
Abstract
The hydrogenation, and competitive hydrogenation, of toluene, ethylbenzene, propylbenzene and the xylenes has been studied over a rhodium catalyst in the liquid phase at 323 K and 3 bar(g). The reactivity of the aromatics gave an order of para-xylene > ortho-xylene > meta-xylene > toluene > ethylbenzene ≫ propylbenzene. Kinetic analysis revealed that the order of reaction in hydrogen was typically first order while the reaction order in toluene was zero order and negative half order for ethylbenzene. The reaction order for propylbenzene and the xylenes was negative first order. Apparent activation energies were calculated and all were in the range 26–46 kJ mol−1. Competitive hydrogenation between toluene, ethylbenzene and propylbenzene revealed that the propylbenzene was the most strongly adsorbed aromatic in agreement with the strongly negative reaction order. The xylenes gave an order of reactivity of para > ortho > meta following the increasing negative reaction order. Reactions with deuterium revealed an inverse kinetic isotope effect, most likely related to the change in hybridization of the carbon from sp2 to sp3, for all reactions, except that of ortho-xylene. Rapid exchange of the methyl group hydrogens was observed with all the xylenes, whereas total exchange was noted with toluene. The generation of trans-1,2-dimethylcyclohexane was explained by the formation of two intermediates, 1,2-dimethylcyclohexene and 1,6-dimethylcyclohexene, which give the cis-1,2-dimethylcyclohexane and trans-1,2-dimethylcyclohexane, respectively.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrogenation technology in chemicals production is ubiquitous. It has often been considered mature technology with little scope for new developments: an area that is not fashionable. However, this belies the difficulties and unknowns that are still extant in hydrogenation. In 1905, Sabatier and Senderence [1] reported the first catalytic hydrogenation of benzene. They ‘attacked’ the benzene ring with hydrogen at atmospheric pressure and temperatures between 343 and 473 K over a nickel catalyst and succeeded in converting it to cyclohexane [1]. This was the first example of hydrogenation of an aromatic ring. Nowadays aromatic hydrogenation is a major industrial process with around 4.6 million tons of benzene hydrogenated to cyclohexane each year.
The catalytic hydrogenation of alkylbenzenes to saturated cyclic products is also an important reaction although it does not get the same interest. It is used in lowering the aromatic content in diesel fuels for environmental reasons [2]. Much of the aromatic content in fuels comes from pyrolysis gasoline (PyGas), which is a by-product of high temperature naphtha cracking to produce ethylene and propylene. It is a mixture of highly unsaturated hydrocarbons and contains considerable amounts of aromatics, normally 40–80% (benzene, toluene and xylene), together with paraffins, olefins and diolefins. The composition depends on the feedstock and operating conditions and hence varies from plant to plant. With an aromatic content of around 65 wt% pygas has been used as a gasoline blend due to its high octane number [3]. However with new legalization set to lower the aromatic content in gasoline [4] further hydrogenation of the aromatics especially as a mixture will be required.
The hydrogenation of alkylbenzenes has only been subject to limited study and even that is focused on toluene and the xylenes [5,6,7,8,9,10]. To find studies of ethylbenzene or propylbenzene hydrogenation we must go back to 1945 [11]. Although there is a more recent study on ethylbenzene hydrogenation [12] examining the kinetics in the gas phase, no other literature on propylbenzene could be found. The xylenes, which have been examined slightly more extensively, are still poorly considered [5,6,7,8,9,10] compared to the hydrogenation of benzene, which has been the subject of numerous studies [13].
The hydrogenation of alkylbenzenes is affected by the length and position of the substituent on the benzene ring [5, 6, 11]. In general the rate of hydrogenation decreases as the length of the substituent increases, while for the xylenes the para-isomer has been found to be the most active [5, 6, 9]. Toppineen et al. [14, 15]. investigated in detail the hydrogenation of five aromatics (di- and tri-substituted alkyl benzenes, xylenes, mesitylene, and p-cymene) over Ni/Al2O3. They also concluded that the reaction rate is affected by the number, length and position of the substituent. They found that the hydrogenation rate increased as the number of substituents decreased (mesitylene < xylenes < toluene < benzene) and the reaction rate was found to increase as the length of substituent decreased (cumene < ethylbenzene < toluene < benzene). For the xylenes they also found the para-isomer to be the most reactive.
Many of the kinetic studies have been gas phase studies at moderate temperatures [8, 9], while the early work in the liquid phase did not undertake any kinetic analysis. Also there is a dearth of information on rhodium as the active phase, with no study examining the xylenes or ethyl- or propylbenzene [5, 16]. In this study we have undertaken a basic kinetic analysis of the hydrogenation of toluene, ethylbenzene, propylbenzene and the xylenes over a rhodium catalyst at low temperatures, examined the competitive hydrogenation and probed the mechanism of exchange and hydrogenation using deuterium and deuterated toluene.
Experimental
The catalyst used throughout this study was a 2.5% Rh/silica supplied by Johnson Matthey (code: M01074). The silica support was supplied by Davison Catalysts and the catalysts, which were supplied and characterized by Johnson Matthey, were prepared using an incipient-wetness method using aqueous rhodium chloride salts. The catalysts were dried overnight at 333 K and reduced in flowing hydrogen at 473 K for 2 h before being cooled and exposed to air. The surface area (311 m2 g−1) and pore size (13.9 nm) were measured using standard BET methodology, while metal surface area (4.7 m2 g−1, dispersion 43%; average metal crystallite size 2.6 nm) was measured by hydrogen chemisorption with reproducibility of ±0.5 m2 g−1 (all characterization data supplied by Johnson Matthey).
The hydrogenation reactions were performed in a 500 cm3 Büchi autoclave stirred tank reactor supplied with an oil heating jacket. The temperature was measured in the liquid slurry with accuracy of ±0.1 K and controlled by a high temperature oil circulator to ±0.5 K. The reactor was equipped with a variable speed stirrer connected to a magnetic drive that could be controlled to ±5 rpm. The pressure and gas flow was controlled by a Büchi press-flow gas controller with an accuracy of ±0.01 bar(g) and measurement of the consumed hydrogen to 0.1 mmol. The gas controller was used to achieve a constant hydrogen pressure in the autoclave. The reactions were performed as follows, in a typical hydrogenation experiment catalyst (100 mg) was charged to the autoclave with 320 ml of isopropanol (IPA) and heated to the reduction temperature under slow stirring (300 rpm). In situ reduction of the catalyst was performed at 343 K by sparging hydrogen gas at a flow rate of 280 cm3 min−1 through the mixture for 0.5 h while stirring at 300 rpm. After the reduction process was completed, the stirrer was turned off and the reactor was purged with nitrogen twice and pressurized to 1 bar(g) pressure. The gas controller was used to measure the flow of hydrogen, deuterium or inert gas to the reactor and also to measure the hydrogen/deuterium consumed in the reaction. The contents of the reactor were then heated to the desired reaction temperature under slow stirring. After reaching the desired temperature (303–333 K) the stirrer was turned off and 1.0 cm3 of toluene (Fischer, 99% 9.42 mmol), d8-toluene (Sigma-Aldrich, 99% D, 9.43 mmol) ethylbenzene (Sigma-Aldrich, 99.5% 8.17 mmol), propylbenzene (Sigma-Aldrich, 98% 7.18 mmol), ortho-xylene (Sigma-Aldrich, 99%, 8.30 mmol), meta-xylene Sigma-Aldrich, 99%, 8.11 mmol) or para-xylene (Sigma-Aldrich, 99%, 8.12 mmol) in 10 ml of isopropanol was added into the reactor vessel through the reactor inlet to give a total reaction volume of 330 ml. The solution was stirred at 1000 rpm to allow for mixing. The stirrer was then turned off and the reactor pressurized with N2 to 1 bar(g) before a sample was withdrawn (2.5 ml). The reactor was de-pressurized, before being purged twice with hydrogen then pressurized with hydrogen to the desired reaction pressure (1–5 bar(g)). Once the vessel was pressurized the reaction was started by switching the stirrer on at 1000 rpm: this was taken as the zero time of the reaction. The progress of the reaction was followed by withdrawing samples of 2.5 cm3 at different time intervals during the reaction. The moles of hydrogen consumed during the reaction were also monitored and recorded. Mass balance was 100 ± 5%. Liquid samples were analyzed using a Thermo Finnigan Focus GC equipped with an AS 3000 autosampler. The column used was an HP-1701, 30 m × 0.25 mm × 1 µm film thickness. GC–MS of the selected samples were performed on a Shimadzu GC-2010 equipped with an AOC-20i auto-injector unit and an GCMS-OP2010S detector. The column was either a Zebron ZB-5MS 30 m × 0.25 mm × 0.25 µm film thickness used only for the analysis of the reaction intermediates or a Petrocol™ Supleco Analytical fused silica capillary column of 100 m × 0.25 mm × 0.5 µm film thickness. The detected reaction intermediates mass spectrograms were compared in the Nist05.lib using Lib2Nist v1.0.23.
D-NMR were performed on a Bruker 500 Ultra Shield-NMR system using a custom pulse and acquisition AU-program provided by the Bruker company using the Deuterium-lock channel as the data channel.
Results
Detailed reaction profiles, determination of rate constants, activation energy plots, order of reaction plots and analysis are all reported in the Supplementary Information. The summary data is presented in the paper along with key results.
Single substrate hydrogenation
The hydrogenation of toluene, ethylbenzene, propylbenzene, ortho-xylene, meta-xylene and para-xylene was carried out as outlined in the experimental section. The effect of temperature (303–343 K), hydrogen pressure (2–5 bar(g)) and aromatic concentration (3.6–11.1 mmol) was investigated to determine apparent activation energies and orders of reaction. The data are summarized in Table 1.
The hydrogenation of toluene, ethylbenzene and propylbenzene resulted in the formation of a single product, the cyclohexyl derivative. A small quantity of the relevant cyclohexene (<2%) was formed during the reaction as an intermediate. However the xylenes formed cis- and trans-dimethylcyclohexyl isomers with intermediate dimethylcyclohexenyls. A reaction profile for each xylene isomer is shown in Figs. 1, 2, and 3. At 323 K the cis:trans ratio of the dialkylcyclohexanes at 100% xylene conversion was 8.1:1 for ortho-xylene, 3.5:1 for meta-xylene and 2.1:1 for para-xylene. The effect of temperature on cis/trans ratio is shown in Fig. 4. Hydrogen pressure and xylene concentration had no effect on the cis/trans ratio.

Hydrogenation of ortho-xylene (323 K, 3 bar(g), ~8 mmol)

Hydrogenation of para-xylene (323 K, 3 bar(g), ~8 mmol)

Hydrogenation of meta-xylene (323 K, 3 bar(g), ~8 mmol)

Change of cis/trans ratio with temperature (3 bar(g), ~8 mmol). DMCH dimethylcyclohexane. 1,2-DMCH is formed from ortho-xylene, 1,3-DMCH is formed from meta-xylene and 1,4-DMCH is formed from para-xylene
Competitive reactions
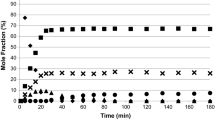
The competitive hydrogenations of toluene, ethylbenzene and propylbenzene were investigated. The compounds were tested in pairs and also with all three reactants. Fig. 5 shows the conversion with time for the alkylbenzenes as single reactants and when hydrogenated together. The first order rate constants are reported in Table 2. A similar process was carried out for the xylenes and the first order rate constants are reported in Table 3. Due to an overlap between cis-1,4-dimethylcyclohexane and trans-1,3-dimethylcyclohexane cis:trans ratios for meta- and para-xylene were not obtained when both were present however ratios were obtained for ortho-xylene products and are reported in Table 4.

Hydrogenation of the alkylbenzenes (a) individually and (b) as a 1:1:1 mixture. Conditions 3 bar(g) hydrogen, 323 K, ~8 mmol alkylbenzene. a Conversion of the alkylbenzenes as single reactants, b conversion of the alkylbenzenes hydrogenated together
Deuterium reactions
Reactions were performed using deuterium rather than hydrogen for the reduction and the hydrogenation. Experiments were also performed using d8-toluene with hydrogen and deuterium. The first order rate constants for the hydrogen and deuterium reactions are reported in Table 5. Apart from ortho-xylene, all the reactants register an inverse kinetic isotope effect.
The toluene system was further examined using d8-toluene with hydrogen and deuterium. Fig. 6 shows the reaction profiles of four reactions; (a) toluene hydrogenation under hydrogen pressure (k = 18.8 × 10−3 min−1), (b) toluene hydrogenation under deuterium (k = 65.6 × 10−3 min−1), (c) d8-toluene hydrogenation under hydrogen (k = 55.0 × 10−3 min−1) and (d) deuterated-toluene hydrogenation under deuterium (k = 67.4 × 10−3 min−1). Clearly reactions involving deuterium and deuterated-toluene, i.e. reactions (b), (c) and (d) were faster than reaction (a), which was the protiated form.


Reaction profiles for a hydrogenation of toluene with hydrogen, b hydrogenation of toluene with deuterium, c hydrogenation of d8-toulene with hydrogen and d hydrogenation of d8-toluene with deuterium. Conditions 323 K, 3 bar(g), ~8 mmol
A number of samples were collected from toluene and deuterated-toluene reactions with deuterium and hydrogen to be analyzed by NMR. One sample was taken at the start of the toluene/deuterium reaction. Another sample was taken from the reaction of deuterated-toluene with hydrogen after 10 min. A reference sample of deuterated-toluene was also run (not shown). The spectra are shown in Figs. 7 and 8.

D-NMR spectrum of toluene hydrogenation with D2 at t = 0 min

D-NMR spectrum of d8-toluene hydrogenation with H21 at t = 10 min
The xylenes were also hydrogenated using deuterium and the products analyzed by D-NMR and GC–MS. Both D-NMR and GC–MS analysis confirmed incorporation of deuterium in the methyl groups but not the aromatic ring prior to hydrogenation for all the xylenes. The ratio of the deuterium incorporation into the xylenes is shown in Fig. 9.

Ratio of isotopically exchanged methyl groups in ortho-, meta- and para-xylene after 15 min reaction. Conditions 323 K, 3 bar(g) deuterium, ~8 mmol
Discussion
From the results presented in Table 1 it can be seen that as the alkyl chain gets longer, the apparent activation energy increases and the order in organic becomes more negative, moving from zero order to negative first order. An apparent activation energy for toluene hydrogenation of 30 kJ mol−1 has been reported over an Rh/MgO catalyst [5] in excellent agreement with our value. As expected as the alkyl chain length is increased the rate of hydrogenation decreases. It has been proposed that this decrease in rate is due the inductive effect of the alkyl chain increasing the electron density of the ring and hence resulting in a stronger π-bond to the surface [8, 9, 16, 17], therefore suggesting that it is the strength of the aromatic adsorption that is inhibiting the reaction. Our results confirm this hypothesis: the reaction order changes from zero-order with toluene to negative half-order with ethylbenzene to negative first-order with propylbenzene (Table 1) indicating a stronger adsorption as the alkyl chain increases in length. (The stronger adsorption will also be confirmed from the competitive hydrogenation reactions to be discussed later.) The apparent activation energy for ethylbenzene hydrogenation was determined to be 46 kJ mol−1, while for the hydrogenation of propylbenzene a value of 39 kJ mol−1 was calculated.
The rate of hydrogenation of the xylenes gives an order para ≈ ortho > meta (Table 1; Figs. 1, 2, 3), which is different from the order reported in the literature over platinum and nickel catalysts (para > meta > ortho) [9, 11]. It is also noticeable that the rate of hydrogenation for the xylenes is faster than that of toluene (meta-xylene, the least active xylene, and toluene have similar rates, Table 1). This is not what is generally reported in the literature, where toluene is usually more reactive than the xylenes, although para-xylene has been reported [11] to show a similar activity to toluene. The apparent activation energy calculated for ortho- and meta-xylene reveals a similar value (~ 26 kJ mol−1) a value that is much lower than that found by Keane and Patterson over nickel (~54 kJ mol−1) [9] and by Rahaman and Vannice over palladium (~51 kJ mol−1) [6]. Para-xylene in contrast gave a value of 42 kJ mol−1, which is similar to that found in the literature over Rh/MgO [5] of 36 kJ mol−1. In the work of Keane and Patterson [8, 9] a compensation effect was observed and our data would also show a similar trend. However more recent work on understanding this relationship [18,19,20] has shown that such behaviour does not represent a true correlation. A full reasoning can be found in the references [18,19,20]. Previous studies [5, 6] have looked for a relationship between ionization potential and activation energy but we do not see any trend.
The cis:trans ratio of the dialkylcyclohexanes is shown in Fig. 4 as a function of temperature. For the 1,4-dimethylcyclohexanes, the trans-isomer is the most stable and as the temperature is raised so the ease of formation of the trans-isomer increases resulting in a steady decrease in cis:trans ratio. For the 1,3-dimethylcyclohexanes it is the cis-isomer that is the most stable and the ratio is almost twice that found for the 1,4-dimethylcyclohexanes (Figs. 2, 3). It is also clear that two intermediate dimethylcyclohexenes are formed; 3,5-dimethylcyclohex-1-ene, with this cyclohexene isomer the methyl groups are the furthest away from the surface minimizing steric hindrance, and the more stable 1,3-dimethylcyclohex-1-ene where both methyl groups will be close to the surface. Nevertheless as the temperature is increased the ratio decreases at the same rate as the 1,4-isomers (Fig. 4). The behavior of the 1,2-dimethylcyclohexanes with temperature is different. The trans-isomer is more stable but at low temperatures there is a significant cis excess (Fig. 1). This high cis:trans ratio, which has been observed with other rhodium catalysts [21], appears to be due to the hydrogenation of the 1,2-dimethylcyclohex-1-ene intermediate with all four positions surrounding the olefinic bond occupied by an alkyl species. As the temperature is raised the cis:trans ratio drops more rapidly than with the other systems until 323 K after which temperature the decrease in cis:trans ratio matches that of the 1,4- and 1,3-cyclohexanes (Fig. 4). Note that there is no evidence for isomerization between species under reaction conditions. Therefore to yield the thermodynamically more stable trans-isomer the cyclohexene intermediate must desorb and re-adsorb with the sterically unfavorable cis configuration to the surface. The closer the methyl substituent is located to the double bond the higher the steric hindrance for the adsorbed surface species yielding the thermodynamically more stable trans product.
The competitive reactions between the alkylbenzenes were highly revealing. The competitive reaction between toluene and ethylbenzene reveals an increase in hydrogenation rate for both species (Table 2). A previous study by Toppinen et al. [15]. gave different results in that no increase in rate was observed but the general behavior was similar. This enhanced rate behaviour is unusual but has been observed before in the competitive reaction of pentynes [22] and pentenes [23]. In both cases it was suggested that each species had a unique adsorption site that the other reactant could not influence and that the hydrogen flux could potentially be increased by enhanced hydrogen transfer from a hydrocarbonaceous deposit. Competitive hydrogenation of benzene and toluene has also been reported [10] and it was suggested that the sites for adsorption and hydrogenation of benzene and toluene were distinct from each other. It is noticeable that that the toluene/ethylbenzene reaction is the only pairing that results in an enhanced rate, all others reduce the rate of toluene and/or ethylbenzene hydrogenation (Table 2). The reduction in rate caused by propylbenzene agrees with the kinetic analysis (Table 1), which suggested that propylbenzene is much more strongly adsorbed than either toluene or ethylbenzene. In every case where propylbenzene is present the rates of hydrogenation for toluene and ethylbenzene are reduced by at least 58%, in contrast, the rates of propylbenzene hydrogenation increase (Table 2). In these reactions a reduction in the strength of propylbenzene adsorption would allow an enhancement in rate as in the absence of a second reactant, propylbenzene is an inhibitor.
The competitive reactions between the xylenes show completely different behavior (Table 3). In each competitive reaction the rate of hydrogenation was always decreased for all reactants. If the strength of bonding for the xylenes was similar then this behavior would be expected, given that the reaction order for the xylenes is typically negative first order (Table 1), as we would have increased the xylene concentration by a factor of two or three. The variations in the activity from the competitive reactions suggest that the strength of bonding is ortho > para > meta. This agrees with the order of reactivity found by Toppinen et al. [15] in their study of xylene competitive hydrogenation. The higher cis:trans ratio for the 1,2-dimethylcyclohexanes is expected as the higher aromatic concentration would tend to inhibit re-adsorption to allow for isomerization.
When hydrogenation reactions are performed using deuterium rather than hydrogen two processes may occur, exchange and hydrogenation. The exchange process of toluene and the xylenes have been well studied [24, 25]. In general the alkyl groups exchange before the ring hydrogens. However, although well studied, the exchange process has not been examined over rhodium. With toluene the ratio of the aliphatic:aromatic peaks in the reaction setting is not the same as in the reference sample. In the reference sample the ratio of peaks is 1.8 whereas in the reaction sample the ratio is 2.5 (Fig. 7, Supplementary Information). Therefore the rate of exchange of the protons in the methyl group is faster than that of the aromatic protons, which contrasts with the exchange over palladium [25], where only the methyl groups were exchanged but is more similar to nickel where both sets of hydrogen exchanged but the rate of methyl group exchange was over an order of magnitude faster [24]. The reverse process, hydrogen exchange with d8-toluene appears to be slower (Fig. 8) possibly displaying a kinetic isotope effect, with d8-toluene still present 10 min into the reaction. The exchange of the xylenes shows only exchange into the two methyl groups similar to that found with palladium [25]. Para-xylene has the highest quantity of d-0 species (Fig. 9) and this is similar to the distribution found over nickel for the exchange process [24].
The hydrogenation of toluene and the xylenes using deuterium results in a kinetic isotope effect. All the reactants except ortho-xylene exhibit an inverse kinetic isotope effect (KIE) (Table 5). This is likely a secondary inverse KIE which can be observed when there is a change in hybridization of the carbon (C–H) from sp2 to sp3 as would be the case in hydrogenation of the aromatic ring. The difference with ortho-xylene and identifying 1,2-dimethylcyclohexene as the main intermediate raises the question of whether the rate determining step is different from that of toluene and meta- and para-xylene.
Two dimethylcyclohexenes were detected and when the cis:trans ratio of the 1,2-dimethylcyclohexanes is compared with the ratio of the 1,6:1,2-dimethyl cyclohexenes (Fig. 10) it can be seen that there is a relationship, suggesting that one intermediate produces the cis-product while the other produces the trans-product. Indeed this can be confirmed with a simple model, addition of hydrogen from below to 1,2-dimethylcyclohexene generates cis-1,2-dimethylcyclohexane, whereas a similar addition to 1,6-dimethylcyclohexene results in trans-1,2-dimethylcyclohexane.

Relationship between dimethylcyclohexene intermediates and cis- and trans-1,2-dimethlcyclohexanes, showing line of best fit
Conclusions
In this study the hydrogenation of toluene, ethylbenzene, propylbenzene and the xylenes has been investigated over a rhodium catalyst for the first time. Kinetic analysis revealed that the order of reaction in hydrogen was typically first order while the reaction order in the aromatic varied between zero order and negative first order. Activation energies were determined but no trends were evident. The competitive hydrogenation between toluene, ethylbenzene and propylbenzene revealed that the propylbenzene was the most strongly adsorbed aromatic in agreement with the strongly negative reaction order. The xylenes gave an order of reactivity of para > ortho > meta following the increasing negative reaction order. Reactions with deuterium revealed an inverse kinetic isotope effect (KIE) for all reactions, except that of ortho-xylene, suggesting that hydrogen bond breaking or making is not rate limiting. The cause of the inverse KIE is most likely related to the change in hybridization of the carbon (C–H) from sp2 to sp3. Rapid exchange of the methyl group hydrogens was observed with all the xylenes, whereas total exchange was noted with toluene. The generation of trans-1,2-dimethylcyclohexane was explained by the formation of two intermediates, 1,2-dimethylcyclohexene and 1,6-dimethylcyclohexene, which give the cis-1,2-dimethylcyclohexane and trans-1,2-dimethylcyclohexane respectively.
References
Sabatier P, Senderens JB (1905) Ann Chim Phys 4:368
Stanislaus A, Cooper BH (1994) Catal Rev 36:75–123
Bader J-M, Rowlands G (2012) Hydrocarbon Proc 91:41–45
Zhou Z, Zeng T, Cheng Z, Yuan W (2010) Ind Eng Chem Res 49:11112–11118
Volter J, Hermann M, Heise K (1968) J Catal 12:307–313
Rahaman MV, Vannice MA (1991) J Catal 127:251–266
Rahaman MV, Vannice MA (1991) J Catal 127:267–275
Keane MA, Patterson PM (1996) J Chem Soc, Faraday Trans 92:1413–1421
Keane MA, Patterson PM (1999) Ind Eng Chem Res 38:1295–1305
Orozco JM, Webb G (1983) Appl Catal 6:67–84
Smith HA, Pennekamp EFH (1945) JACS 67:276–278
Smeds S, Murzin D, Salmi T (1995) Appl Catal A 125:271–291
Bond GC (2005) Metal-catalysed reactions of hydrocarbons. Springer, New York
Toppinen S, Rantakyla T-K, Salmi T, Aittamaa J (1996) Ind Eng Chem Res 35:4424–4433
Toppinen S, Rantakyla T-K, Salmi T, Aittamaa J (1997) Ind Eng Chem Res 36:2101–2109
Phuong TT, Massardier J, Gallezot P (1986) J Catal 102:456–459
Moyes RB, Wells PB (1973) Adv Catal 23:121–156
McBane G (1998) J Chem Ed 75:919–922
Lente G (2015) Deterministic kinetics in chemistry and systems biology. Springer, Heidelberg, pp 121–122
Parmon VN (2016) Reac Kinet Mech Cat 118:165–178
Barthe L, Denicourt-Nowicki A, Roucoux A, Philippot K, Chaudret B, Hemat M (2009) Catal Commun 10:1235–1239
Hamilton CA, Jackson SD, Kelly GJ, Spence RR, de Bruin D (2002) Appl Catal A 237:201–209
Canning AS, Jackson SD, Monaghan A, Wright T (2006) Catal Today 116:22–29
Crawford E, Kemball C (1962) Trans Faraday Soc 58:2452–2467
Williams PG, Than C, Rabbani S, Long MA, Garnett JL (1995) J. Label Compd Radiopharma 36:1–14
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Alshehri, F., Weinert, H.M. & Jackson, S.D. Hydrogenation of alkylaromatics over Rh/silica. Reac Kinet Mech Cat 122, 699–714 (2017). https://doi.org/10.1007/s11144-017-1251-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-017-1251-6