
The effects of Zr, Mo and Zr, Ta powder mixtures during spark-plasma sintering of compositions at pressing loading of 60 MPa in the range 1200 – 1600°C on the phase composition, microstructure, grain sizes of crystalline phases, relative density, linear shrinkage, physical-mechanical properties, and linear correlation of elasticity modulus and fracture toughness of mullite–β-SiAlON–c-BN samples are shown in the present work. Synthesized powders of β-SiAlON and c-BN are characterized by extensive crystallization of β-SiAlON and c-BN, respectively. Sintered samples with Zr, Mo and Zr, Ta mixtures show extensive mullite formation, active growth of β-SiAlON, and less extensive growth of c-BN in the range 1200 – 1600°C. Active growth of crystalline β-Mo, Zr, Mo, and Mo2Zr phases is noticeable in the sample with Zr, Mo mixtures but extensive growth of crystalline β-Ta, Zr, α-Zr, Ta, β-Ta, Ta3Zr, and Ta3Zr2 is observed in the sample with Zr, Ta mixtures with an increase of temperature. The Zr, Mo mixture favors the formation of a more uniformly and densely sintered microstructure of the ceramic phase, round metallic Mo and β-Mo, Zr particles, more reinforced ceramic-metallic boundary areas, and metallic phases and facilitates the reduction of crystalline-phase grain sizes in the range 1400 – 1600°C. As a result, the composition with a Zr, Mo mixture sinters more uniformly and gradually. The corresponding sample shows larger values of physical-mechanical properties, higher cracking resistance with an insignificant quantity of microcracks, and a larger linear correlation of the elasticity modulus and fracture toughness in the range 1200 – 1600°C.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Brittleness of boundary areas in oxide–oxide-free crystalline phases of various solid and impact-resistant ceramics is a common problem that is important for studying the mechanisms and extent of sintering of heterogeneous powders; the formation of phases, microstructure, and properties in boundary layers; and, as a result, the development of new types of these materials [1,2,3,4]. This problem is caused by the uneven diffusion at the boundaries of sintered particles of heterogeneous ceramic components because of the different diffusion coefficients in the powders being sintered as the temperature and pressing load increase [3, 4]. As a result, dislocations that cause microcracks of various trajectories and lengths, degrade the elastic properties, and increase the brittleness arise in the boundary layers of the crystalline phases. This reduces the compaction and weakens the boundary structures of the heterogeneous ceramic particles [3, 4].
Various methods are employed to solve these problems, e.g., addition to mixtures being spark-plasma sintered of oxide and oxide-free powders of sialon [5]; oxide components, in particular Y2O3 and Dy2O3 that form low-melting eutectics and solid solutions with oxide and oxide-free powders [6, 7]; cubic ZrO2 [8]; and TiC–ZrC solid solution [9]. These additives stimulate differently the diffusion of the oxide and oxide-free components at the boundaries of the heterogeneous sintered particles in the liquid and solid phases [5,6,7,8,9]. These methods are not always applicable in practice because of the specifics and side processes of the used additives, which effect the extent and uniformity of the sintering of mixtures of oxide and oxide-free powders, the compaction and strength of the boundary structure of the crystalline phases, and the physicomechanical properties of the materials [5,6,7,8,9].
However, a method involving addition of mixtures of metal powders, e.g., Ni, Co or Ni, Mo, to mixtures of oxide-free components being sintered is widely applied [10,11,12,13]. The main feature of the method is the formation of solid solutions of metallic phases and intermetallic compounds in a Ni and/or Co melt [10,11,12,13]. Transformation of the metal powders into oxide-free components is avoided in this process [10,11,12,13], in contrast to sintered mixtures of oxide-free components and a metal powder where the oxide-free component formed from the metal powder crumbles, disintegrates, or compacts and strengthens the boundary structures of the crystalline phases depending on its properties and, as a result, affects differently the ratio of brittle and elastic properties, elastic modulus, hardness, and impact viscosity of the materials [14, 15]. Sintering of mixtures of oxide-free components with mixtures of Ni, Co and Ni, Mo powders regulates the relationship of the shapes, sizes, sintering of particles of solid solutions of ceramic and metallic phases and intermetallic compounds, and the ratio of brittleness/compaction and strengthening and crack resistance of boundary layers of the ceramic and metallic phases [10,11,12,13].
The aim of the work was to study the effects of Zr, Mo and Zr, Ta additives on the phase composition, microstructure, grain size of crystalline phases, relative density, linear shrinkage, physicomechanical properties, and linear correlation of the elastic modulus and impact viscosity of mullite–β-SiAlON–c-BN samples during their spark-plasma sintering with pressing load 60 MPa at 1200 – 1600°C.
Experimental
Mixtures of Al2O3 and SiO2 powders were prepared using Al2O3 (Aldrich, Belgium; 97.5% pure) and SiO2 (Merck, Germany; 97.5% pure). These components were weighed in the weight proportion corresponding to the mullite stoichiometry (3:2) and mixed in a planetary ball mill (Retsch PM 400) for ~10 min. β-SiAlON and c-BN powders were synthesized in a plasma-chemical apparatus under vacuum at 1600°C for 1 h using the starting components (Table 1).
β-SiAlON and c-BN were synthesized according to the reactions:
β-SiAlON and c-BN powders in mixtures with Zr, Mo and Zr, Ta additives were mixed in weight proportions (Table 2) in a planetary ball mill (Retsch PM 400) for ~10 min to produce two different groups of homogeneous mixtures (Table 2).
The obtained mixture of Al2O3 and SiO2 powders was mixed with the two prepared groups of β-SiAlON/c-BN/Zr, Mo and β-SiAlON/c-BN/Zr, Ta powder mixtures in a planetary ball mill (Resch PM 400) for ~10 min.
The obtained mixtures of components were loaded into a graphite press-mold of diameter 30 mm and underwent spark-plasma sintering (SPS, Sumimoto, Model SPS 825, CE; Dr. Sinter, Japan) under vacuum (6 Pa) with pressing load 60 MPa for 2 min at 1200 – 1600°C at heating rate 100°C/min.
The phase composition, microstructure, relative density ρrel, linear shrinkage Δl, elastic modulus E, Vickers hardness HV, and impact viscosity K1c of the synthesized powders and sintered samples were determined by the previously published methods [4]. The theoretical density of the components (g/cm3) were mullite 3.17; β-Si3Al3O3N5 3.09, c-BN 3.49, h-BN 2.1, α-Mo, Zr 2.46, β-Mo, Zr 3.52, α-Ta, Zr 6.52, β-Ta, Zr 7.68, β-Zr-Ta 2.95, α-Zr, Ta 4.0, Mo 10.28, Ta 16.69, Mo2Zr 3.89, Ta3Zr 3.97, and Ta3Zr2 4.34.
Results and Discussion
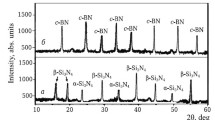
The phase composition of the synthesized sialon and c-BN powders (Fig. 1) consisted mainly of strong diffraction maxima of β-SiAlON with an insignificant amount of unreacted AlN and strong diffraction maxima of c-BN.

Phase composition of β-SiAlON (a) and c-BN powders (b) synthesized by plasma-chemical method at 1600°C: β-SiAlON, sialon; AlN, aluminum nitride; c-BN, cubic boron nitride.
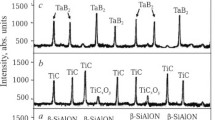
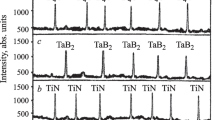
Figure 2 shows the phase compositions of samples prepared by SPS at 1200 – 1600°C.

Phase composition of M30SiAlON10BN30Zr30Mo (a) and M30SiAlON10BN30Zr30Ta samples (b) sintered at 1200 – 1600°C: M) mullite (3Al2O3·2SiO2); h-BN) hexagonal boron nitride; α-Mo, Zr, α-Ta, Zr) solid solution of hexagonal zirconium; β-Mo, Zr, β-Ta, Zr) solid solution of cubic zirconium; α-Zr, Ta) solid solution of cubic tantalum; β-Zr, Ta) solid solution of tetragonal tantalum; Mo, molybdenum; α-Ta) cubic tantalum; β-Ta) tetragonal tantalum; Mo0.5Zr) molybdenum zirconium; Me2Zr) dimolybdenum zirconium; Ta3Zr) tritantalum zirconium; Ta3Zr2) tritantalum dizirconium.
Samples with Zr, Mo and Zr, Ta additives were characterized by extensive mullite formation at 1200 – 1600°C. This was caused by rapid formation of mullite structures with the stoichiometric composition during the reaction of Al2O3 and SiO2. Also, β-SiAlON was observed to form approximately equal to the increase of mullite at 1200 – 1600°C because of structuring of sialon during its viscous flow. However, c-BN formed less extensively than mullite and β-SiAlON at 1200 – 1600°C because of the dense (cubic) structure of this component with distinct covalent bonds and rapid diffusion and structuring of c-BN in the solid. Relatively rapid formation of h-BN and the weakened intensity of c-BN diffraction maxima related to this were noticeable in the samples at 1400 – 1600°C. This was due to a partial phase transition c-BN → h-BN. A reaction of β-SiAlON with c-BN and h-BN was not observed. This was indicated by the lack of additional diffraction maxima at 1200 – 1600°C.
Crystalline α- and β-Mo, Zr, Mo, and Mo2Zr phases were observed in the M30SiAlON10BN30Zr30Mo sample at 1200 – 1600°C. Mainly weak diffraction maxima of less dense α-Mo, Zr and crystalline Mo appeared at 1200°C because of the low solubility of Zr in Mo and poor crystallization of Mo in the solid. A phase transition during which less dense crystalline α-Mo, Zr rearranged into the denser crystalline β-Mo, Zr in the solid because of the increased solubility of Zr in α-Mo, Zr and Mo up to 1400°C was noted. Simultaneously, crystalline Mo2Zr formed because of dissolution of intermediate Mo0.5Zr and crystalline Mo in α-Mo, Zr at 1200 – 1300°C with formation of the Mo2Zr structure at 1400°C. A certain amount of β-Mo, Zr formed at 1400°C because of a solid-state reaction of α-Mo, Zr and Mo2Zr at 1200 – 1400°C. The appearance of weak diffraction maxima of α/β-Mo, Zr at 2θ of 20.7 and 49.5° and their slight ingrowth at 2θ of 27.5, 44.6, and 54.6° at 1400°C were indicative of this. These processes corresponded to the temperature range of a eutectoid in the Mo–Zr system two-phase equilibrium diagram [16]. Crystalline β-Mo, Zr and Mo2Zr phases grew more extensively at 1500 – 1600°C. This was explained by the increasing liquid-phase solubility of Zr and Mo, respectively, in β-Mo, Zr with rapid formation of crystalline β-Mo, Zr and Mo2Zr phases. The ingrowth of crystalline β-Mo, Zr was also explained by the reaction of Mo2Zr and the liquid phase. A crystalline Mo phase was missing in the sample with Zr, Mo above 1400°C (Fig. 2a). This indicated that Mo was fully converted into crystalline β-Mo, Zr and Mo2Zr phases at 1400 – 1600°C (Fig. 2a). These formation mechanisms of crystalline β-Mo, Zr and Mo2Zr phases corresponded to the eutectic temperature ranges 1543 – 1603 and 1583 – 1607°C in the Mo–Zr system two-phase equilibrium diagram [16].
The M30SiAlON10BN30Zr30Ta sample showed crystalline α- and β-Ta, Zr, α- and β-Zr, Ta, and β- and α-Ta phases at 1200 – 1600°C. Less dense structures of crystalline α-Ta, Zr, β-Zr, Ta, and β-Ta were insignificantly developed at 1200°C. This was explained by the limited solubility of Zr in β-Ta and of β-Ta in Zr and poor crystallization of β-Ta in the solid. A phase transition at which less dense crystalline α-Ta, Zr, β-Zr, Ta, and β-Ta rearranged into denser β-Ta, Zr, α-Zr, Ta, and α-Ta in the solid because of transformations in the Zr and Ta crystal structures [17] and the increased solubility of Zr in α-Ta, Zr, β-Ta, and β-Zr, Ta up to 1400°C at 60 MPa was noticeable in the sample with the Zr, Ta mixture. The most rapid development of β-Ta, Zr, α-Zr, Ta, and α-Ta was noted at 1500 – 1600°C because of the increasing solubility of Zr in β-Ta, Zr and α-Ta in α-Zr, Ta, respectively. Also, Ta3Zr and Ta3Zr2 crystallized rapidly at 1400 – 1600°C. The reaction of crystalline Ta3Zr and Ta3Zr2 and the liquid phase promoted rapid development of crystalline β-Ta, Zr and α-Zr, Ta phases. Ta3Zr formed at 1400°C and continued up to 1500°C while Ta3Zr2 formed at 1500°C and continued up to 1600°C. Crystalline Ta3Zr formed because of liquid-phase dissolution of Zr and β/α-Ta in α/β-Ta, Zr and β/α-Zr, Ta, respectively, while Ta3Zr2 formed because of liquid-phase dissolution of Ta3Zr in β-Ta, Zr and α-Zr, Ta with recrystallization of Ta3Zr2. This corresponded to the eutectic temperature range in the Ta–Zr system two-phase equilibrium diagram [17]. Similar appearance of diffraction maxima of crystalline Mo, Mo2Zr, α-Ta, Ta3Zr, and Ta3Zr2 at 1200 – 1600°C was noted. This was explained by the approximately equal structure formation of these components because of dense Mo, α-Ta, and intermetallic compound structures. Simultaneously, reactions of mullite, β-SiAlON, and c-BN with Zr, Mo and Zr, Ta mixtures and intermetallic compounds were not observed because decomposition products of mullite and sialon and oxidation products of c-BN, mixtures of metal powders, and intermetallic compounds were missing at 1200 – 1600°C.
Figure 3 shows the microstructures of the samples prepared by SPS at 1500°C. The microstructure of the M30SiAlON10BN30Zr30Mo sample (Fig. 3a) was more uniformly and densely sintered with a small amount of weakly sintered areas and had smaller pores than that of the M30SiAlON10BN30Zr30Ta sample (Fig. 3b). This was explained by the different effects of diffusion processes in the metallic phases of the Mo, Zr and Zr, Ta metal powder mixtures on sintering of the main components. However, the samples had uniformly and densely sintered microstructures of the ceramic phase with almost no pores and contained different amounts of agglomerated particles (Fig. 3a1 and b1). The uniform sintering of the compositions because of viscous flow of sialon and the almost complete solid-state sintering of c-BN and h-BN particles were responsible for this. The microstructure of the metallic Mo and Mo2Zr phases consisted of round particles of crystalline Mo about 1 μm in size and soft agglomerates of crystalline Mo2Zr about 2 – 3.5 μm in size (Fig. 3a2). This was explained by dissolution of a solid solution of Mo in β-Mo2Zr and development of the Mo2Zr structure from the liquid phase at 1500 – 1600°C (Fig. 2a).

Microstructure of 30SiAlON10BN30Zr30Mo: a) general view; a1) ceramic phase, a2) metallic phase containing crystalline Mo and intermetallic Mo2Zr; a3) solid solution β-Mo, Zr, and M30SiAlON10BN30Zr30Ta samples: b) general view; b1) ceramic phase; b2) metallic phase containing crystalline α-Ta and intermetallic Ta3Zr and Ta3Zr2; b3) solid solution β-Ta, Zr, sintered at 1500°C.
The microstructure of metallic α-Ta and Ta3Zr phases appeared as weakly sintered nonuniform compacted crystalline formations of α-Ta and Ta3Zr about 2 – 3 mm in size as compared to the monolithic crystalline Ta3Zr2 structure about 8 μm in size that consisted mainly of densely fused crystalline Ta3Zr formations and less dense sintered α-Ta particles (Fig. 3b2). This structure of Ta3Zr2 was explained by the corresponding formation of crystalline Ta3Zr2 (Fig. 2b). In general, the structural differences in crystalline α-Ta, Ta3Zr, and Ta3Zr2 were due to formation features of these crystalline phases during the various diffusion processes in the composition being sintered (Fig. 2b). Significant pores between crystalline formations of α-Ta and Ta3Zr2 and Ta3Zr and Ta3Zr2 were observed (Fig. 3b2) and resulted from the various diffusion processes. Hence, the crystal structure of Ta3Zr2 was harder than those of α-Ta and Ta3Zr. Also, β-Mo, Zr and β-Ta, Zr phases formed different microstructures (Fig. 3a3 and b3). The microstructure of β-Mo, Zr was more uniform and densely sintered to round β-Mo, Zr particles (Fig. 3a3), in contrast to crystalline β-Ta, Zr formations of variously agglomerated particles of various shapes (Fig. 3b3).
Figures 4 – 8 show grain sizes of crystalline phases at 1400 – 1600°C; ρrel and Δl at 1200 – 1600°C, the microstructure of boundary areas of oxide and oxide-free crystalline phases at 1500°C, the physicomechanical properties at 1200 – 1600°C, and photographs of indentations at 1500°C.

Grain sizes of crystalline phases in M30SiAlON10BN30Zr30Mo (a) and M30SiAlON10BN30Zr30Ta samples (b) at 1400 – 1600°C:■) 1400°C; ) 1500°C;
) 1500°C;  ) 1600°C.
) 1600°C.

Changes of ρrel (■) and Δl ( ) of M30SiAlON10BN30Zr30Mo (a) and M30SiAlON10BN30Zr30Ta samples (b) at 1200 – 1600°C.
) of M30SiAlON10BN30Zr30Mo (a) and M30SiAlON10BN30Zr30Ta samples (b) at 1200 – 1600°C.
The M30SiAlON10BN30Zr30Mo composition was uniformly sintered at 1200 – 1600°C because of extensive development of crystalline β-Mo, Zr and Mo2Zr phases from the liquid phase (Fig. 2a), formation in general of a uniform and densely sintered microstructure (Fig. 3a), the dense microstructure of the ceramic phase with fewer agglomerated particles (Fig. 3a1), formation of weakly fused and compacted crystalline Mo2Zr formations as soft agglomerates (Fig. 3a2), and development of polydisperse grains of the crystalline phases at 1400 – 1600°C (Fig. 4).
Simultaneously, the development of this sintering was affected by variation of c-BN grain sizes (Fig. 4) associated with formation of h-BN at 1400°C (Fig. 2a). A decrease of mullite and β-SiAlON grain sizes (Fig. 4) had little effect on the extent of sintering of this composition because c-BN grains grew rapidly and the large sizes of Mo2Zr grains (Fig. 4) to a certain degree slowed diffusion at the boundaries of sintered mullite and β-SiAlON grains and c-BN/h-BN, mullite, β-SiAlON, and Mo2Zr grains and sintering of c-BN/h-BN and Mo2Zr grain boundaries. This correlated with results from an analysis of the microstructure of boundary areas of the crystalline phases (Fig. 6a – a3).

Microstructure of boundary areas of mullite, β-SiAlON, c-BN, h-BN, β-Mo, Zr, Mo2Zr, β-Ta, Zr, Ta3Zr, and Ta3Zr2 of M30SiAlON10BN30Zr30Mo (a – a3) and M30SiAlON10BN30Zr30Ta samples (b – b3) sintered at 1500°C.
The M30SiAlON10BN30Zr30Ta composition was nonuniformly sintered at 1200 – 1600°C. Sintering of the composition increased up to 1400°C and decreased at 1400 – 1600°C. The increased sintering up to 1400°C resulted from solid-state mutual dissolution of Zr in β-Ta and β-Ta in Zr at 1200 – 1300°C with initiation of solid-state mutual dissolution of Zr in α-Ta and α-Ta in Zr, dissolution of Zr and β/α-Ta in solid solutions α-Ta, Zr and β/α-Zr, Ta at 1400°C, and development of crystalline c-BN (Fig. 2b). However, the increase in the sintering of this composition up to 1400°C was much less than the increased sintering of the M30SiAlON10BN30Zr30Mo composition because the in general nonuniformly sintered microstructure contained more pores (Fig. 3b) and was less dense than the microstructure of the ceramic phase with many agglomerated particles (Fig. 3b1). The sintering decreased at 1400 – 1600°C because the phase transition of c-BN into h-BN was more rapid (Fig. 2b) and variously compacted crystalline α-Ta, Ta3Zr, and Ta3Zr2 structures formed from partially recrystallized α-Ta, Ta3Zr and fully recrystallized Ta3Zr2 in the liquid phase consisting of tightly fused agglomerates in the metallic phase (Fig. 3b2); variously sintered and agglomerated β-Ta, Zr particles of various shapes formed (Fig. 3b3); α-Ta grains grew smoothly; β-Ta, Zr gains grew more rapidly; and Ta3Zr and Ta3Zr2 grains grew especially extensively with development of a monodisperse composition of the crystalline phases at 1400 – 1600°C (Fig. 4). As a result, the formed α-Ta, Zr and β/α-Zr, Ta crystalline phases had less of an effect on the stimulation of sintering of this composition.
The change of the physicomechanical properties of the samples correlated with sintering of these compositions at 1200 – 1600°C. The elastic properties of the M30SiAlON10BN30Zr30Mo sample increased uniformly and strongly, which correlated with increases of K1c and HV of the sample at 1200 – 1600°C. However, the increases of K1c and HV differed in this temperature range because of the formation of nonuniformly and incompletely compacted crystalline Mo2Zr formations as soft small agglomerates in the metallic phase (Fig. 3a2), a significant increase in the sizes of c-BN grains, and the large sizes of the Mo2Zr grains (Fig. 4). As a result, bonding at the contact boundaries of c-BN/h-BN and Mo2Zr grains decreased with the development in these sections of brittle properties and the reduction of elastic properties. This diminished the resistance of the samples to the effects of an external load. This sample was characterized by lower crack resistance at 1500°C as compared to that at 1300°C (Fig. 8a and a1). In general, the correlating results of these properties were explained by the presence of round β-Mo, Zr particles (Fig. 3a3), the polydisperse grain composition of the crystalline phases (Fig. 4), and the presence of a thin intermediate layer of h-BN at the boundary areas of the oxide–oxide-free and oxide-free crystalline phases (Fig. 6a – a3) that strengthened the sample structure. As a result, the above side effects were fully compensated and microcracks propagated to a limited extent and discontinuously in several directions around the indentation (Fig. 8a1) along a winding trajectory (Fig. 8a1-0) with slow and halting movement of the microcracks along grain boundaries of the crystalline phases. In general, this was related to the interaction of the propagated microcracks and indicated shapes, particle sizes, and/or local stress areas around these particles. As a result, stresses were evenly dissipated from of the propagated microcracks because of the developed elastic properties at the particle contact boundaries that were caused by the strengthening effect at the boundary areas of oxide–oxide-free and oxide-free crystalline phases (Fig. 6a – a3), the polydisperse grain composition (Fig. 4), and, correspondingly, hardening of the boundary areas of the crystalline phases and the structure that increased the resistance of the sample to the action of the external load.
The M30SiAlON10BN30Zr30Ta sample showed nonuniform development of physicomechanical properties, in particular, a smooth growth up to 1400°C and a rapid decrease at 1400 – 1600°C. The physicomechanical properties grew smoothly because of the formation of α-Ta, Zr and β-Zr, Ta phases that strengthened the sample structure during a phase transition of the less dense structure of these solid solutions into denser structures β-Ta, Zr, α-Zr, Ta, and crystalline c-BN in the solid (Fig. 2b) and a relatively polydisperse composition of the crystalline phase grains at 1400°C (Fig. 4) in the sample structure. A slight embrittlement of the sample structure was observed in the photograph of the indentation with an indication of the linear trajectory of microcrack propagation at 1300°C (Fig. 8b ). The reason for this was the formation of less dense crystalline α-Ta, Zr and β-Zr, Ta phases (Fig. 2b). However, the rapid decrease of the physicomechanical properties was explained by the embrittling effect of the formed variously compacted crystalline α-Ta, Ta3Zr, and Ta3Zr2 structures consisting of densely fused agglomerates in the metallic phase (Fig. 3, b2); considerable growth of crystalline phase grains; the practically monodisperse composition of the crystalline phase grains (Fig. 4); and the wide boundary layers of h-BN and α-Ta at the boundary areas of the oxide–oxide-free and oxide-free crystalline phases (Fig. 6b – b2). As a result, the boundary areas of these crystalline phases were significantly weakened (Fig. 6b – b3). This increased the brittleness and decreased the elastic properties and resistance of the sample to the action of the external load. This sample was characterized by lower crack resistance with a linear trajectory of microcrack propagation at 1500°C (Fig. 8b1), on the path of movement of which Ta3Zr and Ta3Zr2 particles were situated (Fig. 8b1-0). In general, a microcrack advanced along the surface of Ta3Zr and Ta3Zr2 particles and not in the particle bulk with insignificant splitting of them because of the formation on the surface of Ta3Zr and Ta3Zr2 particles of brittle boundary layers of h-BN and α-Ta as compared to denser and harder crystalline Ta3Zr and Ta3Zr2 structures (Fig. 3b2). These results corresponded to formation of crystalline Ta3Zr and Ta3Zr2 structures (Fig. 3b2) and microstructures of boundary areas of oxide–oxide-free and oxide-free crystalline phases (Fig. 6b – b3). Therefore, propagating microcracks interacted less vigorously with particles of the crystalline phases of the monodisperse grain composition, i.e., stresses in front of microcracks propagating rapidly along grain boundaries of crystalline phases were unevenly and incompletely dissipated.
Figure 9 shows the linear correlation of E and K1c of sintered samples. A comparison of the quantities R2 of the samples showed an insignificant difference in this quantity (~0.01) that was greater in the M30SiAlON10BN30Zr30Mo sample. Simultaneously, the deviations of the lines relative to the E and K1c values of these samples were practically identical at 1200 – 1600°C. The positions of the lines of the samples relative to each other were approximately the same at 1200 – 1600°C.
The correlation of the properties of M30SiAlON10BN30Zr30Mo relative to the line was somewhat poorer at 1300, 1500, and 1600°C as compared to the more accurate correlation of the properties relative to the line at 1200 and 1400°C. This result at 1300°C was explained by the weak sample structure because of the less dense crystalline α-Mo, Zr phase and the poorly developed c-BN phase (Fig. 2a); at 1500 and 1600°C, by the nonuniformly and incompletely compacted microstructure caused by the formation of weakly fused and poorly compacted crystalline Mo2Zr formations as soft agglomerates in the metallic phase (Fig. 3a2), activation of c-BN grain growth, the large sizes of Mo2Zr grains (Fig. 4), and development of slight brittleness at boundaries of oxide–oxide-free and oxide-free crystalline phases because of a thin intermediate layer of h-BN in the boundary areas of the crystalline phases (Fig. 6a – a3). The poorer correlation of the sample properties to the line had substantially no effect on the value of R2, i.e., on the elastic properties and impact viscosity (Fig. 7). This was due to the formation of round β-Mo, Zr particles (Fig. 3a3), the polydisperse composition of the crystalline phase grains (Fig. 4), and the twisting propagation trajectory of a few microcracks (Fig. 8a1 and a1-0), which in general compensated the above side processes and strengthened the sample structure. The better correlation of the sample properties with the line at 1200 and 1400°C was related to the smaller influence and initiation of development of the above processes during sintering of this composition.

Changes of E and K1c (a, ■ and  , respectively) and HV (b) of M30SiAlON10BN30Zr30Mo (1) and M30SiAlON10BN30Zr30Ta samples (2) at 1200 – 1600°C.
, respectively) and HV (b) of M30SiAlON10BN30Zr30Mo (1) and M30SiAlON10BN30Zr30Ta samples (2) at 1200 – 1600°C.

Photographs of indentations with changing HV of sintered M30SiAlON10BN30Zr30Mo: a) at 1300°C; a1) at 1500°C, and M30SiAlON10BN30Zr30Ta samples: b) at 1300°C; b1) at 1500°C) with an indication of the propagation trajectories of microcracks (a1-0) and (b1-0) at 1500°C, respectively.

Linear correlation of E and K1c of samples at 1200 – 1600°C.
Simultaneously, the correlation of the M30SiAlON10BN30Zr30Ta sample properties with the line at 1300, 1500, and 1600°C as compared to the more accurate correlation of the properties with the line at 1200 and 1400°C vanished. The less accurate correlation of the sample properties with the line at 1300°C was related to its weak structure because of the less dense α-Ta, Zr, β-Zr, Ta, and poorly developed c-BN phases (Fig. 2b). However, the poorer correlation of the sample properties with the line at 1500 and 1600°C was due to a nonuniform, agglomerated, and porous microstructure caused by formation of variously compacted crystalline α-Ta, Ta3Zr, and Ta3Zr2 structures consisting of densely fused agglomerates in the metallic phase (Fig. 3b2), the formation of a monodisperse composition of crystalline phase grains (Fig. 4), development of significant brittleness in the boundary areas of oxide–oxide-free and oxide-free crystalline phases caused by the broad boundary layers of h-BN and α-Ta (Fig. 6b – b2), and the linear propagation trajectory of many microcracks (Fig. 8b1 and b1-0). These reasons and the less accurate correlation of the sample properties with the line related to them decreased the R2 parameter. This indicated that the sample elastic properties decreased and the brittleness increased (Fig. 7). However, the more accurate correlation of the sample properties with the line at 1200 and 1400°C was correspondingly related to the smaller influence and initiation of development of the above processes during sintering of this composition.
Conclusion
Mixtures of Zr, Mo and Zr, Ta powders during SPS of compositions with pressing load 60 MPa at 1200 – 1600°C were shown to affect the phase composition, microstructure, crystalline phase grain size, ρrel, Δl, physicomechanical properties, and linear correlation of E and K1c of the viscosity of mullite–β-SiAlON–c-BN samples. The synthesized powders of β-SiAlON and c-BN were characterized by extensive crystallization of β-SiAlON and c-BN, respectively.
Extensive mullite formation, rapid growth of β-SiAlON, and less extensive development of c-BN at 1200 – 1600°C was observed in sintered samples. The M30SiAlON10BN30Zr30Mo sample had noticeably rapid development of crystalline β-Mo, Zr, Mo, and Mo2Zr phases; M30SiAlON10BN30Zr30Ta, noticeably extensive ingrowth of crystalline β-Ta, Zr, α-Zr, Ta, α-Ta, Ta3Zr, and Ta3Zr2 phases as the temperature increased. The Zr, Mo mixture stimulated formation of more uniform and dense sintering of the ceramic phase microstructure; particles of metallic Mo and round β-Mo, Zr; weakly fused crystalline Mo2Zr formations, and stronger boundary areas of ceramic-metallic and metallic phases and helped to reduce the grain sizes of mullite, β-SiAlON, c-BN, and crystalline Mo, Mo2Zr, β-Mo, Zr phases at 1400 – 1600°C. As a result, the M30SiAlON10BN30Zr30Mo composition was more uniformly sintered. This sample showed high physicomechanical properties, high crack resistance with an insignificant number of microcracks, and a good linear correlation of E and K1c at 1200 – 1600°C
References
M. Hotta and T. Goto, “Densification and microstructure of Al2O3–cBN composites prepared by spark-plasma sintering,” J. Ceram. Soc. Jpn., 116(6), 744 – 748 (2008).
M. Hotta and T. Goto, “Densification, phase transformation and hardness of mullite–cubic BN composites prepared by spark plasma sintering,” J. Ceram. Soc. Jpn., 118(2), 157 – 160 (2010).
D. Chakravarty and G. Sundararajan, “Microstructure, mechanical properties and machining performance of spark plasma sintered Al2O3–ZrO2–TiCN nanocomposites,” J. Eur. Ceram. Soc., 33(13/14), 2597 – 2607 (2013).
A. V. Hmelov, “Preparation of mullite–TiC–TiN materials by a plasma spark method and their properties,” Refract. Ind Ceram., 58(4), 418 – 425 (2017.
A. V. Hmelov, “Producing and properties of mullite–sialon– ZrB2 materials obtained using a spark-plasma technique,” Refract. Ind. Ceram., 59(6), 633 – 641 (2019).
S. Guo and Y. Kagawa, “High-strength zirconium diboridebased ceramic composites consolidated by low temperature hot pressing,” Sci. Technol. Adv Mater., 13(4), 1 – 6 (2012).
A. V. Hmelov, “Sintering a mixture of powders in the Al2O3–SiO2–β-SiAlON–TiC–Dy2O3 system by the sparkplasma method with high compaction loading,” Refract. Ind. Ceram., 60(3), 284 – 290 (2019).
A. V. Hmelov, “Mullite–TiC–c-BN–c-ZrO2 materials produced by spark-plasma sintering and their properties,” Refract. Ind. Ceram., 60(1), 86 – 91 (2019).
A. V. Hmelov, “Strengthening oxide–oxide-free materials by incorporation of TiC–ZrC solid solutions into their structure during spark plasma sintering of initial powder mixtures under high compression load,” Refract. Ind. Ceram., 60(5), 486 – 494 (2020).
X. Ren, Z. Peng, Y. Peng, and C. Wang, “Spark plasma sintered WC–Ni carbides with various contents of ZrC nanopowder,” Key Eng. Mater., 591(1), 75 – 78 (2014).
V. Verma and M. Kumar, “Processing of TiCN–WC–Ni/Co cermets via conventional and spark plasma sintering technique,” Trans. Indian Inst. Met., 70(3), 843 – 853 (2017).
T. Yang, C. Huang, H. Liu, and B. Zou, “Effect of (Ni, Mo) and (W, Ti)C on the microstructure and mechanical properties of TiB2 ceramic tool materials,” Mater. Sci. Forum, 723(4), 233 – 237 (2012).
G. Zhang, W. Xiong, Q. Yang, and Z. Yao, “Effect of Mo addition on microstructure and mechanical properties of (Ti, W)C solid solution based cermets,” Int. J. Refract. Met. Hard Mater., 43, 77 – 82 (2014).
R. Vedant, “Development of ZrB2–B4C–Me ceramic-matrix composite for high temperature applications,” Thesis, Natl. Inst. Technol. Rourkela, 2014, pp. 1 – 61.
A. Purwar, R. Mukherjee, K. Ravikumar, and S. Ariharan, “Development of ZrB2–SiC–Ti by multi-stage spark plasma sintering at 1600°C,” J. Ceram. Soc. Jpn., 124(4), 393 – 402 (2016).
M. Zinkevich and N. Mattern, “Thermodynamic assessment of the Mo–Zr system,” J. Phase Equilib., 23(2), 156 – 162 (2002).
A. F. Guillermet, “Phase diagram and thermochemical properties of the Ta–Zr system. An assessment based on Gibbs energy modelling,” J. Alloys Compd., 226(1/2), 174 – 184 (1995).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated from Novye Ogneupory, No. 6, pp. 27 – 36, June, 2020.
Rights and permissions
About this article

Cite this article
Hmelov, A.V. Development of Dense Materials by Plasma-Spark Sintering of Oxide–Oxide-Free Components with Different Mixtures of Metal Powders. Refract Ind Ceram 61, 313–321 (2020). https://doi.org/10.1007/s11148-020-00479-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11148-020-00479-z