
Abstract
In this work, the nanostructure catalyst of Co–Ru/CNTs is prepared by chemical reduction technique. Then, a set of catalytic experiments are designed and conducted for the Fischer–Tropsch synthesis (FTS) using the synthesized catalyst in a fixed bed reactor. The physical and chemical properties of the support and the synthesized catalyst were determined using the BET, XRD, H2–TPR, TEM, and H2-chemisorption characterization techniques. Based on the alkyl mechanism and using the Langmuir–Hinshelwood–Hougen–Watson (LHHW) isotherm, a kinetic model is developed for FTS. In most of the previous kinetic models, the primary reactions have merely been used, but in the current derivation of the developed kinetic model, the secondary reactions (adsorption, hydrogenation and chain-growth) and re-adsorption of primary olefins at the secondary active sites are considered. The present comprehensive kinetic model is applied for the product distribution such that the rate equations parameters are acquired via optimization. To estimate the kinetic model parameters, FTS was accomplished via a series of tests under the operating conditions as pressure (P): 10–20 bar, temperature (T): 483–513 K, gas hourly space velocity (GHSV): 1400–2400 h−1 and the H2/CO ratio of 1–2. The rationality and significance of the suggested model were checked through the statistical and correlation tests. The obtained results indicated that the outcomes of the current kinetic model were in good agreement with the experimental data. Using the present kinetic model, the average absolute deviations (AAD%) for the prediction of methane, ethylene and heavier hydrocarbons (C5+) formation rates are obtained as 7.06%, 11.57% and 14.74%.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
At present, the most significant energy supplies which account for more than sixty percent of the global total primary energy reservoirs are natural gas and petroleum [1]. Moreover, the worldwide energy demand is expected to grow as the world economy rises, particularly in the transportation sector that demands more fuel [1, 2]. On the other hand, the growth of energy market and limitation of oil availability, the growth of natural gas reservoirs and coal supplies encourage the industry and academia to pay more attention to the alternative liquid fuels [1, 3].
Fischer–Tropsch synthesis (FTS) has been known as a crucial chemical conversion technology to produce chemicals and synthetic hydrocarbon fuels from synthesis gas. The major sources of synthesis gas are natural gas, coal and biomass [4,5,6]. Natural gas is converted to synthetic gas through steam reforming while the conversion of coal and biomass to synthetic gas is carried out by gasification [6, 7]. Due to the production of linear high cetane number diesel fuels with low contents of sulfur and aromatics, Fischer–Tropsch synthesis (FTS) is an eco-friendly fuel production process [6].
A variety of catalysts with different support and active phase materials have been developed so far. Metal oxides and carbon materials are mostly used as support material in FTS catalysts. Based on the previous studies [8, 9], the carbon nanotubes (CNTs) as support material has been used in the preparation of a FTS catalyst that leads to an enhancement in liquid selectivity of the FTS, a decrease in metal particle size, and an increase in catalyst reducibility.
FTS is a catalytic reaction in which the product distribution is strongly based on the configuration of reaction sites over catalyst surface as well as the nature of active phase and support [7]. The products of the FTS are gasoline, olefins, diesel, kerosene, oxygenates and water. The oxygenate products are not accounted as valuable byproducts of the FTS and have not often been detected in the products [10]. It has been accepted that the typical kinetics of FTS is similar to a chain of polymerization reactions so that it can be justified to attain a higher selectivity towards the production of the heavier hydrocarbons including paraffinic products (C5+) [4, 6]. However, the detailed mechanism of FTS is not fully known yet due to its complexity [10, 11]. Nevertheless, the numerous researchers believed that the adsorption of reactants (CO and H2), chain initiation, chain-growth, chain termination, and desorption of products from the catalyst surface are accounted as the four steps of Fischer–Tropsch synthesis [7, 10]. On the other hand, the two procedures have been used for the kinetic modeling of the FTS reactions in the various studies [7, 12, 13], so that in the first approach, the modeling has been focused on the amount of transformation of the syngas (CO conversion) into the products. Using this type of kinetics modeling of Fischer-Tropsch synthesis, which is known as the lumped kinetic (consumption of the feed) so that the distribution of the products is not considered owing to the large range of FTS reactions. Based on the CO conversion method, the varieties of the kinetic modeling on Co-based catalysts have been suggested for FTS [9, 13, 14]. Elbashir and Roberts [13] proposed a mechanistic model using the Langmuir–Hinshelwood–Hougen–Watson (LHHW) approach for a Co-based catalyst. Moreover, for Co–Ru–K/CNT catalyst, Trepanier et al. [9] used the Fischer–Tropsch reaction rate equations based on LHHW and the seven various two-parameter kinetic models in a fixed-bed reactor (FBR). In another work, Mogalicherla and Elbashir [14] developed a model for the CO conversion based on the LHHW scheme over Co/Al2O3 catalyst in a FBR. Moreover, Haghtalab et al. [15] developed a new model for the FTS kinetic using the Fe–Cu–K/SiO2 catalyst in the slurry bubble column (SBC) reactor based on the Langmuir–Freundlich (LF) approach and demonstrated that the proposed model gave the better results compared to those acquired by LHHW.
Alternatively, in the second approach of the FTS reaction modeling, the product distribution including kinds of paraffin, olefins, acids and oxygenates have been taken into account [7, 16,17,18,19]. Despite numerous works on FTS kinetics, a versatile model has not been developed so far to demonstrate an exact mechanism of the products distribution that includes all the steps, i.e. adsorption, chain initiation, chain-growth, chain termination, and desorption. Based on some studies [7, 16, 18], several mechanisms such as alkyl, alkenyl, alkylidene, enol and CO insertion have been applied to develop a comprehensive kinetic model of a FTS. According to the outcomes of these works, one can conclude that owing to the more accurate prediction of olefins formation, the alkylidene mechanism is more appropriate for the Fe-based catalyst, while the alkyl mechanism is suitable for Co/Ru-based catalysts due to the better estimation of high paraffin formation rates. Accordingly, Qian et al. [19] introduced a new mechanism for the product distribution of the FTS based on CO insertion into alkyl-metal bond by using a Co-based catalyst in a fixed-bed reactor. Moreover, Nabipoor and Haghtalab [17] studied the product distribution of a FTS using the dual mechanism theory, which predicts the FTS products using the alkenyl and alkyl mechanisms. On the other hand, several researchers [7, 19] have applied the different models of product distribution in FTS reaction such as the particle size effect theory, Anderson–Schulz–Flory (ASF) law and dual site theory using the Co-based catalyst. The most popular model that has been used particularly for hydrocarbons is Anderson–Schulz–Flory (ASF) law, which is the simplest product distribution model; so that it demonstrates the dependency of the termination, and the chain-growth parameter on carbon number of the products [20]. In spite of soundness of the mentioned theories, particularly the ASF law, a significant deviation has been observed from the experimental results [21]. During the recent decades, researchers have investigated to clarify the deviation of experimental results from ASF law by using the several approaches. These methods comprise the experimental procedures, unsteady state conditions of reactor, the vapor–liquid equilibrium (VLE) phenomenon, catalyst deactivation, heat and mass transfer limitations, hydrogenation and chain-growth in olefins secondary reactions, the operating conditions of FTS, and the olefins re-adsorption at primary and secondary sites etc. [7, 17, 21]. However, in the most studies on the FTS kinetics modeling, the two major factors are secondary reactions and re-adsorption of olefins that lead to the deviation of products distribution from those predicted by the ASF law [17]. It is worth mentioning that the more often deviation from the ASF model has been reported for the formation of methane (CH4), ethane (C2H6) and heavier hydrocarbons (C5+) [21]. A comprehensive kinetic model allows one to consider both feed gas conversion and product distribution simultaneously in a FTS. Nevertheless, due to the FTS reactions complexity and cumbersome derivation and calculation procedures, the comprehensive kinetic modeling has been less developed in the previous works [7, 17,18,19, 22] in compared to those models that are considering the product distribution and CO conversion separately. Several works [7, 11, 22] have been carried out to obtain a comprehensive kinetic model for FTS. Visconti et al. [22] presented a comprehensive FTS kinetic model over Co-based catalyst supported on alumina using the alkyl mechanism and they considered the olefins re-adsorption at the primary active sites. Moreover, using Co–Ru/γ-Al2O3 catalyst, Mosayebi and Haghtalab [7] developed a comprehensive model for the FTS kinetics based on combining the alkyl and alkenyl mechanisms without considering the secondary reactions and olefins re-adsorption.
In the present work, a new comprehensive kinetics model is developed for Fischer–Tropsch kinetics. Thus, using a Co–Ru/CNTs nanostructure catalyst, a set of FTS experiments were carried out at steady state conditions to provide the necessary data of the syngas conversion as well as the selectivity of products to correlate the proposed kinetic model parameters. Moreover, in the development of the present model, we take into account the olefins re-adsorption and secondary reactions (hydrogenation and chain propagation) at the secondary active sites.
Experimental
Preparation of the Co–Ru/CNTs catalyst
Prior to the preparation of the present catalyst, the multiwall carbon Nanotube (MWCNT > 95%) support was treated by nitric acid (HNO3) to modify its surface from hydrophobic to hydrophilic. Consequently, the Co–Ru/CNTs catalyst was prepared using a surface displacement reaction as follows. The deposition of cobalt (Co) nanocrystals at CNTs were performed via the cobalt chloride (CoCl2·6H2O) reduction through sodium borohydride (NaBH4) solution in the presence of the ethanol–water solution. At first, we disperse 0.5 g of the acid-treated CNTs in the 100 mL water–ethanol solution for 30 min using an ultrasonic system. Then, 0.202 g of the CoCl2.6H2O aqueous solution was added dropwise into the suspension and is stirred for 120 min. Under stirring at room temperature, a prepared solution of NaBH4 (0.004 g in 10 mL of 0.5 M NaOH solution) was added dropwise to the suspension as a reducing agent. After mixing of the suspension for 10 h, the Co nanocrystals were deposited at the CNTs support. In the second step, the prepared suspension was heated to 453 K, then, under stirring, 10 ml of the RuCl3 aqueous solution was added dropwise to the suspension. Afterward, four milliliters of hydrazine hydrate (N2H4) solution as the second reducing agent was added dropwise into the suspension for 6 h and the final solution was left to settle down the solid particles. Consequently, we filtered the solid sample via a filtration paper and washed several times using the DDI water and ethanol. Then, the prepared Co–Ru/CNTs catalyst was dried at 383 k in a vacuum oven for 8 h. Moreover, for the removal of the impurities and to achieve the characteristic conditions of CNTs [9, 23], the synthesized catalyst was calcined at 350 °C for 5 h under argon flow.
The characterization techniques
We dissolved a 5 mg of the calcined catalyst in 5 mL HCl/HNO3 in order to determine the elemental contents of the prepared catalyst. Following complete dissolution of the catalyst in the acidic solution, one can use inductively coupled plasma atomic emission spectroscopy (ICP-AES) instrument to analyze the Co and Ru loading contents of the samples. In addition, the X-ray Diffraction (XRD) characterization of the synthesized catalyst was carried out via an X-ray diffractometer (X’Pert MPD, Philips Co.) in a Cu Kα radiation employing mode (λ = 1.54 Å). The average Co3O4 crystallite size was measured by the Sherrer equation as
Here λ and B are the wavelength of X-ray and the full width half maximum (FWHM) of the Co3O4 (at diffraction peak of 2θ = 36.8°). Besides XRD analysis, the surface area, mean particle size and the pore volume of the prepared catalyst and the CNTs support were also attained by a Belsorp mini II system through N2 physisorption.
The Micromeritic TPD-TPR 290 apparatus was used to determine the amount of chemisorbed H2 in the catalyst at 373 K following the reduction treatment. Details of H2 chemisorption calculations can be found elsewhere [24, 25]. Accordingly, Co metal crystallites size (d) could be obtained using hydrogen chemisorption data as
Moreover, the Quantachrome CHEMBET-3000 system was used to perform the temperature-programmed reduction (TPR) analysis. The catalyst was placed in a tubular reactor and a purge gas flow of N2 was passed through it at 453 K for 30 min to remove moisture and impurities. Afterward, under flowing of N2, the catalyst was cooled down to 298 K and then a mixed-flow of H2 (5 V %) and argon (95 V %) was passed through the catalyst. Finally, the temperature was increased to 1173 K by a heating ramp of 10 K/min. We obtained the morphology of the prepared catalyst and particle size distribution using the transmission electron microscopy (TEM) analysis, and a Philips CM30 apparatus, respectively. In addition, the energy-dispersive JEOL X-ray spectroscopy (EDX) (JED-2300) was used to determine the elemental analysis of the calcined catalyst.
Fischer–Tropsch synthesis reaction setup
The tests of the FTS were carried out in a fixed-bed (F-B) reactor (L = 70 cm; I.D. = 1 cm) that were packed by the spherical quartz granules as a diluent in the case of enhancing the residence time as shown in Fig S1 (supplementary information). As presented in this figure, the syngas (H2 + CO) enters at the top of the tubular fixed-bed reactor and then a peripheral heating furnace heated the so-call reactor. The syngas passes through the first region of the reactor to be preheated and subsequently enters the reaction zone, which has already been loaded by the prepared catalyst (0.2 g). Prior to the FTS reaction tests to take place, the catalyst should be activated through reduction treatment, which is conducted under hydrogen flow at 723 K. The FTS tests were performed at the various operating conditions in the range of 483–513 K, 10–20 bar, a space velocity of 1400–2400 h−1 and the H2/CO molar ratio of 1–2. It took at least 10 h to carry out each experiment at the given operating conditions. After achieving the steady state, the products were collected during a period of 20 h. It should be noted that each experiment was performed with the fresh catalyst at the given operating conditions, thus no need for activating the catalysts. The FTS process products were separately collected as the liquid phase using the hot and cold traps at 90 °C and 0 °C, respectively, and gas phase is also collected from the gas effluent. To analyze the collected gas and liquid products from the outlet of the reactor, an Agilent 7890A gas analyzer and an Agilent DHA analyzer were utilized, respectively. Nine olefins (C2H4–C10H20) and ten kinds of paraffin (CH4–C10H22) were experimentally detected and the composition data of the products were used for the estimation of the model parameters. The carbon monoxide (CO) conversion and mass balance were calculated as
Here \({\text{F}}_{{{\text{CO}}_{\text{in}} }}\), \({\text{F}}_{{{\text{CO}}_{\text{out}} }}\), \({\text{M}}\left( {\text{CO}} \right)_{\text{in}}\), \({\text{M}}\left( {\text{CO}} \right)_{\text{out}}\), and \({\text{M}}({\text{C}}_{{\text{i}}} {\text{H}}_{{\text{j}}} )_{{{\text{out}}}}\) are the molar flow of CO in reactor inlet (mol/s), the molar flow of CO in reactor outlet (mol/s), the moles of CO input, the moles of CO output, and the moles of hydrocarbon products with carbon number i, respectively. In addition, the products formation rates can be obtained as
Here rj, F, yj, W, P, wj, Mwj and t are the formation rate of component j (mol/kg s), molar flow of the product in gas phase (mol/s), mole fraction of component j in the gas phase, the catalyst weight (kg), mass of produced hydrocarbon in the liquid phase (productivity), weight fraction of component j in the liquid phase, molecular weight of component j and time consuming of taking place the Fischer–Tropsch reactions.
Results and discussion
Characterization of the prepared catalyst
The ICP analysis showed that the real contents of Co and Ru were 10.07 and 0.84 wt%, respectively, while the nominal cobalt and ruthenium contents in the calcined catalyst were fixed as 10 and 1 wt%. The XRD patterns of the synthesized catalyst are shown in Fig S2 (the supplementary information). As seen, the six peaks at 19.2°, 31.5°, 36.8°, 45.8°, 59.2° and 65.2° have corresponded to the cobalt oxide (Co3O4) [26,27,28,29,30]. Moreover, the intensity of peaks related to CNTs support is distinct in the XRD spectrum (Fig S2-b). As shown, the peaks around 26° and 43° are assigned to the graphite layers (CNTs) [30]. Moreover, in these XRD profiles, the distinct peaks at 28.3°, 35.2° and 54.7° have corresponded to the RuO2 (see Fig S2a) [4, 28]. The mean sizes of crystallite Co3O4 were determined via the Scherrer equation (Eq. 1) according to the characteristic diffraction peaks at 36.8° (9.24 nm). Co0 and Co3O4 crystallite size are related by d(Co0) = 0.75 d(Co3O4) [7, 30]. Accordingly, the average size of the elemental Co nanoparticles (i.e. Co0) was calculated as 6.93 nm. In addition, in compared to the attained results by XRD, the size of elemental cobalt nanoparticles was determined by H2 chemisorption as 7.8 nm that is higher than the value obtained by XRD before reduction treatment (see Table 1). The results via BET analysis for the calcined catalyst and CNTs support are also given in Table 1. As observed, the BET surface area and porosity for the loaded catalyst give a lower value than those obtained for the carbon nanotubes, which is ascribed to some pore blockage owing to the active particles at the CNTs support during the catalyst preparation [28, 31, 32]. Moreover, the EDX pattern of the calcined catalyst is shown in Fig S3b (the supplementary information). The results display that the catalyst composed of O, Ru, Co and C atoms. It should be noted that the O signal in the spectrum is attributed to the oxidation of the surface metal atoms.
The morphological characteristics of the Co–Ru/CNTs catalyst were evaluated by TEM so that the existence of the predominant spherical shape nanoparticles, which are well distributed at the external surface and insides the tubes, are observed in the supplementary information (Fig. S3a). Moreover, distribution of the nanoparticle size is illustrated in the supplementary information (Fig. S3c) and the mean particle size of Co–Ru/CNTs Nanocatalyst was measured by TEM test according to the literature [7]. As seen, the average nanoparticle size of the synthesized Co–Ru/CNTs Nanocatalyst was 9.8 nm (see supplementary information, Fig. S3c), which was nearly similar with the obtained results by XRD analysis (Table 1). Meanwhile, the results of the TPR test for the prepared catalyst are shown in Fig. 1. In this figure, the two obvious peaks were seen and could be ascribed to the reduction of Co3O4 to CoO (first peak) and CoO to Co0 (second peak) at 610 and 698 K, respectively, which was in agreement with the literature [28, 30,31,32,33]. Based on the various works [8, 9, 28, 31], CNTs presented an easier reduction route in compared to alumina, because carbon nanotubes introduced as inert support that did not show any peak related to the oxide compounds formation. Accordingly, more Co atoms were accessible in carbon nanotubes supported catalysts to participate in FTS in comparison with the Al2O3 supported catalysts [28, 31].

The H2-TPR profile of the prepared catalyst for FTS
The comprehensive kinetic model
The model assumptions
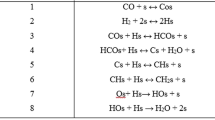
To develop the present kinetic model, the elementary reactions were considered as given in Table 2. To simplify the model, the following assumptions were taking into account:
-
Using the Co–Ru/CNTs catalyst, most of the products are liquid and gaseous hydrocarbons (paraffin and olefins) are produced such that the formation reactions of olefins and kinds of paraffin are taking place over the similar active sites. The main reaction of the Fischer–Tropsch synthesis could be written as
-
Due to the presence of Ru and Co active species, the water–gas-shift (WGS) reaction was neglected [4, 7].
-
Based on the higher tendency of Co and Ru metals (as a catalyst) towards paraffin production, the alkyl mechanisms were used for development of the present kinetics model.
-
A separate reaction was considered for methane product formation because this product cannot be taken into account by the alkyl mechanisms.
-
The Langmuir–Hinshelwood method was used to develop the present kinetics modeling.
-
Quasi-equilibrium assumption was considered for steps 1–2, and 8–10 in Table 2, while the rate determining steps (RDS) were applied for chain initiation, growth and termination reactions of 3–7, and 11–13 in Table 2 for olefins and paraffin formation [7, 34].
-
The reaction rate constants of olefins and kinds of paraffin formation were considered independent of carbon number except for methane.
-
To develop the kinetic model, the olefins re-adsorption, hydrogenation reactions and chain growth on the second type of active site were also considered.
-
For the Fischer–Tropsch synthesis, which reported using the catalysts with mesh smaller than 250 µm (particle size) [7, 27], the resistance of heat and mass transfer were assumed to be negligible in order to simplify the present model.
-
In the present study, deactivation of the catalyst has been neglected, because at each experiment a fresh catalyst was used for FTS reactions.
Thus, based on the above assumptions, the mechanism as depicted in Fig. 2 was used.

The proposed mechanism for FTS reaction (‘‘\(\uppsi\)” and “θ” are the active sites at catalyst surface)
Development of the kinetic model
The rate equations for production of methyl and alkyl at the steady state conditions can be written as:
Here, based on the above assumptions, the surface fraction coverage of the intermediates at the primary active sites are expressed in terms of the partial pressures of the synthesis gases as:
By rearranging Eq. 8 and using Eqs. 10 and 11, the following relation for the methyl surface fraction in the primary sites is obtained:
Similarly by rearranging Eq. 9, one can obtain as:
On the other hand, we define a chain growth factor (αn) as
Then, by substituting Eqs. 10 and 11 into Eq. 14, one can obtain:
On the other hand, by substituting Eqs. 12 and 15 in Eq. 13, one can write:
Moreover, using the secondary reactions, we can write the rate equation for production of alkene at the quasi-steady state conditions as:
Here the surface fractions for the secondary adsorption of the components are obtained as:
By rearrangement of Eq. 17, the surface fraction of the olefin is written as
To write Eq. 21 in a simple form, the following relations are written:
Thus, using Eqs. 22–24, one can write Eq. 21 in a simple expression as:
By applying an iteration technique for Eq. 25, a generalized relation is obtained as:
Finally, the intermediates surface fractions at the primary and secondary active sites of catalyst are obtained, respectively, by the surface balance equations as:
It should be noted that adsorption of the other intermediates at the surface of catalyst was neglected. By substitution of Eqs. 10 and 11 into Eq. 27, the surface fraction of the primary active sites, which are not occupied, is obtained as:
Similarly, the surface fraction of the secondary free active sites could be written as
Now we can write the kinetic rate equations for the methane, olefins and paraffin products as
Here the “+” and “−” signs indicate the forward and backward reactions. Finally, by substituting the surface fractions of the intermediates, i.e. Equations 10, 11, 18, 19, 29 and 30, in Eqs. 31–33, the kinetic rate equations are obtained as:
The reactor model
In this study, a fixed bed reactor was used for FTS reaction so that the flow assumed to be axial and the dispersion effects were neglected. Thus, by an assumption of a steady state ideal plug flow in this reactor, one can write the reactor model as:
Here Fj, W, NR, Ri, \(\upsigma_{\text{ji}}\), NC are the mole flow rates of jth component in the axial direction of the reactor (mol/s), the weight of catalyst loaded in each experiment (kg), the total number of the considered reactions including synthesis gas consummation rate and hydrocarbons formation rate, the rate of ith reaction (mol/kg s), the stoichiometric coefficient of the jth component in the ith reaction, and the total number of the species, respectively. The stoichiometric coefficients matrix for the products and reactants for Fischer–Tropsch synthesis can be found in the literature [7]. Moreover, the overall material balance of the hydrocarbons can also be written as:
Here \({\text{F}}_{\text{CO}}^{0}\) (mol/s) denotes the CO flow rate in the feed, A is the cross-sectional surface area of the reactor, r is the reaction rate (mol/kg s) and ρB is the catalyst bed density (kg/l). The yield of hydrocarbons (yHC) is defined as:
For more detail information on the material balance of the hydrocarbons, one can refer to the literature [27]. By assuming ideality of the gas phase, the partial pressure of the jth component in the proposed kinetic model can be determined as:
Here yj is the mole fraction of j component and PT is the total pressure in the reactor (bar).
The optimization method of kinetic parameters
For the optimization of the parameters in the proposed kinetic model, the initial value problem (IVP) could be written as
Here, \({\mathbf{F}}\left( {\text{x}} \right) = \left[ {{\text{F}}_{1} \left( {\text{x}} \right), \ldots ,{\text{F}}_{{{\text{N}}_{\text{C}} }} \left( {\text{x}} \right)} \right]^{\text{T}}\) is the state of the system and \({\mathbf{p}} = \left[ {{\text{p}}_{ 1} ,\ldots , {\text{p}}_{21} } \right]^{\text{T}}\) is the vector of the unknown parameters. The objective function for the FTS system (O) that should be minimized is written as:
Here \({\text{F}}_{\text{j,i}}\) and \(\widetilde{F}_{\text{j,i}}\) represent the experimental and calculated mole flow of the jth response (in the ith experiment), respectively. At this work, for optimization of the kinetic model parameters, MATLAB function of fmincon along with sequential quadratic programming (sqp) algorithm was applied. Furthermore, to solve the IVP, the MATLAB function ode45, which is based on an explicit Runge–Kutta formula, was used. It should be noted that in Eq. 42, Nexp and Nres stand for the number of the experiments and responses in the system, respectively. For optimization of the present kinetic model parameters, 22 responses (Nres) including the output concentrations of nine olefins (CnH2n, n = 2, 3,…, 10), ten kinds of paraffin (CnH2n+2, n = 1, 2, …, 10), as the most important FTS products, along with H2, CO and the overall concentration of C5+ were used. Moreover, to measure the reliability of the current model and the estimated parameters (CO conversion and product distribution), the statistical analysis was performed using the Statistical Analysis System program (SAS Institute Inc., Cary, NC). The precision of the present model with respect to the experimental data could be determined via the mean absolute relative residuals (MARR) formula which is written as:
Here Nc denotes the number of the components in the system. To measure the agreement between the calculated values and the experimental responses, the relative residual (RR) is used as
Moreover, the percent of the average absolute deviation (AAD%) between the experimental and calculated data is used as:
In addition, the reaction rate constant in terms of temperature is expressed by the Arrhenius relation through the following relation:
Here kj stands for the rate constant of the j reaction (mol kg−1 s−1) and kj,0 presents pre-exponential parameter of the j reaction (mol kg−1 s−1) and also Ej is the activation energy of the j reaction (kJ mol−1).
Kinetic modeling results
For the optimization of the kinetic model parameters, the fifteen experimental sets were considered and conducted at the various operating conditions so that the responses results are given in Table 3. The optimized values of the kinetic model parameters for the Co–Ru/CNTs bimetallic structure catalyst were given in Table 4. As seen in this table, the standard errors of all parameters were determined based on the methods have been used in the literature [35]. Based on the results given in Table 4, one can conclude that the RDS assumptions for the chain growth reactions and termination are rational because the values of the calculated activation energies for these reactions are high so that their values are in good agreement with those given in the literature [7, 35]. Using the present modeling, the activation energy values of the reaction constants for the primary (E3) and the secondary (E11) reactions are 71.6 kJ/mol and 99.8 kJ/mol, respectively. Consequently, one can deduce that the primary chain growth reactions are faster than the secondary chain reactions. It is considered that the activation energy for the kinds of paraffin formations was 82.9 kJ/mol, which was higher than 76.3 kJ/mol for the formation of methane. Thus, one can conclude that the heavier hydrocarbon fractions are less probable for production than the light ones during the FTS reaction due to the rising activation energy barriers, which results in a decrease of chain-growth factor by increasing the product carbon number [7]. In general, the activation energy for the olefins formation (91.8 kJ/mol) is higher than the values were obtained for the formation of the kinds of paraffin so that allows one to deduce the olefins selectivity is much lower than paraffin over a Co–Ru/CNTs catalyst. In the previous kinetic works [7, 34, 36, 37], the values of the activation energy for the FTS over the various catalysts were presented in the range of 70–114 kJ/mol, while in the present study, the values of the activation energy are in the range of 71.6–111.8 kJ/mol. Moreover, by assuming ideality for the gas phase the MARR value was 15.3%. Based on LHHW methodology, Todic et al. [11] proposed a kinetic model for FTS using carbide mechanism over a Co–Re/Al2O3 catalyst in a stirred tank slurry reactor (STSR) so that a value of 26.6% was reported for MARR. In another work, detailed kinetic modeling of FTS over the similar catalyst was derived by Todic et al. [38] so that a slight improvement in accuracy of the regression (MARR = 23.5%) was obtained.
Meanwhile, one can see the results of the statistical and correlation tests for the formation of the products in the present FTS as shown in Table 5 so that the F and ρ2 values show that the proposed kinetic model and the estimated values of the parameters are significant from standpoint of statistical view. Moreover, the results of the calculated CO conversion versus the experimental values and the average absolute deviation percent (AAD%) between the calculated CO conversion and experiment are displayed in Fig. 3 using the present kinetic modeling. As seen, a value of 16.89 for AAD% was obtained. It should be noted that the maximum relative residual (RR) between the experimental and calculated values (using Eq. 44) is 22%, which it shows the correlation of the experimental data by the present kinetic model is relatively good. Moreover, using the proposed kinetic model the calculated formations rates by Eqs. 5 and 6 for the various components such as methane, ethylene and C5+ are compared with the experimental data as shown in Fig. 4. As seen, it is clear that the results of the formation rates are more accurate than the calculated CO conversion so that the AAD% values for methane (CH4), ethylene (C2H4) and heavier hydrocarbons (C5+) formation rates are 7.06, 11.57 and 14.74, respectively. Moreover, the maximum RR values for these three components formation rates are 13%, 18% and 20%, respectively. Based on the literature [7, 19, 37], most of the relative residuals percent (RR%) of the components between calculated and experimental values are within 20%. Hence, one may conclude that in this study the results of the present comprehensive kinetic modeling are reasonable. Chang et al. [37] developed a comprehensive model for a slurry bubble column reactor in which the average absolute deviations (AAD%) of the predicted CO conversion and C5+ products were about 15% and 20%, respectively. Fig. 5 shows the results of the present kinetic modeling for the product distributions of kinds of paraffin and olefins in terms of carbon number in the FTS using the Co–Ru/CNTs catalyst. As shown in Fig. 5, at the carbon number less than eight, the calculated weight percent of the paraffin products are in better agreement with the experimental data that indicates a good consistency with those given in the literature [34, 36, 37]. On the other hand, the calculated olefins distributions give better results for carbon numbers less than seven that shows the accuracy of the present model is relatively good whereas for carbon numbers above seven, more deviation is observed. Thus, based on our survey in the literature [19, 22], one may conclude that in general the results of the different models show that the accuracy of the calculated values of the product formation rates are better at carbon numbers less than seven so that at the higher carbon numbers, more deviation from the experimental data have been observed for both paraffin and olefins using the cobalt catalysts. Hence, it can be deduced that at the lower temperatures and pressures the obtained results are more accurate as shown in Fig. 5a, b, particularly for the kinds of paraffin.

The calculated conversion of CO against the experimental conversion values for FTS using the Co–Ru/CNTs Nano-structure catalyst (The upper and lower lines show ± AAD%, the experimental conditions are given in Table 2)

The calculated hydrocarbons formation rate versus its experimental rate for FTS using the Co–Ru/CNTs Nano-structure catalyst for a: methane; b: ethylene; and c: C5+ (The upper and lower lines show ± AAD%, the operating conditions of the experimental rate values are given in Table 2)

Comparison of the calculated and experimental product distribution in different operating conditions, a and b: T = 493 K, GHSV = 1900 h−1, P = 10 bar, CO/H2 = 1; c and d: T = 513 K, GHSV = 2400 h−1, P = 20 bar, CO/H2 = 0.5
The olefin/paraffin ratio (O/P) of products was also considered as a parameter in the present FTS studies. As reported in the literature [38,39,40], The O/P ratio of FTS versus product carbon number passes through a maximum that shows a close agreement with the present results as shown in Fig S4 (in the supplementary information). The location and the peak of the so-called maximum strongly depends on catalyst type, particle size, structure, diffusion limits, and the reaction conditions [39]. The O/P ratio is usually used as a measure of the tendency of a catalyst with its process parameters to either olefin or paraffin production [40].
Conclusions
In the present study, the Fischer–Tropsch synthesis catalytic tests were conducted over a Co–Ru/CNTs nanostructure catalyst in a fixed-bed reactor. A set of experiments was carried out at the different operating conditions of temperature, pressure, gas hourly space velocity (GHSV), and syngas ratio (H2/CO). Based on the alkyl mechanism, a comprehensive kinetic model was developed for FTS reaction using the prepared catalyst by taking into account the secondary reactions (hydrogenation and chain-growth) and re-adsorption of primary olefins at the secondary sites. The statistical and correlation tests were used for rational investigation of the proposed kinetic model. Accordingly, the obtained results demonstrated that the developed model and the estimated kinetic parameters were statistically significant. In this work, the values of estimated activation energies were compatible with those given by the previous studies. Finally, the results showed that the present FTS kinetic modeling could well correlate the product distribution and the CO conversion.
Abbreviations
- FTS:
-
Fischer–Tropsch synthesis
- LHHW:
-
Langmuir–Hinshelwood–Hougen–Watson
- CNTs:
-
Carbon nanotubes
- ASF:
-
Anderson–Schulz–Flor
- F-B:
-
Fixed-bed reactor
- TPR:
-
Temperature-programmed reduction
- XRD:
-
X-ray diffractometer
- TEM:
-
Transmission electron microscopy
- RDS:
-
Rate-determining step
- FWHM:
-
Full width half maximum
- R:
-
Universal gas constant (8.314 × 10−5 bar m3/mol K)
- x:
-
Position within the catalyst bed
- T:
-
Reaction temperature (K)
- t:
-
Time consuming for Fischer–Tropsch reaction (s)
- Mw,j :
-
Molecular weight of component j
- P:
-
Productivity (kg) (mass of produced hydrocarbon in liquid phase product)
- F:
-
Molar flow of product in gas phase (mol/s)
- rj :
-
Formation rate of component j (mol/kg s)
- yj :
-
Molar fraction of component j in gas phase
- wj :
-
Weight fraction of component j in the liquid phase
- W:
-
The catalyst weight (kg)
- FCO,in :
-
Molar flow of carbon monoxide in the reactor inlet (mol/s)
- FCO,out :
-
Molar flow of carbon monoxide in the reactor outlet (mol/s)
- B:
-
FWHM of the Co3O4 at diffraction peak of 2θ = 36.8
- MC,out :
-
Mass of output carbon
- MC,in :
-
Mass of input carbon
- NC :
-
Total number of the species
- Fj :
-
Mole flow rates of jth component (mol/s)
- Ri :
-
Rate of ith reaction (mol/kg s)
- NR :
-
Number of total considered reactions
- XCO :
-
CO conversion (%)
- PT :
-
Total pressure in the reactor (bar)
- Pj :
-
Partial pressure of j component (bar)
- O:
-
Objective function of FTS reaction
- K1 :
-
Equilibrium constant for the H2 adsorption on the primary active site
- E:
-
Reaction activation energy (kJ/mol)
- k3 :
-
Rate constant of chain growth in FTS mechanism for primary active site (mol/kg s)
- k3,0 :
-
Pre-exponential factor of chain growth in FTS mechanism for primary active site (mol/kg s)
- k5 :
-
Rate constant of the formation of methane (mol/kg s)
- k5,0 :
-
Pre-exponential factor of the formation of methane (mol/kg s)
- k4 :
-
Rate constant of the formation of paraffins on primary active site (mol/kg s)
- k4,0 :
-
Pre-exponential factor of the formation of paraffins on primary active site (mol/kg s)
- k6 :
-
Rate constant of the formation of olefins (mol/kg s)
- k6,0 :
-
Pre-exponential factor of the formation of olefins (mol/kg s)
- K7 :
-
Equilibrium constant for the CO adsorption on the secondary active site
- k10 :
-
Rate constant for the forward reaction of olefin re-adsorption (mol/kg s bar)
- k10,0 :
-
Pre-exponential factor for the forward reaction of olefin re-adsorption (mol/kg s bar)
- k−10 :
-
Rate constant for the reverse reaction of olefin re-adsorption (mol/kg s)
- k−10,0 :
-
Pre-exponential factor for the reverse reaction of olefin re-adsorption (mol/kg s)
- k11 :
-
Rate constant of chain growth in FTS mechanism for secondary active site (mol/kg s)
- k11,0 :
-
Pre-exponential factor of chain growth in FTS mechanism for secondary active site (mol/kg s)
- k12 :
-
Rate constant of the formation of paraffins on secondary active site (mol/kg s bar)
- k12,0 :
-
Pre-exponential factor of the formation of paraffins on secondary active site (mol/kg s bar)
- ψ:
-
Primary active site on catalyst surface
- θ:
-
Secondary active site on catalyst surface
- σji :
-
Stoichiometric coefficient of jth component in ith reaction
- αn :
-
Chain growth factor of FTS reaction for carbon number (n > 1)
References
Pardo-Tarifa F, Cabrera S, Sanchez-Dominguez M, Boutonnet M (2017) Int J Hydrogen Energy 42:9754–9765
Mohr SH, Wang J, Ellem G, Ward J, Giurco D (2015) Fuel 141:120–135
Abas N, Kalair A, Khan N (2015) Futures 69:31–49
Liu Y, Li Zh, Zhang Y (2016) Reac Kinet Mech Cat 119:457–468
Bukur DB, Todic B, Elbashir NO (2016) Catal Today 275:66–75
Dry ME (1982) J Mol Catal 17:133–144
Mosayebi A, Haghtalab A (2015) Chem Eng J 259:191–204
Farzad S, Haghtalab A, Rashidi A (2013) J Energy Chem 22:573–581
Trepanier M, Dorval Dion CA, Dalai AK (2011) Can J Chem Eng 89:1441–1450
Sari A, Zamani Y, Taheri SA (2009) Fuel Process Technol 90:1305–1313
Todic B, Bhatelia T, Froment GF, Ma W, Jacobs G, Davis BH, Bukur DB (2013) Ind Eng Chem Res 52:669–679
Shiva M, Atashi H, Tabrizi F, Mirzaei AA, Zare A (2013) Fuel Process Technol 106:631–640
Elbashir NO, Roberts CB (2004) Prepr-Am Chem Soc Div Pet Chem 49:57–160
Mogalicherla AK, Elbashir NO (2011) Energy Fuels 25:878–889
Haghtalab A, Nabipour M, Farzad S (2011) Fuel Process Technol 34:546–553
Fabiano A, Fernandes N (2005) Chem Eng Technol 28:1–9
Nabipoor M, Haghtalab A (2013) Chem Eng Commun 200:1170–1186
Zhang X, Liu Y, Liu G, Tao K, Jin Q, Meng F, Wang D, Tsubaki N (2012) Fuel 92:122–129
Qian W, Zhang H, Ying W, Fang D (2013) Chem Eng J 228:526–534
Vaniice AM, Bell AT (1981) J Catal 70:418–432
Vander laan GP, Beenackers ACM (1999) Ind Eng Chem 38:1277–1290
Visconti CG, Tronconi E, Lietti L, Zennaro R, Forzatti P (2007) Chem Eng Sci 62:5338–5343
Shariati J, RamazaniSaadatabadi A, Khorasheh F (2012) J Macromol Sci A 49:749–757
Tavasoli A, Karimi S, Taghavi S, Zolfaghari Z, Amirfirouzkouhi H (2012) J Energy Chem 21:605–613
Karimi S, Tavasoli A, Mortazavi Y, Karimi A (2015) Appl Catal A Gen 499:188–196
Irandoust A, Haghtalab A (2017) Catal Lett 147:2967–2981
Irankhah A, Haghtalab A (2008) Chem Eng Technol 31:525–536
Shariati J, Haghtalab A, Mosayebi A (2019) J Energy Chem 28:9–22
Da Silva JF, Braganca LFFPG, Pais da Silva MI (2018) Reac Kinet Mech Cat 124:563–574
Trépanier M, Tavasoli A, Dalai AK, Abatzoglou N (2009) Appl Catal A Gen 353:193–202
Tavasoli A, Sadagiani K, Khorashe F, Seifkordi AA, Rohani AA, Nakhaeipour A (2008) Fuel Process Technol 89:491–498
Xie Z, Frank B, Huang X, Schlögl R, Trunschke A (2016) Catal Lett 146:2417–2424
Phaahlamohlaka TN, Kumi DO, Dlamini MW, Forbes R, Jewell LL, Billing DG, Coville NJ (2017) ACS Catal 7:1568–1578
Teng BT, Chang J, Zhang CH, Cao DB, Yang J, Liu Y, Guo XH, Xiang HW, Li YW (2006) Appl Catal A 301:39–50
Lente G (2015) Deterministic kinetics in chemistry and systems biology. Springer, New York
Mosayebi A, Abedini R (2017) Int J Hydrogen Energy 42:27013–27023
Chang J, Bai L, Teng B, Li Y (2007) Chem Eng Sci 62:4983–4991
Todic B, Ma W, Jacobs G, Davis BH, Bukur DB (2014) Catal Today 228:32–39
NakhaeiPour A, Housaindokht MR (2013) J Nat Gas Sci Eng 14:204–210
Rao PVR, Shafer WD, Jacobs G, Martinelli M, Sparks DE, Davis BH (2017) RSC Adv 7:7793–7800
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Haghtalab, A., Shariati, J. & Mosayebi, A. Experimental and kinetic modeling of Fischer–Tropsch synthesis over nano structure catalyst of Co–Ru/carbon nanotube. Reac Kinet Mech Cat 126, 1003–1026 (2019). https://doi.org/10.1007/s11144-019-01535-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-019-01535-7