
Abstract
Limitations in physical fitness, a consistent finding in individuals with both type I and type 2 diabetes mellitus, correlate strongly with cardiovascular and all-cause mortality. These limitations may significantly contribute to the persistent excess cardiovascular mortality affecting this group. Exercise impairments in VO2 peak and VO2 kinetics manifest early on in diabetes, even with good glycemic control and in the absence of clinically apparent complications. Subclinical cardiac dysfunction is often present but does not fully explain the observed defect in exercise capacity in persons with diabetes. In part, the cardiac limitations are secondary to decreased perfusion with exercise challenge. This is a reversible defect. Similarly, in the skeletal muscle, impairments in nutritive blood flow correlate with slowed (or inefficient) exercise kinetics and decreased exercise capacity. Several correlations highlight the likelihood of endothelial-specific impairments as mediators of exercise dysfunction in diabetes, including insulin resistance, endothelial dysfunction, decreased myocardial perfusion, slowed tissue hemoglobin oxygen saturation, and impairment in mitochondrial function. Both exercise training and therapies targeted at improving insulin sensitivity and endothelial function improve physical fitness in subjects with type 2 diabetes. Optimization of exercise functions in people with diabetes has implications for diabetes prevention and reductions in mortality risk. Understanding the molecular details of endothelial dysfunction in diabetes may provide specific therapeutic targets for the remediation of this defect. Rat models to test this hypothesis are under study.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
People with T2D are typically sedentary and overweight. Over the last 5 decades there has been a substantive decrease in occupational physical activity which correlates with the current obesity epidemic [1]. A recent position paper suggested that childhood obesity and physical inactivity predict that children born today will have a shorter life expectancy than their parents [2]. Of particular importance for diabetes is the consistent observation that people with both type I (T1D) and type 2 diabetes mellitus (T2D) have decreased physical fitness as measured by VO2 peak compared to similarly obese and or sedentary subjects [3–6]. This defect has the potential to contribute importantly to the persistent excess cardiovascular and all-cause mortality observed in people with diabetes despite aggressive cardiovascular risk factor intervention.
Cardiovascular disease is the leading cause of mortality in people with diabetes. This is true even in light of the overall decrease in cardiovascular mortality with improved cardiovascular risk factor modification over the past 10 years. Specifically, population based prevalence of cardiovascular disease has decreased remarkably in the last 10 years, yet excess cardiovascular mortality related to diabetes remains 3–5 fold higher [7] . This persistent mortality gap may be, in part, secondary to inadequate achievement of cardiovascular risk factor reduction [8]. It is also possible that additional risk factors contribute to the excess cardiovascular and all-cause mortality observed in diabetes. Our laboratory and others have identified that children and adults with type I and type 2 diabetes mellitus have defects in functional exercise capacity [3–6]. A primary manifestation of this defect is that people with diabetes have decreased VO2 peak and slowed kinetics for oxygen uptake and heart rate. This is a clinically relevant impairment in light of the strong relationship between cardiovascular fitness and all-cause mortality [9, 10]. In addition to and in association with the decreased VO2 peak and VO2 kinetics, we have demonstrated a number of phenotypic characteristics of diabetes including insulin resistance (IR), endothelial dysfunction, decreased myocardial perfusion, slowed tissue hemoglobin oxygen saturation and, most recently, impairment in mitochondrial function [9, 10] Green and Nadeau personal communication. In this review article, we will outline the importance of cardiovascular fitness and exercise for prevention of diabetes mortality, review data from our group and others that clarify the clinical parameters associated with defects in functional exercise capacity, outline the role of cardiac and muscle perfusion observed in diabetes, discuss proof of concept clinical interventions targeting IR and endothelial dysfunction for improvement in exercise capacity, and close with recent mechanistic observations which highlight the likelihood of endothelial-specific impairments as mediators of exercise dysfunction and diabetes.
2 Physical activity, physical fitness and mortality
Evidence endorsing a strong relationship between physical fitness and mortality is compelling. These data have led both the Centers for Disease Control (CDC) and the American Heart Association (AHA) to present position statements stating that physical inactivity is a leading cause of death and that primary prevention of heart disease requires increased physical fitness [11, 12]. In pivotal studies, Wei et al. demonstrated a strong relationship between physical fitness and mortality in men with and without diabetes [9, 10]. A recent position paper suggested that childhood obesity and physical inactivity predict that children born today will have a shorter life expectancy than their parents [2]. Of note, physical activity also predicts cardiovascular and all-cause mortality, but is a less potent predictor than physical fitness [13, 14]. Of particular importance for diabetes is the consistent observation that people with both type I (T1D) and type 2 diabetes mellitus (T2D) have decreased physical fitness as measured by VO2 peak compared to similarly obese and or sedentary subjects [3–6]. This defect has the potential to contribute importantly to the persistent excess cardiovascular and all-cause mortality observed in people with diabetes despite aggressive cardiovascular risk factor intervention
Extensive epidemiological and observational data indicate a correlation between physical activity and cardiovascular all-cause mortality in people with T2D [9, 10, 15–20]. People with diabetes in the US had an increase in all-cause mortality related to modifiable risk factors: A1c ≥8 % (HR 1.65), self-reported no regular physical activity (HR 1.58) and current tobacco use (1.77) [18]. In two large epidemiological studies examining the relationship between physical activity and cardiovascular all-cause mortality, physical activity was associated with decreased CVD and all-cause mortality in T2D (n = 5859, prospective cohort) and predicted decreased CVD and all-cause mortality in T2D across all levels of glycemic control (n = 10,352 NHANES III study sample) [19, 20]. Physical fitness defined as exercise capacity measured by VO2 peak or VO2 max is a more potent predictor of cardiovascular and all-cause mortality than physical activity (4–6). In a cohort of men referred for exercise testing, exercise capacity was the top predictor of CV mortality (n = 6213, prospective cohort) [17]. This observation is further supported by three studies demonstrating that cardiorespiratory fitness is predictor of mortality in men across all BMI groups (n = 25,714, prospective observational) and men with T2D (n = 1263, n = 3148) [9, 10, 15]. Lee et al. conducted a prospective observational comparison of cardiorespiratory fitness to physical activity for mortality protection. Cardiorespiratory fitness was more strongly associated with mortality than physical activity (n = 50,244) [16]. However in contrast to the strong mostly observational data supporting the importance of physical activity/fitness with regard to mortality, the NIH multicenter Look Ahead trial recently was halted because it failed to show a difference in the rate of nonfatal myocardial infarction, nonfatal stroke, death, or hospitalization for angina among patients randomized to an intensive lifestyle intervention versus a control arm consisting of education alone (communication from Look Ahead leadership). The reasons for this are unknown but may relate to the finding that event rates were too low in both groups to enable detecting a significant difference or that physical activity/physical fitness prior is critical earlier in the disease process. Based upon the epidemiological and cohort data both the CDC and the AHA consider lack of physical activity as a risk factor for heart disease.
3 Exercise in diabetes prevention
The likelihood of developing diabetes is substantially less in those who participate in exercise versus those who report being sedentary, even in high risk individuals and after adjustment for major risk factors (e.g. BMI, family history, etc.). Likewise, higher levels of fitness are inversely related to diabetes incidence. Women who reported engaging in vigorous physical activity ≥1x/week had a 16 % decreased risk of developing T2D over 8-year follow-up compared to more sedentary women after adjustment for BMI and family history of diabetes (n = 87,253, ages 34–59) [21]. Exercise intervention, whether aerobic, resistance, or the combination, can improve cardiorespiratory fitness in those with T2D. For example, lifestyle intervention counseling (including exercise, at least 150 min/week) in patients with impaired glucose tolerance reduced progression to T2D by 58 % (n = 552, RCT, ~3 years) [22, 23]. Hamman and others reported that even in the absence of weight loss, physical activity alone still prevented diabetes [24].
Self-report from people with risk for the development of diabetes demonstrated a decreased risk of developing diabetes in those reporting participation in exercise activity versus being sedentary. Here again, physical activity (self-reported) was associated with reduced risk of developing T2D across BMI categories and in abdominally lean or obese men and women (n = 16,154, median 12.3 years follow-up) [25]. Physical fitness is also inversely associated with incident diabetes (n = 23,444 men, 18 year follow-up, mail-back survey and exercise testing) [26]. Taken all together, individuals at risk for diabetes who increase physical activity and enhance their cardiorespiratory fitness can decrease their likelihood of developing diabetes with its associated increased risk of mortality.
4 Effects of exercise training programs
In contrast to the well-established relationship between physical activity, physical fitness and the development of diabetes, the impact on outcomes of exercise training program targeting physical activity or physical fitness are fewer and varied. Most, but not all, participants in prospective exercise interventions improved their cardiovascular risk profiles with exercise interventions [27]. Our group has demonstrated a significant improvement in VO2 peak and exercise kinetics with supervised exercise training [28]. Larger studies including HERITAGE Family Study (n = 130), DREW (n = 326), INFLAME (n = 70), STRRIDE (n = 303); have demonstrated improvement in fitness with various combinations of aerobic and resistance training [29–33]. To date, these prospective interventional studies other than the Look Ahead trial have not been appropriately powered to assess the relationship between training and cardiovascular and all-cause mortality. Despite this limitation with interventional trials, Booth et al. were able to synthesize observations from two large prospective cohort studies where physical fitness was characterized at baseline and 6 months to 8 years later [34]. This analysis took advantage of the fact that individuals in these prospective cohorts had fitness measured and some subjects changed their level of fitness with time [14, 35]. Figure 1 illustrates the synthesis of data from these two studies suggesting that fitness decreases cardiovascular and all-cause mortality. Of greatest interest, changing fitness level either from fit-to-unfit or unfit-to-fit, as defined in the trials by maximal METS (A MET is an index of aerobic exercise capacity: one MET unit = resting metabolic rate) confers a survival advantage suggesting a legacy effect of fitness [34]. Over the last 5 decades there has been a substantive decrease in occupational physical activity which correlates with our obesity epidemic [1]. To summarize, physical activity and physical fitness are not equivalent; both physical activity and physical fitness confer a survival advantage in prospective cohort and epidemiological studies; not all interventions with physical activity improve physical fitness and physical fitness at any point in time appears to confer a survival advantage. Therefore, decreased physical fitness in diabetes poses an important risk for cardiovascular and all-cause mortality.

Fitness and relative risk of death: Relative risk of death is expressed as a function of the maintenance of, or the change in, cardiorespiratory fitness (CRF) level occurring between two CRF determinations that were made years apart. Deaths were recorded for 5 or 8 years following the second test. Results for maintenance or change in CRF are divided into four groups including: 1) stayed unfit; 2) became fit after being unfit; 3) became unfit after being fit; and 4) stayed fit. Three sets of data using this experimental strategy were obtained from two publications. The publications by Blair’s group provided data sets for all-cause mortality (shown in rectangles) and cardiovascular disease (shown in hexagons) for men [13, 14, 87]. The publication by Kokkonis and Myers had a male data set (shown in ovals) [15]. Note the essential similarity of data sets. Figure and legend reprinted with permission
5 Defects in functional exercise capacity in type 2 diabetes
We and others have demonstrated a consistent defect in maximal exercise capacity in subjects with T2D. Of particular importance, children and adults with uncomplicated T1D andT2D demonstrate decreased functional exercise capacity as assessed by VO2 peak and other measures of cardiovascular exercise performance. In this section, we will review the compelling clinical data supporting a consistent defect and cardiovascular fitness in subjects with diabetes. This will be followed by a review of the body of work generated by our lab and others that forms the basis for our working model that endothelial dysfunction and substrate delivery are primary mediators of the defect and functional exercise capacity observed in T2D.
6 Exercise deficit
Defects in functional exercise capacity in subjects with T2D were first identified in the 1980s [36]. It was observed that people with T2D have impaired submaximal and maximal exercise performance that is independent of the effects of obesity, and this impairment is present even in the absence of clinically apparent cardiovascular (CV) disease [3–5, 36–42]. Fang and colleagues observed a correlation between exercise capacity associated with impaired glycemic control, subclinical LV dysfunction and impaired heart rate recovery (170 pt with T2D vs. 56 controls) [43]. Decreased peak exercise capacity in overweight and obese T2D subjects was associated with older age, increased waist circumference and BMI, longer duration of DM, increased A1C, a history of CV disease, metabolic syndrome, B-blocker use and African American ethnicity [44]. Our group has reported an association IR as assessed by euglycemic hyperinsulinemic clamp, endothelial dysfunction as measured by decreased flow mediated vasodilatation after hyperemia, decreased blood flow by plethysmography, abnormal tissue hemoglobin oxygen saturation (STO2) assessed using near infrared spectroscopy, diastolic dysfunction and decreased cardiac perfusion and more recently decreased muscle mitochondrial function (unpublished). It is of importance to note that these defects are consistently observed in subjects who are similarly sedentary and overweight to the subjects with diabetes (consistent with diabetes per se as an important contributor to this defect). Based on these correlations we have a working model positing endothelial dysfunction as a critical component of exercise dysfunction and diabetes (Fig. 2).

Working model of the role of endothelial dysfunction in exercise defect in diabetes: Proposed factors contributing to exercise dysfunction in T2D: T2D and prediabetic conditions such as insulin resistance are associated with an abnormal metabolic environment that induces endothelial, cardiac, and peripheral dysfunction. Target organ dysfunction feeds back to exacerbate these defects. Ultimately these factors result in defects in functional exercise capacity
7 Insulin resistance correlates with defects in functional exercise capacity
A relationship between IR and VO2 max was described by Reaven in the 1980s [37, 45, 46]. We and others have confirmed and expanded upon this relationship. Individuals with IR and/or T2D manifest decreased maximal oxygen consumption and/or submaximal exercise capacity [37, 40, 47, 48]. In addition, oxygen consumption at submaximal workloads is decreased during graded exercise in T2D compared to healthy controls [40]. Furthermore, individuals with T2D have slower oxygen uptake kinetics at the beginning of constant load exercise [4, 40, 49]. VO2 kinetics are a marker of the ability to oxygenate the body upon exercise initiation at submaximal levels of exertion. Lower VO2 peak and slowed VO2 kinetics have been found to correlate with decreased insulin sensitivity as measured using the euglycemic hyperinsulinemic clamp in our work.
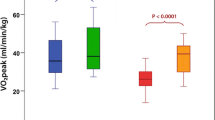
The relationship between IR and decreased functional exercise capacity, observed in T2D, is also present in individuals with type 1 diabetes (T1D). Although T1D is primarily a disease of insulin deficiency, we and others have demonstrated IR (IR) in T1D [50–53]. In the Pittsburgh Epidemiology of Diabetes Complications study and the DCCT, estimated IR predicted CVD events [54, 55]. Eighty seven subjects participating in the NIH CACTI study, direct measures of IR correlate with CAC. These results and others suggest that IR is a correlate of CVD in T1D as well [55, 56]. Decreased insulin sensitivity in T1D is present in adolescents as well as adults. Decreased insulin sensitivity T1D population does not have the typical metabolic syndrome phenotype associated with IR in other populations, making IR harder to recognize and quantify in this population and raising the possibility that the underlying mechanism of IR in T1D may be distinct [55, 56]. Regardless of the etiology of IR in T1D there is a significant correlation between IR and decreased functional exercise capacity (Fig. 3).

Insulin resistance correlates with VO2 Peak: The relationship of insulin resistance to VO2 peak was assessed a graded cycle ergometer (Lode, Groningen, The Netherlands) protocol to exhaustion and insulin sensitivity was assessed with a 3-h hyperinsulinemic-euglycemic clamp (80 mU/m2 min insulin). Glucose disposal rate was expressed as milligrams per kilogram body mass and milligrams per kilogram fat-free mass. Reprinted with permission J Clin Endocrinol Metab, 95(2), 513–521 [6]
8 Intervention augments functional exercise capacity in T2D
Identification of a correlation between decreased insulin sensitivity and defects functional exercise capacity in diabetes suggested an opportunity for intervention with insulin sensitizing agents. We hypothesized that a thiazolidinedione would improve exercise function by improving insulin sensitivity [57]. Twenty patients with uncomplicated T2D were randomized in a double-blinded study to receive either 4 mg/day of rosiglitazone or matching placebo after baseline measurements which included endothelial function (by brachial ultrasound), VO2peak and insulin sensitivity with reassessment after 4 months of treatment. The two groups of patients did not differ at baseline in any measure. Patients treated with rosiglitazone had significantly improved VO2peak (by 7 %)(Table 1) as well as improved brachial artery diameter (BAD) and insulin sensitivity [57]. Rosiglitazone increased the BAD response to cuff inflation by 83 % over the baseline response (from 0.024 ± 0.03 cm to 0.044 ± 0.03 cm, P < 0.05) while no significant increase seen in the response of placebo-treated controls (from 0.010 ± 0.02 cm to 0.017 ± 0.02 cm, P = NS). Change in VO2peak correlated with changes in BAD and IR in the rosiglitazone treated group only. These data demonstrate that an agent that improves insulin sensitivity can improve exercise function in T2D.
Exercise training also augments functional exercise capacity in T2D. Our group has demonstrated that formal exercise training improved VO2 peak in subjects with diabetes as well as obese and lean sedentary controls. The favorable response to these 2 interventions indicates that clarification of the underlying physiology determining decreased functional exercise capacity in people with diabetes can be used to target interventions. In the next section, we will discuss the contributions of abnormal central (cardiac) and systemic responses contributing to defective exercise capacity in diabetes.
9 Cardiac abnormalities and abnormal exercise capacity in T2D
Subclinical and potentially reversible cardiac dysfunction, particularly left ventricular diastolic dysfunction (LVDD), during submaximal exercise is frequently seen in well-controlled, recently diagnosed, sedentary subjects with T2D (even in the absence of clinically evident coronary artery disease or microvascular complications). Poirier et al. demonstrated that the presence of LVDD was associated with reduce exercise performance (time and METs achieved) in sedentary men with well-controlled uncomplicated T2D [58]. Similarly, myocardial metabolic derangements assessed using 31P-MRS assessment correlated with LVDD in recently diagnosed uncomplicated men with T2D [59]. Our group demonstrated elevated pulmonary capillary wedge pressure (PCWP; n = 10) during peak exercise in uncomplicated T2D women compared to overweight controls consistent with diastolic dysfunction even though they were relatively recently diagnosed (average T2D diagnosis 3.6 years, similar activity levels, n = 10) [39]. Additionally, myocardial perfusion following exercise is reduced in this group and the decreased perfusion was correlated with the abnormally increased wedge pressure. Importantly, these abnormalities only emerged during exercise and were not evident at rest.
Many possible mechanisms have been proposed for cardiac dysfunction in uncomplicated T2D, including impaired calcium and ion transport, altered glucose and lipid metabolism, mitochondrial dysfunction, decreased perfusion and renin-angiotensin-system activation. The early development of preclinical diabetic cardiomyopathy has also been posited. Novel Cardiac MRI (CMR)-based measurements of extracellular matrix expansion (via “T1 mapping techniques”) are markedly abnormal in T2D even before the development of significant diastolic dysfunction, ventricular hypertrophy or clinical heart failure. Furthermore, these indices are associated with long-term outcomes (beyond ejection fraction) in a general referral population [60]. It is unclear which of these abnormalities are reversible and their contribution to limitations in functional exercise capacity.
A number of factors suggest that diabetic cardiomyopathy alone cannot account for the exercise impairment in T2D and supports the contribution of a perfusion defect. First of all, LVDD correlates with reduced exercise performance, but measurable differences in cardiac output are not a prerequisite. Some investigators have shown that cardiac output significantly increases in T2D subjects at higher intensity exercise compared to controls [38]. We have demonstrated decreased myocardial perfusion with stress (termed decreased cardiac reserve) in uncomplicated T2D consistent with findings of Schindler et al. [61].This group demonstrated that the impairment in cardiac reserve was improved by glucose control with glyburide and metformin. Joshi et al. demonstrated both decreased cardiac perfusion and stroke volume in response to submaximal exercise challenge and recovery in T2D (10 min, bicycle ergometry, constant load of 50 W – mimic 6 min walk test [62]. This defect was not significant in individuals with hemoglobin A1c less than 8 %. Of note, Brassard et al. demonstrated improvement in diastolic dysfunction and normalization (in 5/11 T2D subjects) after 3 months of aerobic exercise training (cycle ergometry, 3×/week, 30–60 min, 60–70 % of VO2max) [63]. The observation that intervention with either exercise training or improved glycemic control can ameliorate the subclinical cardiac dysfunction in T2D, suggests that the contribution of cardiac dysfunction, related to decreased perfusion is a modifiable and targetable defect contributing to decreased cardiovascular fitness in diabetes.
10 Muscle contribution to exercise dysfunction in T2D
The findings outlined above indicate that the cardiac dysfunction does not fully explain the observed defect in exercise capacity in diabetes. We and others have reported decreased functional exercise capacity in the context of preserved cardiac output with exercise, suggesting factors beyond the heart also contribute to exercise dysfunction [38, 39, 62]. In fact, exercise capacity improves following a period of exercise training prior to changes in left ventricular filling dynamics (i.e. normalization of LVDD). These findings would suggest systemic limitations of the exercising skeletal muscle distinct from cardiac dysfunction, whether it is insufficient oxygen supply and/or oxygen utilization, to contribute to the decline in exercise capacity. Of particular interest for this review article, experimental evidence from our group and others indicates decreased skeletal muscle perfusion at baseline and in response to insulin in subjects with T2D. Skeletal muscle is quantitatively the most important tissue involved in maintaining glucose homeostasis and is a major site of IR in T2D patients [64]. Both the muscle vasculature and the skeletal muscle fibers are insulin resistant in T2D. Muscle IR leads to decreased glucose uptake, decreased glycogen stores and decreased efficiency of ATP production [65, 66]. Scheuermann-Freestone, using MRS, found that T2D patients with apparently normal cardiac function had impaired myocardial and skeletal muscle energy metabolism related to changes in circulating metabolic substrates before, during and after exercise [67]. IR is associated with decreased insulin stimulated vasodilation, decreased capillary recruitment and decreased nutritive muscle blood flow [68–70]. Our group reported that the slowed oxygen uptake kinetics upon initiation of constant workload exercise is associated with slowed muscle perfusion as assessed by near infrared spectroscopy (NIRS). Concomitant VO2 and determination of the ratio of oxygenated and deoxygenated hemoglobin measurement permitted inference of regional blood flow (via Fick principle), demonstrating delay response time of increasing blood flow at exercise onset in T2D subjects [3]. Figure 4. Using a different assessment, Sanchez et al. demonstrated impaired skeletal muscle blood flow in T2D and obese-matched controls (56 % lower MRI signal intensity with maximum voluntary isometric contraction) [71]. There is evidence for both structural and functional abnormalities contributing to decrease skeletal muscle blood flow in diabetes. These include decreased capillary recruitment in response to insulin, decreased capillarization and capillary basement membrane thickening [68, 69, 72, 73]. Capillary recruitment in the setting of exercise is normal in uncomplicated T2D and impaired in subjects with T2D and a microvascular complications [74]. To summarize, in people with both complicated an uncomplicated T2D, there are impairments muscle substrate delivery and utilization presumed secondary to decreased cardiac function, decreased blood flow in conduit vessels, increased precapillary vasoconstriction and abnormal endothelium-mediated vasodilatation, and decreased capillary recruitment. As such, nutritive blood flow appears to be an important contributor to the impaired response of the skeletal muscle to exercise in people with T2D.

Exercise kinetics correlate with slowed muscle O2 delivery: Muscle deoxyhemoglobin ([HHb]) concentration kinetics in a representative healthy subject and a subject with T2D at rest and following the onset of moderate exercise. Exercise begins at time = 0. Note pronounced overshoot of HHb in the T2D subject early in the exercise transition. This response indicates a greater reliance upon local muscle oxygen extraction (e.g. due to slowed increase in microvascular blood flow) early in exercise that is not observed in healthy muscle. Published in Bauer, TA et al. Diabetes Care. 2007 Nov;30(11):2880–5 [3]
In healthy subjects, exercise training induces muscle angiogenesis and vasculogenesis. Seven consecutive days of aerobic training in uncomplicated well-controlled T2D (n = 11, supervised exercise, 60 min/day at 60–75 % HRR) led to higher femoral blood flow measurements from baseline during oral glucose tolerance testing [75]. A similar exercise intervention in healthy subjects led to increased muscle capillarization and mitochondrial density as assessed by mitochondrial enzyme content and activity [76]. The capillarization response to exercise training is also present in people with diabetes and metabolic syndrome (Goldberg ADA abstracts) suggesting a potential reversible defect in muscle blood flow in this population. Increased skeletal muscle substrate demand and hypoxia are two potential mechanisms for exercise mediated angiogenesis and vasculogenesis [77]. Two studies suggest that hypoxia enhances the adaptive response to exercise training in people with T2D [78, 79]. MacKenzie et al. demonstrated that a single-episode of moderate-intensity (~50 % VO2max) normoxic exercise and resting hypoxic exposure each improved insulin sensitivity in T2D; this improvement was augmented when exercise and hypoxia were combined [78]. This may in part be mediated by increased effective muscle blood flow. This is an area of ongoing investigation in the field.
11 Summary of human data
Physical activity and, to a greater extent, physical fitness predict cardiovascular and all-cause mortality in subjects with and without diabetes. In subjects who vacillate between physical fitness and lack of physical fitness, the period of fitness will have a lasting effect on mortality risk. Adults and children with T1D and T2D have decreased physical fitness relative to control subjects matched for: age, body weight, physical activity and co-morbidities. Characterization of the physiological parameters predicting decrease physical fitness in diabetes have revealed a significant correlation with IR, endothelial dysfunction, decreased cardiac perfusion, LVDD, slowed muscle oxygenation with exercise and decreased tissue perfusion. Endothelial dysfunction, specifically in nitric oxide mediated nutritive blood flow to the heart and vasculature is a consistently correlate with the exercise defect. Therapies targeted at improving insulin sensitivity and endothelial function improve physical fitness in subjects with T2D in the absence of exercise training.
12 Potential molecular mechanisms: future targets
The defect in functional exercise capacity observed in T2D is improved with both exercise training and insulin sensitizer therapy with thiazolidinediones. These proof-of-concept pilot studies in humans with T2D demonstrate that targeting the defects of IR, endothelial dysfunction or decreased tissue perfusion with pharmacological therapy can improve physical fitness. Exercise is a complex intervention that has been rigorously studied in the heart, skeletal muscle and brain. Exercise stimulates mitochondrial biogenesis and angiogenesis through Nitric oxide synthases (NOS), the nicotinamide adenine dicucleotide (NAD)-dependent histone deacetylase SIRT1 or both. Deficiency in NOS enzymes or exposure to the NOS inhibitor N G-nitro-l-arginine methyl ester (L-NAME) can decrease the mitochondrial adaptation to exercise intervention [80–82]. In healthy human subjects, 7 days of bed rest can eliminate exercise induced mRNA expression of the regulators of mitochondrial biogenesis and vasculogenesis [83]. High fat feeding can abolish stimulation of AMPKα2 muscle response to exercise training [84]. Exercise failed to decrease oxidant load or mitochondrial damage in hypercholesterolemic mice [85]. Taken together, these individual findings suggest a pattern wherein metabolic disease, specifically endothelial dysfunction, leads to failed homeostatic response to stimuli.
We sought to begin to understand the molecular basis decreased fitness in diabetes by examining the vascular response to an exercise intervention. We exposed control, hypertensive and hypertensive diabetic rats to an 8 day exercise intervention and examined the adaptive response in aortic lysates [86]. In control Sprague Dawley rats, we observed a robust induction of mitochondrial protein expression which was absent in the hypertensive and diabetic rat models. We further observed no induction of endothelial nitric oxide synthase. Ongoing work in our laboratory indicates that therapies which block NOS interfere with the adaptive response to exercise training and therapies which target endothelial NOS for restore vascular signaling to mitochondria and enhance functional exercise capacity when combined with exercise training in diabetic rodents (Reusch, Miller, Keller, Knaub unpublished). Characterization of the molecular signaling defects interfering with optimal tissue target training should present opportunities for pharmacological interventions to augment fitness in persons with diabetes and other forms of cardiovascular disease.
References
Church TS, Thomas DM, Tudor-Locke C, Katzmarzyk PT, Earnest CP, Rodarte RQ, et al. Trends over 5 decades in U.S. occupation-related physical activity and their associations with obesity. PLoS One. 2011;6(5):e19657. doi:10.1371/journal.pone.0019657.
Olshansky SJ, Passaro DJ, Hershow RC, Layden J, Carnes BA, Brody J, et al. A potential decline in life expectancy in the United States in the 21st century. N Engl J Med. 2005;352(11):1138–45.
Bauer TA, Reusch JE, Levi M, Regensteiner JG. Skeletal muscle deoxygenation following the onset of moderate exercise suggests slowed microvascular blood flow kinetics in type 2 diabetes. Diabetes Care. 2007
Brandenburg SL, Reusch JE, Bauer TA, Jeffers BW, Hiatt WR, Regensteiner JG. Effects of exercise training on oxygen uptake kinetic responses in women with type 2 diabetes. Diabetes Care. 1999;22(10):1640–6.
Nadeau KJ, Regensteiner JG, Bauer TA, Brown MS, Dorosz JL, Hull A, et al. Insulin resistance in adolescents with type 1 diabetes and its relationship to cardiovascular function. J Clin Endocrinol Metab. 2009
Nadeau KJ, Regensteiner JG, Bauer TA, Brown MS, Dorosz JL, Hull A, et al. Insulin resistance in adolescents with type 1 diabetes and its relationship to cardiovascular function. J Clin Endocrinol Metab. 2010;95(2):513–21.
Vamos EP, Millett C, Parsons C, Aylin P, Majeed A, Bottle A. Nationwide study on trends in hospital admissions for major cardiovascular events and procedures among people with and without diabetes in England, 2004–2009. Diabetes Care. 2012;35(2):265–72. doi:10.2337/dc11-1682.
Center for Disease Contro and Preventionl. Data and statistics: diabetes data & trends. 2011.
Wei M, Gibbons LW, Kampert JB, Nichaman MZ, Blair SN. Low cardiorespiratory fitness and physical inactivity as predictors of mortality in men with type 2 diabetes. Ann Intern Med. 2000;132(8):605–11.
Wei M, Kampert JB, Barlow CE, Nichaman MZ, Gibbons LW, Paffenbarger Jr RS, et al. Relationship between low cardiorespiratory fitness and mortality in normal-weight, overweight, and obese men. JAMA. 1999;282(16):1547–53.
Weintraub WS, Daniels SR, Burke LE, Franklin BA, Goff Jr DC, Hayman LL, et al. Value of primordial and primary prevention for cardiovascular disease: a policy statement from the American Heart Association. Circulation. 2011;124(8):967–90.
Centers for Disease Control and Prevention. Data and statistics: obesity rates among all children in the United States 2010. http://www.cdc.gov/obesity/data/childhood.html.
Lee CD, Blair SN, Jackson AS. Cardiorespiratory fitness, body composition, and all-cause and cardiovascular disease mortality in men. Am J Clin Nutr. 1999;69(3):373–80.
Lee DC, Artero EG, Sui X, Blair SN. Mortality trends in the general population: the importance of cardiorespiratory fitness. J Psychopharmacol. 2010;24(4 Suppl):27–35. doi:10.1177/1359786810382057.
Kokkinos P, Myers J, Nylen E, Panagiotakos DB, Manolis A, Pittaras A, et al. Exercise capacity and all-cause mortality in African American and Caucasian men with type 2 diabetes. Diabetes Care. 2009;32(4):623–8. doi:10.2337/dc08-1876.
Lee DC, Sui X, Ortega FB, Kim YS, Church TS, Winett RA, et al. Comparisons of leisure-time physical activity and cardiorespiratory fitness as predictors of all-cause mortality in men and women. Br J Sports Med. 2011;45(6):504–10. doi:10.1136/bjsm.2009.066209.
Myers J, Prakash M, Froelicher V, Do D, Partington S, Atwood JE. Exercise capacity and mortality among men referred for exercise testing. N Engl J Med. 2002;346(11):793–801. doi:10.1056/NEJMoa011858.
Nelson KM, Boyko EJ, Koepsell T. All-cause mortality risk among a national sample of individuals with diabetes. Diabetes Care. 2010;33(11):2360–4. doi:10.2337/dc10-0846.
Reddigan JI, Riddell MC, Kuk JL. The joint association of physical activity and glycaemic control in predicting cardiovascular death and all-cause mortality in the US population. Diabetologia. 2012;55(3):632–5. doi:10.1007/s00125-011-2374-3.
Sluik D, Buijsse B, Muckelbauer R, Kaaks R, Teucher B, Johnsen NF, et al. Physical activity and mortality in individuals with diabetes mellitus: a prospective study and meta-analysis. Arch Intern Med. 2012;1–11. doi:10.1001/archinternmed.2012.3130.
Manson JE, Rimm EB, Stampfer MJ, Colditz GA, Willett WC, Krolewski AS, et al. Physical activity and incidence of non-insulin-dependent diabetes mellitus in women. Lancet. 1991;338(8770):774–8.
Knowler WC, Barrett-Connor E, Fowler SE, Hamman RF, Lachin JM, Walker EA, et al. Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med. 2002;346(6):393–403. doi:10.1056/NEJMoa012512.
Tuomilehto J, Lindstrom J, Eriksson JG, Valle TT, Hamalainen H, Ilanne-Parikka P, et al. Prevention of type 2 diabetes mellitus by changes in lifestyle among subjects with impaired glucose tolerance. N Engl J Med. 2001;344(18):1343–50. doi:10.1056/NEJM200105033441801.
Hamman RF, Wing RR, Edelstein SL, Lachin JM, Bray GA, Delahanty L, et al. Effect of weight loss with lifestyle intervention on risk of diabetes. Diabetes Care. 2006;29(9):2102–7. doi:10.2337/dc06-0560.
InterAct Consortium. Physical activity reduces the risk of incident type 2 diabetes in general and in abdominally lean and obese men and women: the EPIC-InterAct Study. Diabetologia. 2012;55(7):1944–52. doi:10.1007/s00125-012-2532-2.
Sieverdes JC, Sui X, Lee DC, Church TS, McClain A, Hand GA, et al. Physical activity, cardiorespiratory fitness and the incidence of type 2 diabetes in a prospective study of men. Br J Sports Med. 2010;44(4):238–44. doi:10.1136/bjsm.2009.062117.
Bouchard C, Blair SN, Church TS, Earnest CP, Hagberg JM, Hakkinen K, et al. Adverse metabolic response to regular exercise: is it a rare or common occurrence? PLoS One. 2012;7(5):e37887. doi:10.1371/journal.pone.0037887.
Regensteiner JG, Sippel J, McFarling ET, Wolfel EE, Hiatt WR. Effects of non-insulin-dependent diabetes on oxygen consumption during treadmill exercise. Med Sci Sports Exerc. 1995;27(5):661–7.
Bouchard C, Leon AS, Rao DC, Skinner JS, Wilmore JH, Gagnon J. The HERITAGE family study. Aims, design, and measurement protocol. Med Sci Sports Exerc. 1995;27(5):721–9.
Morss GM, Jordan AN, Skinner JS, Dunn AL, Church TS, Earnest CP, et al. Dose response to exercise in women aged 45–75 yr (DREW): design and rationale. Med Sci Sports Exerc. 2004;36(2):336–44. doi:10.1249/01.MSS.0000113738.06267.E5.
Sisson SB, Katzmarzyk PT, Earnest CP, Bouchard C, Blair SN, Church TS. Volume of exercise and fitness nonresponse in sedentary, postmenopausal women. Med Sci Sports Exerc. 2009;41(3):539–45. doi:10.1249/MSS.0b013e3181896c4e.
Slentz CA, Aiken LB, Houmard JA, Bales CW, Johnson JL, Tanner CJ, et al. Inactivity, exercise, and visceral fat. STRRIDE: a randomized, controlled study of exercise intensity and amount. J Appl Physiol. 2005;99(4):1613–8. doi:10.1152/japplphysiol.00124.2005.
Thompson AM, Mikus CR, Rodarte RQ, Distefano B, Priest EL, Sinclair E, et al. Inflammation and exercise (INFLAME): study rationale, design, and methods. Contemp Clin Trials. 2008;29(3):418–27. doi:10.1016/j.cct.2007.09.009.
Booth FW, Laye MJ, Roberts MD. Lifetime sedentary living accelerates some aspects of secondary aging. J Appl Physiol. 2012;111(5):1497–504.
Kokkinos P, Myers J, Faselis C, Panagiotakos DB, Doumas M, Pittaras A, et al. Exercise capacity and mortality in older men: a 20-year follow-up study. Circulation. 2010;122(8):790–7.
Schneider SH, Amorosa LF, Khachadurian AK, Ruderman NB. Studies on the mechanism of improved glucose control during regular exercise in type 2 (non-insulin-dependent) diabetes. Diabetologia. 1984;26(5):355–60.
Kjaer M, Hollenbeck CB, Frey-Hewitt B, Galbo H, Haskell W, Reaven GM. Glucoregulation and hormonal responses to maximal exercise in non-insulin-dependent diabetes. J Appl Physiol. 1990;68(5):2067–74.
Mac Ananey O, Malone J, Warmington S, O’Shea D, Green S, Egana M. Cardiac output is not related to the slowed O2 uptake kinetics in type 2 diabetes. Med Sci Sports Exerc. 2011;43(6):935–42. doi:10.1249/MSS.0b013e3182061cdb.
Regensteiner JG, Bauer TA, Reusch JE, Quaife RA, Chen MY, Smith SC, et al. Cardiac dysfunction during exercise in uncomplicated type 2 diabetes. Med Sci Sports Exerc. 2009;41(5):977–84. doi:10.1249/MSS.0b013e3181942051.
Regensteiner JG, Sippel J, McFarling ET, Wolfel EE, Hiatt WR. Effects of non-insulin-dependent diabetes on oxygen consumption during treadmill exercise. Med Sci Sports Exerc. 1995;27(6):875–81.
Gusso S, Hofman P, Lalande S, Cutfield W, Robinson E, Baldi JC. Impaired stroke volume and aerobic capacity in female adolescents with type 1 and type 2 diabetes mellitus. Diabetologia. 2008;51(7):1317–20. doi:10.1007/s00125-008-1012-1.
Wilkerson DP, Poole DC, Jones AM, Fulford J, Mawson DM, Ball CI, et al. Older type 2 diabetic males do not exhibit abnormal pulmonary oxygen uptake and muscle oxygen utilization dynamics during submaximal cycling exercise. Am J Physiol Regul Integr Comp Physiol. 2011;300(3):R685–92. doi:10.1152/ajpregu.00479.2010.
Fang ZY, Sharman J, Prins JB, Marwick TH. Determinants of exercise capacity in patients with type 2 diabetes. Diabetes Care. 2005;28(7):1643–8.
Ribisl PM, Lang W, Jaramillo SA, Jakicic JM, Stewart KJ, Bahnson J, et al. Exercise capacity and cardiovascular/metabolic characteristics of overweight and obese individuals with type 2 diabetes: the Look AHEAD clinical trial. Diabetes Care. 2007;30(10):2679–84. doi:10.2337/dc06-2487.
Hollenbeck CB, Chen N, Chen YD, Reaven GM. Relationship between the plasma insulin response to oral glucose and insulin-stimulated glucose utilization in normal subjects. Diabetes. 1984;33(5):460–3.
Laws A, Reaven GM. Effect of physical activity on age-related glucose intolerance. Clin Geriatr Med. 1990;6(4):849–63.
Reusch JE, Regensteiner JG, Watson PA. Novel actions of thiazolidinediones on vascular function and exercise capacity. Am J Med. 2003;115(Suppl 8A):69S–74.
Schneider SH, Khachadurian AK, Amorosa LF, Gavras H, Fineberg SE, Ruderman NB. Abnormal glucoregulation during exercise in type II (non-insulin-dependent) diabetes. Metabolism. 1987;36(12):1161–6.
Regensteiner JG, Bauer TA, Reusch JE, Brandenburg SL, Sippel JM, Vogelsong AM, et al. Abnormal oxygen uptake kinetic responses in women with type II diabetes mellitus. J Appl Physiol. 1998;85(1):310–7.
DeFronzo RA, Hendler R, Simonson D. Insulin resistance is a prominent feature of insulin-dependent diabetes. Diabetes. 1982;31(9):795–801.
Yki-Jarvinen H, Kiviluoto T, Taskinen MR. Insulin resistance is a prominent feature of patients with pancreatogenic diabetes. Metabolism. 1986;35(8):718–27.
Yki-Jarvinen H, Koivisto VA. Natural course of insulin resistance in type I diabetes. N Engl J Med. 1986;315(4):224–30.
Williams KV, Erbey JR, Becker D, Arslanian S, Orchard TJ. Can clinical factors estimate insulin resistance in type 1 diabetes? Diabetes. 2000;49(4):626–32.
Orchard TJ, Olson JC, Erbey JR, Williams K, Forrest KY, Smithline Kinder L, et al. Insulin resistance-related factors, but not glycemia, predict coronary artery disease in type 1 diabetes: 10-year follow-up data from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetes Care. 2003;26(5):1374–9.
Kilpatrick ES, Rigby AS, Atkin SL. Insulin resistance, the metabolic syndrome, and complication risk in type 1 diabetes: “double diabetes” in the Diabetes Control and Complications Trial. Diabetes Care. 2007;30(3):707–12.
Soedamah-Muthu SS, Chaturvedi N, Toeller M, Ferriss B, Reboldi P, Michel G, et al. Risk factors for coronary heart disease in type 1 diabetic patients in Europe: the EURODIAB Prospective Complications Study. Diabetes Care. 2004;27(2):530–7.
Regensteiner JG, Bauer TA, Reusch JE. Rosiglitazone improves exercise capacity in individuals with type 2 diabetes. [Comparative Study, Randomized Controlled Trial, Research Support, N.I.H., Extramural, Research Support, Non-U.S. Gov’t, Research Support, U.S. Gov’t, Non-P.H.S.]. Diabetes Care. 2005;28(12):2877–83.
Poirier P, Garneau C, Bogaty P, Nadeau A, Marois L, Brochu C, et al. Impact of left ventricular diastolic dysfunction on maximal treadmill performance in normotensive subjects with well-controlled type 2 diabetes mellitus. Am J Cardiol. 2000;85(4):473–7.
Diamant M, Lamb HJ, Groeneveld Y, Endert EL, Smit JW, Bax JJ, et al. Diastolic dysfunction is associated with altered myocardial metabolism in asymptomatic normotensive patients with well-controlled type 2 diabetes mellitus. J Am Coll Cardiol. 2003;42(2):328–35.
Wong TC, Piehler K, Meier CG, Testa SM, Klock AM, Aneizi AA, et al. Association between extracellular matrix expansion quantified by cardiovascular magnetic resonance and short-term mortality. Circulation. 2012;126(10):1206–16.
Schindler TH, Facta AD, Prior JO, Cadenas J, Hsueh WA, Quinones MJ, et al. Improvement in coronary vascular dysfunction produced with euglycaemic control in patients with type 2 diabetes. Heart. 2007;93(3):345–9. doi:10.1136/hrt.2006.094128.
Joshi D, Shiwalkar A, Cross MR, Sharma SK, Vachhani A, Dutt C. Continuous, non-invasive measurement of the haemodynamic response to submaximal exercise in patients with diabetes mellitus: evidence of impaired cardiac reserve and peripheral vascular response. Heart. 2010;96(1):36–41. doi:10.1136/hrt.2009.177113.
Brassard P, Legault S, Garneau C, Bogaty P, Dumesnil JG, Poirier P. Normalization of diastolic dysfunction in type 2 diabetics after exercise training. Med Sci Sports Exerc. 2007;39(11):1896–901. doi:10.1249/mss.0b013e318145b642.
DeFronzo RA, Gunnarsson R, Bjorkman O, Olsson M, Wahren J. Effects of insulin on peripheral and splanchnic glucose metabolism in noninsulin-dependent (type II) diabetes mellitus. J Clin Invest. 1985;76(1):149–55.
Befroy DE, Petersen KF, Dufour S, Mason GF, de Graaf RA, Rothman DL, et al. Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes. 2007;56(5):1376–81.
Petersen KF, Dufour S, Befroy D, Garcia R, Shulman GI. Impaired mitochondrial activity in the insulin-resistant offspring of patients with type 2 diabetes. N Engl J Med. 2004;350(7):664–71.
Scheuermann-Freestone M, Madsen PL, Manners D, Blamire AM, Buckingham RE, Styles P, et al. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation. 2003;107(24):3040–6.
Barrett EJ, Eggleston EM, Inyard AC, Wang H, Li G, Chai W, et al. The vascular actions of insulin control its delivery to muscle and regulate the rate-limiting step in skeletal muscle insulin action. Diabetologia. 2009;52(5):752–64. doi:10.1007/s00125-009-1313-z.
Barrett EJ, Rattigan S. Muscle perfusion: its measurement and role in metabolic regulation. Diabetes. 2012;61(11):2661–8.
Liu Z, Liu J, Jahn LA, Fowler DE, Barrett EJ. Infusing lipid raises plasma free fatty acids and induces insulin resistance in muscle microvasculature. J Clin Endocrinol Metab. 2009;94(9):3543–9.
Sanchez OA, Copenhaver EA, Chance MA, Fowler MJ, Towse TF, Kent-Braun JA, et al. Postmaximal contraction blood volume responses are blunted in obese and type 2 diabetic subjects in a muscle-specific manner. Am J Physiol Heart Circ Physiol. 2011;301(2):H418–27.
Williamson JR, Chang K, Frangos M, Hasan KS, Ido Y, Kawamura T, et al. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes. 1993;42(6):801–13.
He J, Watkins S, Kelley DE. Skeletal muscle lipid content and oxidative enzyme activity in relation to muscle fiber type in type 2 diabetes and obesity. Diabetes. 2001;50(4):817–23.
Clark MG. Impaired microvascular perfusion: a consequence of vascular dysfunction and a potential cause of insulin resistance in muscle. Am J Physiol Endocrinol Metab. 2008;295(4):E732–50.
Mikus CR, Fairfax ST, Libla JL, Boyle LJ, Vianna LC, Oberlin DJ, et al. Seven days of aerobic exercise training improves conduit artery blood flow following glucose ingestion in patients with type 2 diabetes. J Appl Physiol. 2011;111(3):657–64.
Green HJ, Burnett M, Kollias H, Ouyang J, Smith I, Tupling S. Can increases in capillarization explain the early adaptations in metabolic regulation in human muscle to short-term training? Can J Physiol Pharmacol. 2012;90(5):557–66.
Mackenzie RW, Watt PW, Maxwell NS. Acute normobaric hypoxia stimulates erythropoietin release. High Alt Med Biol. 2008;9(1):28–37.
Mackenzie R, Maxwell N, Castle P, Brickley G, Watt P. Acute hypoxia and exercise improve insulin sensitivity (S(I) (2*)) in individuals with type 2 diabetes. Diabetes Metab Res Rev. 2011;27(1):94–101.
Mackenzie R, Maxwell N, Castle P, Elliott B, Brickley G, Watt P. Intermittent exercise with and without hypoxia improves insulin sensitivity in individuals with type 2 diabetes. J Clin Endocrinol Metab. 2012;97(4):E546–55.
Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299(5608):896–9.
Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310(5746):314–7.
Gutsaeva DR, Carraway MS, Suliman HB, Demchenko IT, Shitara H, Yonekawa H, et al. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J Neurosci. 2008;28(9):2015–24.
Ringholm S, Bienso RS, Kiilerich K, Guadalupe-Grau A, Aachmann-Andersen NJ, Saltin B, et al. Bed rest reduces metabolic protein content and abolishes exercise-induced mRNA responses in human skeletal muscle. Am J Physiol Endocrinol Metab. 2011;301(4):E649–58.
Lee-Young RS, Ayala JE, Fueger PT, Mayes WH, Kang L, Wasserman DH. Obesity impairs skeletal muscle AMPK signaling during exercise: role of AMPKalpha2 in the regulation of exercise capacity in vivo. Int J Obes (Lond). 2011;35(7):982–9.
Young CG, Knight CA, Vickers KC, Westbrook D, Madamanchi NR, Runge MS, et al. Differential effects of exercise on aortic mitochondria. Am J Physiol Heart Circ Physiol. 2005;288(4):H1683–9.
Knaub LA, McCune S, Chicco AJ, Miller M, Moore RL, Birdsey N et al. Impaired response to exercise intervention in the vasculature in metabolic syndrome. Diab Vasc Dis Res. 2012
Blair SN, Kohl 3rd HW, Barlow CE, Paffenbarger Jr RS, Gibbons LW, Macera CA. Changes in physical fitness and all-cause mortality. A prospective study of healthy and unhealthy men. JAMA. 1995;273(14):1093–8.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reusch, J.E.B., Bridenstine, M. & Regensteiner, J.G. Type 2 diabetes mellitus and exercise impairment. Rev Endocr Metab Disord 14, 77–86 (2013). https://doi.org/10.1007/s11154-012-9234-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-012-9234-4