and leptin resistance: distinguishing cause from effect. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Review]. Trends Endocrinol Metab. 2010;21(11):643–51. doi:10.1016/j.tem.2010.08.002.
Van Heek M, Compton DS, France CF, Tedesco RP, Fawzi AB, Graziano MP, et al. Diet-induced obese mice develop peripheral, but not central, resistance to leptin. [Clinical Trial]. J Clin Invest. 1997;99(3):385–90. doi:10.1172/JCI119171.
Lin S, Thomas TC, Storlien LH, Huang XF. Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. [Research Support, Non-U.S. Gov’t]. Int J Obes Relat Metab Disord. 2000;24(5):639–46.
Banks WA, DiPalma CR, Farrell CL. Impaired transport of leptin across the blood–brain barrier in obesity. [Research Support, U.S. Gov’t, Non-P.H.S. Research Support, U.S. Gov’t, P.H.S.]. Peptides. 1999;20(11):1341–5.
Knight ZA, Hannan KS, Greenberg ML, Friedman JM. Hyperleptinemia is required for the development of leptin resistance. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. PLoS One. 2010;5(6):e11376. doi:10.1371/journal.pone.0011376.
El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS. Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. J Clin Invest. 2000;105(12):1827–32. doi:10.1172/JCI9842.
Milagro FI, Campion J, Garcia-Diaz DF, Goyenechea E, Paternain L, Martinez JA. High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. [Research Support, Non-U.S. Gov’t]. J Physiol Biochem. 2009;65(1):1–9.
De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, et al. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. [Research Support, Non-U.S. Gov’t]. Endocrinology. 2005;146(10):4192–9. doi:10.1210/en.2004-1520.
Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. [Comparative Study Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Nat Med. 2006;12(8):917–24. doi:10.1038/nm1435.
Loh K, Fukushima A, Zhang X, Galic S, Briggs D, Enriori PJ, et al. Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Cell Metab. 2011;14(5):684–99. doi:10.1016/j.cmet.2011.09.011.
Lee HJ, Caldwell HK, Macbeth AH, Tolu SG, Young 3rd WS. A conditional knockout mouse line of the oxytocin receptor. [Research Support, N.I.H., Extramural Research Support, N.I.H., Intramural]. Endocrinology. 2008;149(7):3256–63. doi:10.1210/en.2007-1710.
Hicks C, Jorgensen W, Brown C, Fardell J, Koehbach J, Gruber CW, et al. The nonpeptide oxytocin receptor agonist WAY 267,464: receptor-binding profile, prosocial effects and distribution of c-Fos expression in adolescent rats. [Research Support, Non-U.S. Gov’t]. J Neuroendocrinol. 2012;24(7):1012–29. doi:10.1111/j.1365-2826.2012.02311.x.
Mens WB, Witter A, van Wimersma Greidanus TB. Penetration of neurohypophyseal hormones from plasma into cerebrospinal fluid (CSF): half-times of disappearance of these neuropeptides from CSF. [Research Support, Non-U.S. Gov’t]. Brain Res. 1983;262(1):143–9.
Kendrick KM, Keverne EB, Baldwin BA, Sharman DF. Cerebrospinal fluid levels of acetylcholinesterase, monoamines and oxytocin during labour, parturition, vaginocervical stimulation, lamb separation and suckling in sheep. [Research Support, Non-U.S. Gov’t]. Neuroendocrinology. 1986;44(2):149–56.
Neumann ID, Maloumby R, Beiderbeck DI, Lukas M, Landgraf R. Increased brain and plasma oxytocin after nasal and peripheral administration in rats and mice. Psychoneuroendocrinology. 2013. doi:10.1016/j.psyneuen.2013.03.003.
Emch GS, Hermann GE, Rogers RC. TNF-alpha-induced c-Fos generation in the nucleus of the solitary tract is blocked by NBQX and MK-801. [Research Support, U.S. Gov’t, P.H.S.]. Am J Physiol Regul Integr Comp Physiol. 2001;281(5):R1394–1400.
Maolood N, Meister B. Protein components of the blood–brain barrier (BBB) in the brainstem area postrema-nucleus tractus solitarius region. [Research Support, Non-U.S. Gov’t]. J Chem Neuroanat. 2009;37(3):182–95. doi:10.1016/j.jchemneu.2008.12.007.
Banks WA. Blood–brain barrier and energy balance. [Review]. Obesity (Silver Spring). 2006;14 Suppl 5:234S–7S. doi:10.1038/oby.2006.315.
Banks WA. The blood–brain barrier as a regulatory interface in the gut-brain axes. [Review]. Physiol Behav. 2006;89(4):472–6. doi:10.1016/j.physbeh.2006.07.004.
Ring RH, Malberg JE, Potestio L, Ping J, Boikess S, Luo B, et al. Anxiolytic-like activity of oxytocin in male mice: behavioral and autonomic evidence, therapeutic implications. Psychopharmacology (Berl). 2006;185(2):218–25. doi:10.1007/s00213-005-0293-z.
Vrang N, Larsen PJ, Kristensen P, Tang-Christensen M. Central administration of cocaine-amphetamine-regulated transcript activates hypothalamic neuroendocrine neurons in the rat. [Research Support, Non-U.S. Gov’t]. Endocrinology. 2000;141(2):794–801.
Kristensen P, Judge ME, Thim L, Ribel U, Christjansen KN, Wulff BS, et al. Hypothalamic CART is a new anorectic peptide regulated by leptin. [Research Support, Non-U.S. Gov’t]. Nature. 1998;393(6680):72–6. doi:10.1038/29993.
Olszewski PK, Fredriksson R, Olszewska AM, Stephansson O, Alsio J, Radomska KJ, et al. Hypothalamic FTO is associated with the regulation of energy intake not feeding reward. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. BMC Neurosci. 2009;10:129. doi:10.1186/1471-2202-10-129.
Olszewski PK, Fredriksson R, Eriksson JD, Mitra A, Radomska KJ, Gosnell BA, et al. Fto colocalizes with a satiety mediator oxytocin in the brain and upregulates oxytocin gene expression. [Research Support, Non-U.S. Gov’t]. Biochem Biophys Res Commun. 2011;408(3):422–6. doi:10.1016/j.bbrc.2011.04.037.
Church C, Moir L, McMurray F, Girard C, Banks GT, Teboul L, et al. Overexpression of Fto leads to increased food intake and results in obesity. [Research Support, Non-U.S. Gov’t]. Nat Genet. 2010;42(12):1086–92. doi:10.1038/ng.713.
Fischer J, Koch L, Emmerling C, Vierkotten J, Peters T, Bruning JC, et al. Inactivation of the Fto gene protects from obesity. [Research Support, Non-U.S. Gov’t]. Nature. 2009;458(7240):894–8. doi:10.1038/nature07848.
Church C, Lee S, Bagg EA, McTaggart JS, Deacon R, Gerken T, et al. A mouse model for the metabolic effects of the human fat mass and obesity associated FTO gene. [Research Support, Non-U.S. Gov’t]. PLoS Genet. 2009;5(8):e1000599. doi:10.1371/journal.pgen.1000599.
Blouet C, Schwartz GJ. Brainstem nutrient sensing in the nucleus of the solitary tract inhibits feeding. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Cell Metab. 2012;16(5):579–87. doi:10.1016/j.cmet.2012.10.003.
Shimizu H, Oh IS, Okada S, Mori M. Nesfatin-1: an overview and future clinical application. [Review]. Endocr J. 2009;56(4):537–43.
Leone TC, Lehman JJ, Finck BN, Schaeffer PJ, Wende AR, Boudina S, et al. PGC-1alpha deficiency causes multi-system energy metabolic derangements: muscle dysfunction, abnormal weight control and hepatic steatosis. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. PLoS Biol. 2005;3(4):e101. doi:10.1371/journal.pbio.0030101.
Blechman J, Amir-Zilberstein L, Gutnick A, Ben-Dor S, Levkowitz G. The metabolic regulator PGC-1alpha directly controls the expression of the hypothalamic neuropeptide oxytocin. [Research Support, Non-U.S. Gov’t]. J Neurosci. 2011;31(42):14835–40. doi:10.1523/JNEUROSCI.1798-11.2011.
Atasoy D, Betley JN, Su HH, Sternson SM. Deconstruction of a neural circuit for hunger. [Research Support, Non-U.S. Gov’t]. Nature. 2012;488(7410):172–7. doi:10.1038/nature11270.
Smith MJ, Wise PM. Localization of kappa opioid receptors in oxytocin magnocellular neurons in the paraventricular and supraoptic nuclei. [Research Support, U.S. Gov’t, P.H.S.]. Brain Res. 2001;898(1):162–5.
Olszewski PK, Shi Q, Billington CJ, Levine AS. Opioids affect acquisition of LiCl-induced conditioned taste aversion: involvement of OT and VP systems. [Research Support, U.S. Gov’t, Non-P.H.S. Research Support, U.S. Gov’t, P.H.S.]. Am J Physiol Regul Integr Comp Physiol. 2000;279(4):R1504–1511.
Brunton PJ, Sabatier N, Leng G, Russell JA. Suppressed oxytocin neuron responses to immune challenge in late pregnant rats: a role for endogenous opioids. [Research Support, Non-U.S. Gov’t]. Eur J Neurosci. 2006;23(5):1241–7. doi:10.1111/j.1460-9568.2006.04614.x.
Douglas AJ, Neumann I, Meeren HK, Leng G, Johnstone LE, Munro G, et al. Central endogenous opioid inhibition of supraoptic oxytocin neurons in pregnant rats. [Research Support, Non-U.S. Gov’t]. J Neurosci. 1995;15(7 Pt 1):5049–57.
Kirchgessner AL, Sclafani A, Nilaver G. Histochemical identification of a PVN-hindbrain feeding pathway. Physiol Behav. 1988;42(6):529–43.
Peters JH, McDougall SJ, Kellett DO, Jordan D, Llewellyn-Smith IJ, Andresen MC. Oxytocin enhances cranial visceral afferent synaptic transmission to the solitary tract nucleus. [In Vitro Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. J Neurosci. 2008;28(45):11731–40. doi:10.1523/JNEUROSCI.3419-08.2008.
Sutton GM, Patterson LM, Berthoud HR. Extracellular signal-regulated kinase 1/2 signaling pathway in solitary nucleus mediates cholecystokinin-induced suppression of food intake in rats. [Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, P.H.S.]. J Neurosci. 2004;24(45):10240–7. doi:10.1523/JNEUROSCI.2764-04.2004.
Blevins JE, Chelikani PK, Haver AC, Reidelberger RD. PYY(3–36) induces Fos in the arcuate nucleus and in both catecholaminergic and non-catecholaminergic neurons in the nucleus tractus solitarius of rats. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.]. Peptides. 2008;29(1):112–9. doi:10.1016/j.peptides.2007.11.003.
Valentine JD, Matta SG, Sharp BM. Nicotine-induced cFos expression in the hypothalamic paraventricular nucleus is dependent on brainstem effects: correlations with cFos in catecholaminergic and noncatecholaminergic neurons in the nucleus tractus solitarius. [Research Support, U.S. Gov’t, P.H.S.]. Endocrinology. 1996;137(2):622–30.
Faipoux R, Tome D, Gougis S, Darcel N, Fromentin G. Proteins activate satiety-related neuronal pathways in the brainstem and hypothalamus of rats. [Research Support, Non-U.S. Gov’t]. J Nutr. 2008;138(6):1172–8.
Rinaman L, Verbalis JG, Stricker EM, Hoffman GE. Distribution and neurochemical phenotypes of caudal medullary neurons activated to express cFos following peripheral administration of cholecystokinin. J Comp Neurol. 1993;338(4):475–90. doi:10.1002/cne.903380402.
Blevins JE, Ho JM, Hwang BJ, Thatcher BS, Anekonda VT, Baskin DG (2012) Fourth ventricular administration of oxytocin activates Fos expression in caudal NTS catecholamine neurons. In: Society for Neuroscience, New Orleans, 2012 (2012 Abstract Viewer and Itinerary Planner. Washington, D.C.: Society for Neuroscience, 2012. Online)
Ho JM, Morton GJ, Baskin DG, Blevins JE (2012) Peripheral oxytocin activates Fos expression in NTS catecholamine neurons. In: Society for Neuroscience, New Orleans, 2012 (2012 Abstract Viewer and Itinerary Planner. Washington, D.C.: Society for Neuroscience. Online)
Rothe E, Rinaman L. Central oxytocin (OT) activates hindbrain glucagon-like peptide-1 (GLP-1) neurons in rats. Appetite. 2001;37:160.
Llewellyn-Smith IJ, Kellett DO, Jordan D, Browning KN, Travagli RA. Oxytocin-immunoreactive innervation of identified neurons in the rat dorsal vagal complex. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Neurogastroenterol Motil. 2012;24(3):e136–146. doi:10.1111/j.1365-2982.2011.01851.x.
Williams DL, Schwartz MW, Bastian LS, Blevins JE, Baskin DG. Immunocytochemistry and laser capture microdissection for real-time quantitative PCR identify hindbrain neurons activated by interaction between leptin and cholecystokinin. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S.]. J Histochem Cytochem. 2008;56(3):285–93. doi:10.1369/jhc.7A7331.2007.
Babic T, Townsend RL, Patterson LM, Sutton GM, Zheng H, Berthoud HR. Phenotype of neurons in the nucleus of the solitary tract that express CCK-induced activation of the ERK signaling pathway. [Research Support, N.I.H., Extramural]. Am J Physiol Regul Integr Comp Physiol. 2009;296(4):R845–854. doi:10.1152/ajpregu.90531.2008.
Garfield AS, Patterson C, Skora S, Gribble FM, Reimann F, Evans ML, et al. Neurochemical characterization of body weight-regulating leptin receptor neurons in the nucleus of the solitary tract. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Endocrinology. 2012;153(10):4600–7. doi:10.1210/en.2012-1282.
Davies BT, Wellman PJ. Effects on ingestive behavior in rats of the alpha 1-adrenoceptor agonist cirazoline. [Research Support, Non-U.S. Gov’t]. Eur J Pharmacol. 1992;210(1):11–6.
Wellman PJ. Norepinephrine and the control of food intake. [Review]. Nutrition. 2000;16(10):837–42.
Sands SA, Morilak DA. Expression of alpha1D adrenergic receptor messenger RNA in oxytocin- and corticotropin-releasing hormone-synthesizing neurons in the rat paraventricular nucleus. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Neuroscience. 1999;91(2):639–49.
Maruyama M, Matsumoto H, Fujiwara K, Noguchi J, Kitada C, Fujino M, et al. Prolactin-releasing peptide as a novel stress mediator in the central nervous system. [Research Support, Non-U.S. Gov’t]. Endocrinology. 2001;142(5):2032–8.
Lawrence CB, Celsi F, Brennand J, Luckman SM. Alternative role for prolactin-releasing peptide in the regulation of food intake. Nat Neurosci. 2000;3(7):645–6. doi:10.1038/76597.
Takayanagi Y, Matsumoto H, Nakata M, Mera T, Fukusumi S, Hinuma S, et al. Endogenous prolactin-releasing peptide regulates food intake in rodents. [Research Support, Non-U.S. Gov’t]. J Clin Invest. 2008;118(12):4014–24. doi:10.1172/JCI34682.
Bjursell M, Lenneras M, Goransson M, Elmgren A, Bohlooly YM. GPR10 deficiency in mice results in altered energy expenditure and obesity. Biochem Biophys Res Commun. 2007;363(3):633–8. doi:10.1016/j.bbrc.2007.09.016.
Gu W, Geddes BJ, Zhang C, Foley KP, Stricker-Krongrad A. The prolactin-releasing peptide receptor (GPR10) regulates body weight homeostasis in mice. J Mol Neurosci. 2004;22(1–2):93–103. doi:10.1385/JMN:22:1-2:93.
Larsen PJ, Tang-Christensen M, Holst JJ, Orskov C. Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience. 1997;77(1):257–70.
Rinaman L. Interoceptive stress activates glucagon-like peptide-1 neurons that project to the hypothalamus. [Research Support, U.S. Gov’t, P.H.S.]. Am J Physiol. 1999;277(2 Pt 2):R582–590.
Sawchenko PE, Arias C, Bittencourt JC. Inhibin beta, somatostatin, and enkephalin immunoreactivities coexist in caudal medullary neurons that project to the paraventricular nucleus of the hypothalamus. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. J Comp Neurol. 1990;291(2):269–80. doi:10.1002/cne.902910209.
Hisadome K, Reimann F, Gribble FM, Trapp S. Leptin directly depolarizes preproglucagon neurons in the nucleus tractus solitarius: electrical properties of glucagon-like Peptide 1 neurons. [Research Support, Non-U.S. Gov’t]. Diabetes. 2010;59(8):1890–8. doi:10.2337/db10-0128.
Vrang N, Phifer CB, Corkern MM, Berthoud HR. Gastric distension induces c-Fos in medullary GLP-1/2-containing neurons. [Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]. Am J Physiol Regul Integr Comp Physiol. 2003;285(2):R470–478. doi:10.1152/ajpregu.00732.2002.
Llewellyn-Smith IJ, Reimann F, Gribble FM, Trapp S. Preproglucagon neurons project widely to autonomic control areas in the mouse brain. [Research Support, Non-U.S. Gov’t]. Neuroscience. 2011;180:111–21. doi:10.1016/j.neuroscience.2011.02.023.
McMahon LR, Wellman PJ. PVN infusion of GLP-1-(7–36) amide suppresses feeding but does not induce aversion or alter locomotion in rats. [Research Support, Non-U.S. Gov’t]. Am J Physiol. 1998;274(1 Pt 2):R23–29.
Zueco JA, Esquifino AI, Chowen JA, Alvarez E, Castrillon PO, Blazquez E. Coexpression of glucagon-like peptide-1 (GLP-1) receptor, vasopressin, and oxytocin mRNAs in neurons of the rat hypothalamic supraoptic and paraventricular nuclei: effect of GLP-1(7–36)amide on vasopressin and oxytocin release. [Research Support, Non-U.S. Gov’t]. J Neurochem. 1999;72(1):10–6.
Larsen PJ, Tang-Christensen M, Jessop DS. Central administration of glucagon-like peptide-1 activates hypothalamic neuroendocrine neurons in the rat. [Research Support, Non-U.S. Gov’t]. Endocrinology. 1997;138(10):4445–55.
Kendrick KM (2000) Oxytocin, motherhood and bonding. [Review]. Exp Physiol, 85 Spec No, 111S-124S.
Sandoval DA, Bagnol D, Woods SC, D’Alessio DA, Seeley RJ. Arcuate glucagon-like peptide 1 receptors regulate glucose homeostasis but not food intake. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Diabetes. 2008;57(8):2046–54. doi:10.2337/db07-1824.
Schick RR, Zimmermann JP, vorm Walde T, Schusdziarra V. Peptides that regulate food intake: glucagon-like peptide 1-(7–36) amide acts at lateral and medial hypothalamic sites to suppress feeding in rats. Am J Physiol Regul Integr Comp Physiol. 2003;284(6):R1427–1435.
Scott MM, Williams KW, Rossi J, Lee CE, Elmquist JK. Leptin receptor expression in hindbrain Glp-1 neurons regulates food intake and energy balance in mice. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. J Clin Invest. 2011;121(6):2413–21. doi:10.1172/JCI43703.
Sutton GM, Duos B, Patterson LM, Berthoud HR. Melanocortinergic modulation of cholecystokinin-induced suppression of feeding through extracellular signal-regulated kinase signaling in rat solitary nucleus. [Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, P.H.S.]. Endocrinology. 2005;146(9):3739–47. doi:10.1210/en.2005-0562.
Moos F, Richard P. Paraventricular and supraoptic bursting oxytocin cells in rat are locally regulated by oxytocin and functionally related. [Research Support, Non-U.S. Gov’t]. J Physiol. 1989);408:1–18.
Yamashita H, Okuya S, Inenaga K, Kasai M, Uesugi S, Kannan H, et al. Oxytocin predominantly excites putative oxytocin neurons in the rat supraoptic nucleus in vitro. [In Vitro Research Support, Non-U.S. Gov’t]. Brain Res. 1987;416(2):364–8.
Catheline G, Touquet B, Lombard MC, Poulain DA, Theodosis DT. A study of the role of neuro-glial remodeling in the oxytocin system at lactation. [Research Support, Non-U.S. Gov’t]. Neuroscience. 2006;137(1):309–16. doi:10.1016/j.neuroscience.2005.08.042.
Rossoni E, Feng J, Tirozzi B, Brown D, Leng G, Moos F. Emergent synchronous bursting of oxytocin neuronal network. [Research Support, Non-U.S. Gov’t]. PLoS Comput Biol. 2008;4(7):e1000123. doi:10.1371/journal.pcbi.1000123.
Moos F, Freund-Mercier MJ, Guerne Y, Guerne JM, Stoeckel ME, Richard P. Release of oxytocin and vasopressin by magnocellular nuclei in vitro: specific facilitatory effect of oxytocin on its own release. [Research Support, Non-U.S. Gov’t]. J Endocrinol. 1984;102(1):63–72.
Vankrieken L, Godart A, Thomas K. Oxytocin determination by radioimmunoassay. Gynecol Obstet Invest. 1983;16(3):180–5.
Reidelberger RD, Haver AC, Apenteng BA, Anders KL, Steenson SM. Effects of exendin-4 alone and with peptide YY(3–36) on food intake and body weight in diet-induced obese rats. [Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, Non-P.H.S.]. Obesity (Silver Spring). 2011;19(1):121–7. doi:10.1038/oby.2010.136.
Slattery DA, Neumann ID. Chronic icv oxytocin attenuates the pathological high anxiety state of selectively bred Wistar rats. [Research Support, Non-U.S. Gov’t]. Neuropharmacology. 2010;58(1):56–61. doi:10.1016/j.neuropharm.2009.06.038.
Petersson M, Uvnas-Moberg K. Postnatal oxytocin treatment of spontaneously hypertensive male rats decreases blood pressure and body weight in adulthood. [Research Support, Non-U.S. Gov’t]. Neurosci Lett. 2008;440(2):166–9. doi:10.1016/j.neulet.2008.05.091.
Ondrejcakova M, Barancik M, Bartekova M, Ravingerova T, Jezova D. Prolonged oxytocin treatment in rats affects intracellular signaling and induces myocardial protection against infarction. [Research Support, Non-U.S. Gov’t]. Gen Physiol Biophys. 2012;31(3):261–70. doi:10.4149/gpb_2012_030.
Verbalis JG, McHale CM, Gardiner TW, Stricker EM. Oxytocin and vasopressin secretion in response to stimuli producing learned taste aversions in rats. [Research Support, U.S. Gov’t, P.H.S.]. Behav Neurosci. 1986;100(4):466–75.
Chang SW, Barter JW, Ebitz RB, Watson KK, Platt ML. Inhaled oxytocin amplifies both vicarious reinforcement and self reinforcement in rhesus macaques (Macaca mulatta). [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Proc Natl Acad Sci U S A. 2012;109(3):959–64. doi:10.1073/pnas.1114621109.
Gossen A, Hahn A, Westphal L, Prinz S, Schultz RT, Grunder G, et al. Oxytocin plasma concentrations after single intranasal oxytocin administration - a study in healthy men. [Research Support, Non-U.S. Gov’t]. Neuropeptides. 2012;46(5):211–5. doi:10.1016/j.npep.2012.07.001.
MacDonald E, Dadds MR, Brennan JL, Williams K, Levy F, Cauchi AJ. A review of safety, side-effects and subjective reactions to intranasal oxytocin in human research. [Evaluation Studies Review]. Psychoneuroendocrinology. 2011;36(8):1114–26. doi:10.1016/j.psyneuen.2011.02.015.
Born J, Lange T, Kern W, McGregor GP, Bickel U, Fehm HL. Sniffing neuropeptides: a transnasal approach to the human brain. [Research Support, Non-U.S. Gov’t]. Nat Neurosci. 2002;5(6):514–6. doi:10.1038/nn849.
Dhuria SV, Hanson LR, Frey 2nd WH. Novel vasoconstrictor formulation to enhance intranasal targeting of neuropeptide therapeutics to the central nervous system. [Research Support, Non-U.S. Gov’t]. J Pharmacol Exp Ther. 2009;328(1):312–20. doi:10.1124/jpet.108.145565.
Goldman MB, Gomes AM, Carter CS, Lee R. Divergent effects of two different doses of intranasal oxytocin on facial affect discrimination in schizophrenic patients with and without polydipsia. [Randomized Controlled Trial Research Support, N.I.H., Extramural]. Psychopharmacology (Berl). 2011;216(1):101–10. doi:10.1007/s00213-011-2193-8.
Rubin LH, Carter CS, Drogos L, Pournajafi-Nazarloo H, Sweeney JA, Maki PM. Peripheral oxytocin is associated with reduced symptom severity in schizophrenia. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Schizophr Res. 2010;124(1–3):13–21. doi:10.1016/j.schres.2010.09.014.
Alvares GA, Hickie IB, Guastella AJ. Acute effects of intranasal oxytocin on subjective and behavioral responses to social rejection. [Randomized Controlled Trial Research Support, Non-U.S. Gov’t]. Exp Clin Psychopharmacol. 2010;18(4):316–21. doi:10.1037/a0019719.
Devost D, Zingg HH. Homo- and hetero-dimeric complex formations of the human oxytocin receptor. [Research Support, Non-U.S. Gov’t Review]. J Neuroendocrinol. 2004;16(4):372–7. doi:10.1111/j.0953-8194.2004.01188.x.
Viero C, Shibuya I, Kitamura N, Verkhratsky A, Fujihara H, Katoh A, et al. REVIEW: Oxytocin: Crossing the bridge between basic science and pharmacotherapy. [Research Support, Non-U.S. Gov’t Review]. CNS Neurosci Ther. 2010;16(5):e138–156. doi:10.1111/j.1755-5949.2010.00185.x.
Bales KL, Perkeybile AM, Conley OG, Lee MH, Guoynes CD, Downing GM, et al. Chronic Intranasal Oxytocin Causes Long-Term Impairments in Partner Preference Formation in Male Prairie Voles. Biol Psychiatry. 2012. doi:10.1016/j.biopsych.2012.08.025.
Miller G. Neuroscience. The promise and perils of oxytocin. [News]. Science. 2013;339(6117):267–9. doi:10.1126/science.339.6117.267.
Acknowledgments
This material is based upon work supported by the Office of Research and Development, Medical Research Service, Department of Veterans Affairs (VA). The research in our laboratory has been supported by the Department of VA Merit Review Research Program. The authors are appreciative of the efforts by Vishwanath Anekonda, Benjamin Thompson, Denis Baskin and Bang Hwang for their assistance in critically reviewing this manuscript.
Conflict of Interest
James Blevins and Jacqueline Ho have no conflicts of interest in the data presented in this review.