
Abstract
The novel γ-Al2O3 synthesis by mechanochemical activation of aluminum nitrate hydrate was applied to prepare hydrodesulfurization (HDS) NiMo catalysts. Impregnation techniques using the complex made by dissolution of nitrilotriacetic acid (NTA), ammonium heptamolybdate and nickel nitrate and the Anderson-type heteropolyoxymolybdate ((NH4)4Ni(OH)6Mo6O18) complex were compared with conventional impregnation using subsequent deposition of ammonium heptamolybdate (first) and nickel nitrate (second) with calcination in between. Properties of the support and HDS catalysts were studied by N2 physisorption, Raman spectroscopy, H2 temperature-programmed reduction (H2-TPR), O2 chemisorption and X-ray photoelectron spectroscopy (XPS). Both catalysts prepared from the complexes were at least 1.5-fold more active in the HDS reaction of 1-benzothiophene (360 °C and 1.6 MPa) than the catalyst prepared conventionally. The most promising preparation method for gaining highly active HDS catalyst on the studied γ-Al2O3 was the deposition of (NH4)4Ni(OH)6Mo6O18. This catalyst exhibited about 1.9 and 1.4 higher HDS activity in comparison to the conventionally prepared NiMo and reference commercial NiMo counterparts, respectively.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The hydrodesulfurization (HDS) process continuously faces strict regulations on the sulfur content in liquid fuels on one hand and diminution of available low sulfur feedstock on the other hand. Apart from more severe process conditions or major investments into plants, more efficient catalysts represent convenient ways to solve this challenge. The conventional hydrodesulfurization catalysts, either γ-Al2O3-supported CoMo, NiMo, NiW or unsupported NiMoW sulfides, are therefore comprehensively studied in the recent literature. The support is widely modified or replaced by other oxides or non-oxide materials and deposition of the active phase is thoroughly investigated.
In a previous study, we have reported on mechanochemically prepared mixed Al2O3-CeO2 supports, which led to high HDS activities of NiMo catalysts [1]. This mechanochemical synthesis was also applied for preparation of chemically pure γ-Al2O3 [2]. The first aim of the present work thus focuses on utilization of this simplified method, based on direct precipitation of aluminum nitrate with ammonium carbonate during milling, for preparation of γ-Al2O3 as support of HDS catalysts.
Nonetheless, the properties of HDS catalysts depend significantly on both properties of the support and preparation methods. Within the latter, precursors of active phase are also of great importance. The NiMo phase is often deposited onto supports by conventional impregnation (I) from ammonium heptamolybdate in the first step, which is followed by deposition of nickel nitrate in the second step [3,4,5,6,7,8]. These salts were also replaced by thiomolybdates or acetylacetonates in the reports [9, 10] or [11], respectively. Alternatively, NiMo phase was deposited in one impregnation step. This group of preparation methods includes direct dosage of the NiMo precursors during the sol–gel synthesis of support [12, 13], addition of chelating agents such as citric acid [14,15,16], nitrilotriacetic acid (NTA) [17], ethylenediamine tetraacetic acid [14] or thioglycolic acid [18] to NiMo solutions.
Substantial improvement of the synthesis methods of hydrodesulfurization catalysts were achieved applying heteropoly compounds as precursors of active phase. The Mo and W heteropoly oxoanions of Keggin and Anderson types [19] are interesting precursors of mixed oxides which show some advantages over other systems. Numerous papers report about increased HDS activity of supported Mo, W heteropolycompound based catalysts [20,21,22,23,24,25,26,27,28]. Anderson type heteropoly oxocompounds are especially interesting for synthesis of hydrotreating catalysts because they contain both principal components, active metals (Mo, W) and promoting elements (Co, Ni) in one molecule. The presence of the surface Anderson Co (Ni) heteropoly molybdenum analogues promotes the formation of the sites with high HDS catalytic activity, which is additionally increased when additional introduction of Co (Ni) is applied first to meet optimal ratios of Co/Mo (0.31) [29] and Ni/Mo (0.33) [30] in the final catalysts.
The second aim of the present work is to synthesize highly active HDS catalysts by the exploration of the preparation route of NiMo active components for the mechanochemically prepared Al2O3. Specifically, the novelty of this report lays in comparison of deposition of two type of NiMo complexes: (i) the NTA NiMo complex formed by dissolution of (NH4)6Mo7O24, Ni(NO3)2 and NTA and (ii) the Anderson-type heteropolyoxymolybdate (NH4)4Ni(OH)6Mo6O18 (HPO) complex. The conventional sequential impregnation using (NH4)6Mo7O24 (first) and Ni(NO3)2 (second) is studied as the reference method. The prepared catalysts in their sulfided forms are evaluated by temperature programed reduction (TPR), nitrogen physisorption, oxygen chemisorption, X-ray photoelectron spectroscopy (XPS), Raman spectroscopy and their activities are determined in reaction of 1-benzothiophene HDS at 360 °C and 1.6 MPa.
Experimental
Synthesis of Al2O3
The following procedure was applied for γ-Al2O3 support preparation: aluminum(III) nitrate hexahydrate was placed in a preheated oven at about 80 °C to melt. Then solid NH4HCO3 (molar ratio HCO3−:NO3− was 1.25:1) was added. The mixture was ground using an agate mortar and pestle mill (Fritsch) for 1 h, dried at 60 °C for 20 h, and calcined at 500°C for 4 h. About 50 g of γ-Al2O3 support was obtained. The support fraction with grain size 0.16–0.32 mm was used hereinafter.
Catalysts preparation
The catalysts were prepared by impregnation from the excess of the impregnation solution. Typically, 15 mL of an aqueous solution was used per 3 g of solid. The high surface area of the support allowed preferential deposition of Ni and Mo precursors into pores during drying in vacuum rotary evaporator. The loadings of Ni and Mo were kept the same in the prepared catalysts possessing values 2.0 and 12.0 wt%, respectively, which was confirmed by chemical analysis.
Deposition from NiMo complexes
Ni and Mo were deposited together either applying impregnation using nitrilotriacetic acid (NTA, C6H9NO6) as chelating agent or impregnation with an aqueous solution of the ammonium salt of nickel heteropolyoxomolybdate (HPO), i.e. (NH4)4Ni(OH)6Mo6O18.
Ammonium heptamolybdate (AHM), nickel nitrate and nitrilotriacetic acid (the molar ratio of (Ni + Mo):NTA was 1:1) was dissolved in distilled water. Then the solution was gently stirred with the support at laboratory temperature for 1 h, dried in a vacuum evaporator at 95 °C for 1 h. The sample was not calcined but it was directly sulfided. Avoiding the calcination step is typically used for this preparation route, reported elsewhere [17], because it is favorable for high HDS activity. The sample was sulfided in H2S/H2 mixture (molar ratio of H2S:H2 was 1:10) at 400 °C for 1 h with temperature ramp 6°C min−1. After cooling in N2, the obtained NiMoNTA catalyst was kept in N2 in gas tight ampules.
The heteropolyoxomolybdate (HPO) was synthesized using a previously reported method [31]. To keep the same nominal loadings, i.e. 12 wt% Mo and 2.0 wt% Ni, as in other experiments, an aqueous solution of Ni(NO3)2 was used to pre-load the support with Ni (the pre-loading with Ni was found beneficial in our previous papers [29, 30]). The Ni-modified support was dried in a vacuum evaporator at 95 °C for 1 h and an oven at 105 °C for 4 h, and calcined in air at 350 °C for 2 h at a heating rate of 1.7 °C min−1. The aqueous solution of the HPO was added at 70 °C. The impregnated support was dried at 105 °C for 4 h and calcined at 350 °C for 2 h to decompose the ammonium salt. The catalyst was then sulfided by the aforementioned procedure and labelled as NiMoHPO.
For the matter of Raman spectroscopy, also unsupported forms of sulfides were prepared from both complexes. The preparation procedure was kept the same but the support was not added. The dried form of precipitate of the NTA complex was sulfided. The HPO was calcined and sulfided.
Conventional and reference catalysts
An aqueous solution of ammonium heptamolybdate (AHM) was gently stirred with γ-Al2O3 for 1 h and then the mixture was dried in a vacuum evaporator at 95 °C for 1 h and calcined. The catalyst was labelled as MoI. Then, the aqueous solution of nickel nitrate was stirred with MoI for 1 h and the mixture was dried in a vacuum evaporator at 95 °C for 1 h. The obtained NiMoI was calcined then sulfided (the calcination and sulfidation procedure was described above).
The industrial Mo/Al2O3 (BASF, M8-30, Germany, 10.0 wt% Mo, SBET (sulfidic state) = 249 m2g−1) and NiMo/Al2O3 (Albemarle, KF 846, The Netherlands, 2.4 wt% Ni, 13.8 w % Mo, SBET (sulfidic state) = 218 m2g−1) catalysts were named as MoRef and NiMoRef, respectively, and they were used as reference catalysts in HDS of 1–benzothiophene.
Methods of support and catalysts characterization
Powder X-ray diffraction (XRD) was performed using a Bruker D8 Discover diffractometer equipped with a Si-strip linear LynxEye detector and a Ge primary monochromator providing Cu Kα1 radiation (λ = 0.154056 nm). Data were collected in the 2θ range 5–80°, with a step size of 0.019° and a step time of 1 s.
The textural properties of the supports and of the sulfided catalysts were investigated with a Micromeritics ASAP 2010 apparatus. Prior to the analysis, the oxide precursors were sulfided under the same conditions as described above and evacuated at 350 °C for 4 h before the N2 adsorption. Specific surface area and pore size distributions were determined from nitrogen adsorption–desorption isotherms at − 195 °C. To calculate the specific surface area (SBET), the data were treated by the standard BET method. Total volume of pores VTotal was determined from the amount of N2 adsorbed at P/P0 = 0.98. Surface area of mesopores (SM) and volume of micropores (Vmicro) were calculated from t-plot according to Schneider [32].
The number of unsaturated sites in sulfided Mo and NiMo catalysts was determined by O2 chemisorption. The catalysts were re-sulfided in situ, flushed by helium (Linde AG, 6.0, Germany) at 400 °C for 1 h and cooled in a mixture of dry ice and ethanol. The amount of chemisorbed O2 was determined from pulses of O2 (Linde Gas a.s., Czech Republic) added to the flow of He, which was monitored with a thermal conductivity detector VICI (Valco Instrument Inc., USA) and an HP 3394A integrator (Hewlett Packard, USA).
Temperature programmed reduction (H2-TPR) was performed in the flow of H2:Ar (1:19, 35 cm3 min−1) with a temperature ramp 5 °C min−1 from 40 to 960 °C on a Micromeritics chemisorber AutoChem 2950 HP equipped with a thermal conductivity detector. The catalyst was placed in a quartz ampule and it was ex situ sulfided (details of the sulfidation are given above). The ampule was then filled with nitrogen and transferred into TPR apparatus. A trap cooled at − 72 °C was placed between sample tube and detector to remove water out of reduction medium.
Raman spectra were collected using a dispersive Nicolet Almega XR spectrometer equipped with the Olympus BX51 microscope, excitation laser source (473 nm), and with a maximum incident power of 50 mW with 128 expositions with 0.5 s exposition time. For all experiments, the laser beam was focused by 50 × objective on spot with area around 1 µm2 resulting in maximum irradiance 5 MW/cm2. The measurements were done within 5 min after the sample exposure to ambient atmosphere. No changes in Raman bands were observed during this time limit.
The XPS measurements of the sulfide catalysts were carried out on a spectrometer ESCALAB–MkII (VG Scientific) with a base pressure in the analysis chamber of ~ 10−8 Pa. Glove box was used to load the samples to prevent the sulfides to be oxidized in ambient atmosphere. The spectra were excited with an Al Kα radiation (hν = 1486.6 eV) at total instrumental resolution of ~ 1.0 eV as measured by the FWHM of Ag 3d5/2 photoelectron line.
Catalytic activity
The hydrodesulfurization of 1-benzothiophene (BT) was performed in the gas phase using an integral fixed-bed tubular flow microreactor (i.d. 3 mm) at 360 °C and 1.6 MPa. Prior to the measurements, the catalysts were sulfided in situ with an H2S/H2 flow (1/10) at 400 °C and atmospheric pressure with a temperature ramp of 10 °C min−1 and a dwell time of 1 h. The composition of the feed was kept constant at 16, 200 and 1384 kPa of 1–benzothiophene (BT), decane and hydrogen, respectively. The catalyst sample (0.20–0.03 g) was diluted with an inert α-Al2O3 with particle size fraction 0.16–0.32 mm to form a bed length of 30 mm. The reaction was run at three feed rates of BT including 7.7, 10.3 and 15.5 mmol h−1. The steady state was reached in 30 min after each change in the feed rate. No changes of conversions during the following 1 h were observed. The reaction mixture was analyzed on a Hewlett–Packard gas chromatograph (6890 series) equipped with a capillary column (HP-5, 30 m, 0.53 mm, 1.5 μm). Dihydrobenzothiophene (DHBT) and ethylbenzene (EB) were identified in the reaction products. The scheme of BT HDS is shown in Fig. 1a. The relative compositions a(BT), a(EB) and a(DHBT), BT conversion x(BT) and EB and DHBT yields, y(EB), and y(DHBT) were defined as a(BT) = (1−x(BT)) = n(BT)/n0(BT), a(EB) = y(EB) = n(EB)/n0(BT), a(DHBT) = y(DHBT) = n(DHBT)/n0(BT), where n0(BT), n(BT), n(EB), and n(DHBT) were the initial number of moles of BT, final number of moles of BT, EB, and DHBT, respectively. The rate constant of EB formation, kEB, was calculated using an empirical pseudo-first-order rate equation (a(EB) = 1 − exp (− kEBW/F). The kEB was normalized both per mol of Mo or Ni + Mo expressing intrinsic activity and per gram of catalysts. An example how relative composition depends on space time W/F and how kEB was obtained is given in Fig. 1b.

a Scheme of 1-benzothiophene hydrodesulfurization: (HYD) hydrogenation of benzothiophene to dihydrobenzothiophene, (de-HYD) dehydrogenation of dihydrobenzothiophene to 1-benzothiophene, (HYD-HYG) hydrogenolysis of dihydrobenzothiophene to ethylbenzene (hydrogenation route), (DDS) hydrogenolysis of 1-benzothiophene to ethylbenzene (direct desulfurization route). b Relative composition during reaction of 1-benzothiophene (BT, open circles) hydrodesulfurization over MoI and NiMoHPO catalysts: (DHBT, open triangles) dihydrobenzothiophene, (EB, solid circles) ethylbenzene, (dash lines) the curves calculated using the parallel consecutive scheme (a) of four pseudo-first-order rate reactions (da(BT)/d(W/F) = − kHYDa(BT) − kDDSa(BT) + kde−HYDa(DHBT), da(DHBT)/d(W/F) = − kHYD-HYGa(DHBT) − kde−HYDa(DHBT) + kHYDa(BT), da(EB)/d(W/F) = + kHYD−HYGa(DHBT) + kDDSa(BT)), (solid line) the curve calculated using an empirical pseudo-first-order rate equation a(EB) = 1 − exp (− kEBW/F) to determine total HDS activity kEB (see Table 5)
Results and discussion
Concept of mechanochemical preparation of γ-Al2O3
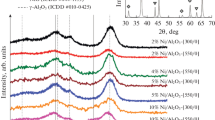
The mechanochemical reaction of aluminum nitrate with NH4HCO3 resulted in a voluminous white precipitate. SEM images (not shown here) showed that the dried precipitate consisted of nearly spherical particles. The finding is in accord with the data published by Djuricic and Pickering [33] who also found spherical particles in fresh precipitate. Agglomeration of primary particles of precipitate in an aqueous suspension usually proceeds to form densely packed agglomerates. The agglomerates often have a narrow particle size distribution with agglomerate diameters up to 1 μm. Crystal structure and phase identification of the mechanochemically prepared precursor, dried at 60 °C, were analyzed from the X-ray diffraction patterns: Diffraction lines of the alumina precursor showed peaks indicating rather low crystallinity of the sample. The main component was aluminum ammonium carbonate hydroxide NH4Al(OH)2CO3 (PDF 71-1314), accompanied with a rest of undecomposed ammonium nitrate (PDF 01-0809) in Fig. 2a. The main chemical reaction of Al nitrate precipitation can be written as follows [34]:

Structural and textural properties of the prepared γ-Al2O3 and a commercial γ-Al2O3: a X-ray diffraction (Cu Kα1 radiation λ = 0.154056 nm), b N2 adsorption–desorption isotherms at − 196 °C, c pore-size distribution
The textural parameters of the support are summarized in Figs. 2b and c and Table 1. The surface area, SBET, of the calcined γ-Al2O3 was 282 m2g−1. It exhibited a type of IV adsorption isotherm (Fig. 2b) typical for mesoporous solids [35]. The comparison with one of typical industrial γ-Al2O3 (HDS-000-1.5, AKZO Chemicals, The Netherlands) in Table 1 and Fig. 2 did not show significant differences in textural and structural properties.
Concept of NiMo nitrilotriacetic acid complex deposition
The impregnation of the prepared γ-Al2O3 using solution of the Ni and Mo precursors and chelating agent nitrilotriacetic acid, NTA, represents well known approach of preparation of highly active HDS catalysts. The positive effect of NTA was generally observed only when NTA-based catalysts were not calcined before the catalytic activity test [17, 36]. The explanation of high HDS activity is often based on the formation of complex of chelating agent with Ni. The complex stabilizes Ni against sulfidation so that Ni is sulfided after Mo. The result is larger number of Ni atoms decorating MoS2 crystals, which is important for activity promotion. In the absence of chelating agent, Ni is sulfided before Mo and this leads to formation of inactive bulk Ni sulfides [17, 37,38,39,40]. The NiMoNTA was therefore directly sulfided after drying (avoiding calcination) and its characteristics are discussed hereinafter.
Concept of nickel heteropolyoxomolybdate deposition
The NiMoHPO catalyst, prepared by impregnation of the γ-Al2O3 support with an aqueous solution of the ammonium salt of nickel heteropolyoxomolybdate, (NH4)4Ni(OH)6Mo6O18 (HPO), was necessary to preload with Ni to obtain a catalyst with 12 wt% of Mo and 2.0 wt% of Ni. The alumina support possessed surface area SBET 282 m2 g−1, therefore, the deposited amount of Mo, 12 wt%, represented surface density 3.3 atom of Mo per square nanometer, which corresponded to our previously reported results about saturated monolayer [41, 42]. The plane molecular structure of the supported NiMo heteropolyoxoanion offers possibility of good contact with the alumina support and homogenous distribution of the active sites on catalytic surface. One explanation could be based on the interaction of preliminary loaded nickel with NH4+ ion of the ammonium heteropolyoxomolybdate. The preliminary loaded Ni has a role of counteraction. In this way, a small amount of preliminary loaded nickel can keep the loaded heteropolyoxoanion on the surface during calcination and sulfidation. Nevertheless, the second explanation of the homogeneous distribution could also be proposed. Despite the fact that Mo occurs in octahedral entities in HPO [24, 28], the tetrahedral Mo anion, MoO42−, can be formed in interaction with the hydroxyl groups of the support to form stable Al–O–Mo bonds. This interaction species is hardly transformed to sulfides during sulfidation. The rest of HPO mostly consisting of octahedral polymolybdates is, on the contrary, well sulfided. HPO precursor thus optimally combines required strong interaction with the support to keep good dispersion and at the same time, good propensity to sulfidation. The comparison of NiMoHPO with other two catalysts is discussed thereinafter in more details.
Textural properties of catalysts
The textural parameters of the sulfide catalysts are summarized in Table 1. The selected nitrogen adsorption–desorption isotherms and pore-size distribution are shown in Fig. 3a and b, respectively. The prepared sulfided catalysts exhibited type IV adsorption isotherm typical for mesoporous solids [35]. The pore-size distribution (Fig. 3b) revealed significant maximum at pore radius about 2.5 nm with somewhat pronounced shoulder at about 2 nm. The character of the isotherm and the pore-size distribution of the prepared γ-Al2O3 remained the same after deposition of NiMo phase (Fig. 3). The values of textural parameters (SBET and surface of mesopores SM) exhibited ascending order in the line NiMoI < NiMoNTA < NiMoHPO < γ-Al2O3 (Table 1). The lowest values of N2 adsorbed were found over NiMoI pointing on the highest interaction of Ni and Mo precursor with the support during preparation procedure leading to partial destruction of the support surface or partial blockage of the pores by the deposited phase. The presence of NTA during preparation hindered the unwanted interaction or pore blockage and resulted in higher values of surface areas. Only partial blocking of micropores during sulfidation of NiMoNTA should be assumed from Table 1. Nevertheless, the high values of both, SBET and surface of mesopores SM, of the catalysts prepared from the complexes clearly showed good stability of the mechanochemically prepared γ-Al2O3. The decrease of SBET and SM after deposition of sulfidic NiMo phase was not accompanied with changes in pore-size distribution, which pointed out on preservation of porous structure of the support. The most promising texture was observed over NiMoHPO. The texture of the reference NiMoRef differed only slightly from that of the prepared catalysts possessing maximum of the pore size distribution curve at pore radius about 3.2 nm.

a N2 adsorption–desorption isotherms − 196°C and b pore-sized distribution of the prepared support and sulfided NiMo catalysts
H2-TPR and chemisorption of O2
H2-TPR profiles and O2 up-takes of freshly sulfided catalysts are summarized in Fig. 4 and Table 2. TPR profiles of the sulfided catalysts in Fig. 4 showed the first reduction peak with the distinctly pronounced maximum, TMAX, at 115–180 °C, which is typical for highly labile sulfur in sulfides [7, 43,44,45,46]. The second peaks with maxima at about 450 °C were less noticeable. The Mo catalysts exhibited the first peak maximum at higher temperatures than the NiMo catalysts as it was also observed previously [47]. The MoRef and MoI catalysts showed TMAX with somewhat declining rank at 180 and 160 °C, respectively. Both Mo catalysts exhibited lower O2 up-takes than NiMos. In contrast, TMAXs of the prepared NiMo catalysts were in the range 115–138 °C with O2 up-takes 20–25 μmol g−1.

H2-TPR profiles of the sulfided catalysts; experimental conditions: sample weight 0.2 g, gas flow 35 cm3 min−1, gas composition H2:Ar = 1:19, and temperature ramp 5 °C min−1
For the novel alumina supported catalysts, the lowest value of 125 °C was found for NiMoHPO. However, the presence of NTA during preparation led to the highest TMAX, H2 consumption and O2 up-takes among the prepared NiMo catalysts, which indicated the largest formation of sulfide phase containing labile sulfur.
The reference Mo and NiMo catalysts exhibited higher hydrogen consumptions and O2 up-takes than the prepared counterparts (despite having lower HDS activities, see below).
Raman spectroscopy
Fig. 5 summarizes Raman spectra of the selected sulfided catalysts, sulfided starting complexes and support. The Mo containing samples exhibited typical bands of MoS2 layers [47] at about 405 and 380 cm−1. Some spectra also contained less intense MoS2 bands [48] at 820 and 446 cm−1. However, the Raman signal of sulfide species was too weak for quantification of a crystallization degree or number of layers of MoS2 [49].

Nevertheless, the other bands at about 960, 940, or 870 cm−1 are ascribed to the interaction of species of polymolybdates with the support [50,51,52], hydrated polymolybdates [50], or dehydrated polymolybdates [50]. Furthermore, the bands at about 995, 820 or 670 cm−1 are ascribed to crystalline MoO3 [53,54,55] or the band at about 870 cm−1 is referred to tetrahedral MoO42− [56]. Some of these species were also found in the sulfide samples.
The majority of these bands were observed over the catalysts prepared by conventional impregnation. The features of crystalline MoO3 could be found in sulfided MoI or NiMoI. It should be nevertheless noted that the band at 820 cm−1 is also the accompanying band of MoS2 and that at 670 cm−1 is of low intensity. In contrast, the band characteristic for polymolybdates at about 940 cm−1 is clearly pronounced in NiMoI. These observations can be explained by deep interaction of Ni and Mo precursors with the support during calcination step. This deep interaction leads to the formation of bulky aluminum polymolybdates, which is resistant to sulfidation. Part of this interaction can proceed in small pores of the support and the partially destroyed walls of Al2O3 prevent sulfidation of the encapsulated MoO3.
In contrast with the conventional NiMoI catalyst, the catalyst prepared with the assistance of NTA was directly sulfided avoiding calcination. The degree of interaction of oxidic Ni and Mo species with the support was thus limited and most of the deposited NiMo species was turned to sulfidic during sulfidation. Apart from the bands of sulfides, only the low intensity band of polymolybdate interaction species was observed at about 960 cm−1. Rather low intensity and wide band of carbon at 1580 cm−1 is not shown in Fig. 5.
In the case of both sulfided starting HPO complex and NiMoHPO catalyst, the band characteristic for tetrahedral MoO42− species was found. It was thus proposed that these species are hardly sulfidable and represent the interaction species of HPO that react with hydroxyl groups of the mechanochemically prepared alumina. These interactions are not so strong and as extensive as observed in NiMoI but they are stronger than in NiMoNTA and for that reason they provide stable dispersion of the deposited phase and high activity catalyst.
XPS spectroscopy
The oxidation states of the elements in the prepared NiMo catalysts were examined by XPS of the catalysts in sulfide forms (Tables 3 and 4). The examples of the recorded Mo 3d and Ni 2p and S 2p spectra are shown in Fig. 6.

a Mo 3d, b Ni 2p and c S 2p spectra of the prepared catalysts (excitation with an Al Kα radiation hν = 1486.6 eV)
A systematic deconvolution of the XPS data was performed. XPS data demonstrate oxidation numbers of molybdenum and nickel and distribution of sulfides on the surface. Various species were identified on the surface (Table 3): Three kinds of Mo species with binding energies of Mo 3d5/2 = 229, 233 and 230 eV were found corresponding to MoS2, MoO3 and MoOxSy, respectively [57, 58]. The surface composition is shown in Table 4. The Ni species were quantified only as a sum in Table 4 because two little recognizable contributions, i.e. sulfidic NiS (Ni 2p3/2 = 854 eV) and Ni2+ in a mixed oxide (Ni 2p3/2 856 eV), were hardly to resolve by the deconvolution. Nevertheless, all S 2p spectra were fitted with two S 2p doublets (sums of the doublets are shown on the fig.), the first peak having a S 2p3/2 BE of 162 eV corresponds to S2− ions in MoS2 and/or NiS phase, the second one at 163 eV for disulfide species and the third one at BE = 170 eV characterizes the presence of the SO42− ions in the surface. The presence of SO42− can be explained by partial oxidation of the sulfidic surface during the samples handling.
It was found that the surface concentrations of Mo species qualitatively well corresponded to Raman spectroscopy results. NiMoI contained the highest amount of Mo6+ among the studied catalysts (Table 4) and Raman spectra showed the bands of MoO3. In contrast, the catalyst prepared from NTA complex contained the most Mo4+ and therefore was well sulfided. Low metal support interaction were demonstrated by the presence of only low intensity band of interaction species in Raman spectra, on one hand, and the highest O2 up-take, on the other hand. The highest Ni/(Ni + Mo) ratio also well documented that Ni was not lost in alumina subsurface by calcination, which was avoided. Moderate surface composition was found in the NiMoHPO catalyst, wherein the highest content of partially sulfided MoOxSy was determined with somewhat low content of both Mo4+ and Mo6+.
It should be noted that the ratio (MoS2)/total Mo, as a measure of a sulfidation degree, in Table 4 might seem controversial in comparison to some previously reported literature data. For example, Ishutenko et al. [27] reported about 77% of MoS2 in similar catalytic systems while 33% is reported here for NiMoHPO. Nevertheless, differentiation between Mo4+ and MoOxSy (both of them have sulfidic character and they are active in HDS) by the deconvolution procedure of XPS data alone should be considered sensitive. If the sulfidation measure is expressed as the ratio of a sum of Mo4+ and MoOxSy to total Mo, then NiMoI, NiMoNTA and NiMoHPO possess values of 59, 85 and 89%, in order. Only 11% of total Mo thus remains in Mo6+ form in the catalyst prepared from HPO, which is in good agreement with the literature data [27].
Activity in HDS of 1-benzothiophene
The activities of the studied catalysts in the reaction of 1-benzothiophene (BT) hydrodesulfurization are summarized in Table 5 as an empirical pseudo-first-order rate constant of ethylbenzene formation kEB. Because the relatively high content of hydrogenation intermediate dihydrobenzothiophene (DHBT) was observed in the reaction mixture, the HDS reaction was additionally described by parallel consecutive scheme of four pseudo-first-order reaction with the rate constants kHYD (hydrogenation), kde-HYD (dehydrogenation), kHYD-HYG (hydrogenation-hydrogenolysis) and kDDS (direct desulfurization). These rate constants are also summarized in Table 5. They report on the relative selectivity between hydrogenation route, HYD, (kHYD, kde-HYD, kHYD-HYG) and direct desulfurization route, DDS, (kDDS) of 1-benzothiophene HDS. The selectivity to DHBT is shown in Fig. 7 as the dependence of yields y(DHBT) on BT conversion x(BT).

Selectivity to dihydrobenzothiophene (DHBT) during 1-benzothiophene (BT) HDS at 360 °C and 1.6 MPa over: MoRef (solid circles), NiMoRef (solid squares), MoI (open circles), NiMoI (open down triangles), NiMoNTA (open diamonds), and NiMoHPO (open squares) catalysts
It was ascertained that the Mo catalysts exhibited the ascending order of the weight-normalized HDS activities MoRef < MoI. Nevertheless, the activities of MoI and MoRef become nearly the same if only the Mo content is considered. The relative selectivity HYD-HYG/DDS was the same over both Mo catalysts (Table 5, Fig. 7). The properties of the novel mechanochemically prepared γ-Al2O3 in the synthesized MoI HDS catalysts were thus qualitatively the same as those of the γ-Al2O3 in the reference catalyst but it enabled deposition of higher loadings of Mo due to higher SBET, SM and high volume of pores VTotal.
Promotion of the MoI catalysts with Ni led to significantly (about 7.2-fold as intrinsic, i.e. normalized per mol of Mo or Ni + Mo, and about ninefold as weigh normalized) higher activity kEB compared to MoI catalyst and significantly different selectivity in HDS of 1-benzothiophene favoring DDS pathway.
Nevertheless, further improvement of HDS activity over the studied alumina was achieved using deposition of the NiMo complexes. First, NTA-assisted deposition of NiMo led to about 1.5-fold higher activities (both intrinsic and weight normalized) in comparison to conventionally prepared NiMoI. The improved sulfidation demonstrated above by high content of Mo4+, high amount of sulfur vacancies (high O2 up-takes), low metal support interaction (only low intensity band of the interaction species being observed in Raman spectroscopy), i.e. typical features of NiMoS Type II sites, can explain this high activity. Second, the deposition of the nickel heteropolyoxomolybdate in NiMoHPO led to additional increase of the activity indexes up to 1.9 times in comparison to NiMoI. Furthermore, NiMoHPO catalyst also exhibited more than about 1.46-fold higher kEBs than commercial NiMoRef.
In terms of the relative selectivity HYD/DDS, all NiMo catalysts significantly differed from Mo catalysts. NiMo catalysts possessed in two order of magnitude higher kDDS while the other constants kHYD, kde-HYD, kHYD-HYG describing the hydrogenation route, HYD, remained within the difference of one order of magnitude. The yields of dihydrobenzothiophene in Fig. 7 documented qualitatively the same. Furthermore, the prepared NiMo catalysts yielded about the same amount of DHBT as the reference industrial NiMoRef and only insignificant decrease in y(DHBT) could be found for NiMoNTA or NiMoHPO. Nickel in the prepared NiMo catalysts thus efficiently promoted the direct desulfurization route of BT HDS.
It was concluded that the impregnation of mechanochemically prepared γ-Al2O3 with (NH4)4Ni(OH)6Mo6O18 was the best of the studied methods to achieve high HDS activities. The presence of optimal amount of tetrahedral MoO42− species that are hardly sulfided and that represent the interaction species with alumina surface in this complex is presumably responsible for high concentration of surface MoOxSy (1.5 at.%) and high HDS activity.
Conclusions
The novel γ-Al2O3 synthesis by mechanochemical activation of aluminum nitrate hydrate was successfully up-scaled for the preparation of NiMo HDS catalysts. The novel γ-Al2O3 exhibited sufficiently high SBET, SM, VTotal of 282, 201 m2 g−1, 0.42 cm3 g−1 to deposit 2.2 and 12 wt% of Ni and Mo, respectively, in highly active form. The complexes of NiMo with NTA or (NH4)4Ni(OH)6Mo6O18 were found to be feasible precursors for preparation of catalysts highly active in hydrodesulfurization reaction of 1-benzothiophene. The NTA assisted deposition resulted in the catalyst with low metal-support interaction and 1.5 higher activity than the conventionally prepared NiMo counterpart. The most promising method of NiMo catalyst preparation for the studied γ-Al2O3 was the deposition of (NH4)4Ni(OH)6Mo6O18. An optimal interaction between this complex and the new alumina, manifested by the presence of MoO42− or MoOxSy species, was found to be beneficial and determining factor of about 1.9 and 1.4 higher HDS activity in comparison to the conventionally prepared NiMo and reference commercial NiMo counterparts, respectively.
References
Jirátová K, Spojakina A, Kaluža L, Palcheva R, Balabánová J, Tyuliev G (2016) Chin J Catal 37:258–267
Jirátová K, Spojakina A, Tyuliev G, Balabánová J, Kaluža L, Palcheva R (2015) In: Kalenda P, Lubojatsky J (eds) Proceedings of the 3rd International Conference on Chemical Technology, Czech Society of Industrial Chemistry, Prague
Giraldo SA, Centeno A (2008) Catal Today 133–135:255–260
Ramirez J, Castillo P, Cedefio L, Cuevas R, Castillo M, Palacios JM, Lopez-Agudo A (1995) Appl Catal A Gen 132:317–334
Perez-Martinez DJ, Eloy P, Gaigneaux EM, Giraldo SA, Centeno A (2010) Appl Catal A Gen 390:59–70
Qian EW, Chen N, Gong SF (2014) J Mol Catal A Chem 387:76–85
Duan A, Li T, Zhao Z, Liu B, Zhou X, Jiang G, Liu J, Wei Y, Pan H (2015) Appl Catal B Environ 165:763–773
Dugulan AI, Van Veen JAR, Hensen EJM (2013) Appl Catal B Environ 142:178–186
Liu B, Chai Y, Wang Y, Zhang T, Liu Y, Liu C (2010) Appl Catal A Gen 388:248–255
Afanasiev P (2006) Appl Catal A Gen 303:110–115
Kaluža L, Gulková D, Vít Z, Zdražil M (2015) Appl Catal B Environ 162:430–436
Baston EP, Franca AB, Neto AVD, Urquieta-Gonzalez EA (2015) Catal Today 246:184–190
Baston EP, Urquieta-Gonzalez EA (2010) Stud Surf Sci Catal 175:671–674
Escobar J, Barrera MC, De Los Reyes JA, Toledo JA, Santes V, Colin JA (2008) J Mol Catal A Chem 287:33–40
Al-Dalama K, Stanislaus A (2011) Thermochim Acta 520:67–74
Budukva SV, Klimov OV, Chesalov YA, Prosvirin IP, Larina TV, Noskov AS (2018) Catal Lett 148:1525–1534
Medici L, Prins R (1996) J Catal 163:38–49
Palcheva R, Kaluža L, Dimitrov L, Tyuliev G, Avdeev G, Jirátová K, Spojakina A (2016) Appl Catal A Gen 520:24–34
Ayala M, Esneyder P, Quintana P, Gonzalez-Garcia G, Diaz C (2015) RSC Adv 5:102652–102662
Spojakina A, Kraleva E, Jiratova K, Petrov L (2005) Appl Catal A Gen 288:10–17
Spojakina A, Jiratova K, Kostova N, Kocianova J, Stamenova M (2003) Kinet Katal 44:813–818
Palcheva R, Spojakina A, Dimitrov L, Jiratova K (2009) Micropor Mesopor Materials 122:128–134
Nikulshin PA, Tomina NN, Pimerzin AA, Kucherov AV, Kogan VM (2010) Catal Today 149:82–90
Nikulshin P, Mozhaev A, Lancelot C, Blanchard P, Payen E, Lamonier C (2016) C R Chimie 19:1276–1285
Nikulshin PA, Salnikov VA, Mozhaev AV, Minaev PP, Kogan VM, Pimerzin AA (2014) J Catal 309:386–396
Nikulshin PA, Mozhaev AV, Ishutenko DI, Minaev PP, Lyashenko AI, Pimerzin AA (2012) Kinet Catal 53:620–631
Ishutenko D, Mozhaev A, Salnikov V, Nikulshin P (2016) Reac Kinet Mech Cat 119:615–627
Blanchard P, Lamonier C, Griboval A, Payen E (2007) Appl Catal A Gen 322:33–45
Palcheva R, Spojakina A, Jiratova K, Kaluza L (2010) Catal Lett 137:216–223
Palcheva R, Kaluza L, Spojakina A, Jiratova K, Tyuliev G (2012) Chin J Catal 33:952–961
Nomiya K, Takahashi T, Shirai T, Miwa M (1987) Polyhedron 6:213–218
Schneider P (1995) Appl Catal A Gen 129:157–165
Djuricic B, Pickering S (1999) J Euro Ceram Society 19(1999):1925–1934
Wu Z, Shen Y, Dong Y, Jiang J (2009) J Alloys Comp 467:600–604
Gregg SJ, Sing KSW (1982) Adsorption, surface area and porosity. Academic Press, London
Koizumi N, Mochizuki T, Yamada M (2009) Catal Today 141:34–42
Cattaneo R, Shido T, Prins R (1999) J Catal 185:199–212
Cattaneo R, Weber T, Shido T, Prins R (2000) J Catal 191:225–236
Ohta Y, Shimizu T, Homma T, Yamada M (1999) Stud Surf Sci Catal 127:161
Coulier L, De Beer VHJ, Van Veen JAR, Niemantsverdriet JW (2001) J Catal 197:26–33
Kaluža L, Zdražil M, Vít Z (2009) React Kinet Catal Lett 97:307–313
Kaluža L, Vít Z, Zdražil M (2005) Appl Catal A Gen 282:247–253
Calais C, Matsubayashi N, Geantet C, Yoshimura Y, Shimada H, Nishijima A, Lacroix M, Breysse M (1998) J Catal 174:130–141
Magnus PJ, Riezebos A, Van Langeveld AD, Mouljin JA (1995) J Catal 151:178–191
McGarvey JB, Kasztelan S (1994) J Catal 148:149–156
Scheffer B, Dekker NJJ, Mangnus PJ, Mouljin JA (1990) J Catal 121:31–46
Lacroix M, Dumonteil C, Breysse M, Kasztelan S (1999) J Catal 185:219–222
Lewis R, Edwards HGM (2001) Handbook of Raman Spectroscopy. Marcel Dekker Inc, New York
Golasa K, Grzeszczyk M, Korona KP, Bozek R, Binder J, Szczytko J, Wysmolek A, Babinski A (2013) Acta Phys Pol A 124:849–851
Mestl G, Srinivasan TKK (1998) Cat Rev Sci Eng 40:451–570
Debecker DP, Stoyanova M, Rodemerck U, Gaigneaux EM (2011) J Mol Catal A 340:65–76
Zingg DS, Makovsky LE, Tischer RE, Brown FR, Hercules DM (1980) J Phys Chem 84:2898–2906
Dufresne P, Payen P, Grimblot J, Bonnelle JP (1981) J Phys Chem 85:2344–2351
Xu Z, Shi X, Zhang Q, Zhai W, Li X, Yao J, Song S, Chen L, Xiao Y, Zhu Q (2014) Tribol Trans 57:1017–1027
Kumari L, Ma YR, Tsai CC, Lin YW, Wu SY, Cheng KW, Liou Y (2007) Nanotechnology 18:115717–115723
Murase K, Ando H, Matsubara E, Hirato T, Awakura Y (2000) J Electrochem Soc 147:2210–2217
Gandubert AD, Krebs E, Legens C, Costa D, Guillaume D, Raybaud P (2008) Catal Today 130:149–159
Gandubert AD, Legens C, Guillaume D, Rebours S, Payen E (2006) Surf Interface Anal 38:206–209
Acknowledgements
Scientific Cooperation Funds of Bulgarian and Czech Academies of Sciences are gratefully acknowledged. L.K. and D.G. appreciate and acknowledge the Czech Science Foundation (project no. 17-22490S) and Albemarle (The Netherlands), Akzo Chemicals (The Netherlands) and BASF (Germany) companies for financial support and providing of reference materials, respectively.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Kaluža, L., Jirátová, K., Tyuliev, G. et al. Hydrodesulfurization NiMo catalysts over gamma-alumina prepared mechanochemically. Reac Kinet Mech Cat 125, 319–337 (2018). https://doi.org/10.1007/s11144-018-1436-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-018-1436-7