
Abstract
The hydroxylation of phenol was investigated using cobalt(II) complexes of bidentate ligands including N,N′-ethylenebis(salicylideneamine)(salen), N,N′-propylenbis(salicylideneamine)(salpn) and N,N′-phenylenebis(salicylideneamine)(salophen) encapsulated in zeolite-Y. The catalysts were characterized by Fourier transform infrared and X-ray diffraction analyses to confirm the complex encapsulation. These complexes catalyze the liquid-phase hydroxylation of phenol, through H2O2, to catechol and hydroquinone as major and minor products, respectively. The activities of all prepared catalysts were tested for the oxidation of phenol and hydrogen peroxide. The overall reaction conditions were optimized to get maximum hydroxylation gain to consider the concentration of substrate and oxidant, amount of catalyst, and the reaction temperature. The zeolite encapsulated complexes of cobalt(II) were found to be catalytically active toward the hydroxilation of phenol. Under the optimized reaction conditions, [Co-(salen)]-Y showed the highest conversion, about 43 %, after 6 h which was followed by [Co-(salpn)]-Y with 39 % conversion and [Co-(salophen)]-Y showing the lowest efficiency with 35 % conversion. The hydroxylation of phenol without catalyst and with Co/zeolite-Y catalyst showed poor results.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The heterogenization of homogeneous catalysts has attracted wide attention regarding their enhanced activity, selectivity, thermal stability, and reusability. These catalysts include the advantages of both solid heterogeneous catalysts and homogeneous catalysts. Zeolites are often regarded as remarkable materials for encapsulation of transition metal complexes acting as homogeneous catalysts within their pores for the heterogenization. These zeolite-encapsulated metal complexes have been extensively used for the selective oxidation of olefins [1–3], cyclohexane [4], and phenols [5–9] etc. The liquid-phase hydroxylation of phenol to hydroquinone and catechol using conventional oxidants like O2 and H2O2 is remarked as an industrial interesting reaction [10, 11]. The mentioned products have several important applications such as photographic film developer, antioxidant, polymerization inhibitor, medicines, organic synthesis, perfumes, etc.
Phenol has been oxidized, by H2O2, to catechol and hydroquinone as the major products in the presence of Co(II), Cu(II), and Zn(II) complexes in the supercages of zeolite Y and ZSM-5 [5]. The oxidation of phenol with hydrogen peroxide has also been studied through miscellaneous metal complexes encapsulated in zeolite-Y, in which most of them have shown higher selectivity towards catechol formation [12–15]. Various metal phthalocyanine complexes encapsulated in zeolite-Y have been studied for the hydroxylation of phenol by H2O2 [7]. These compounds demonstrate that the activity and product selectivity of the encapsulated complexes can be correlated with the redox potential of the central metal atom of the encapsulated phthalocyanine. The catalytic performance of encapsulated copper and manganese salen encapsulated in the cavities of zeolites NaX and NaY in the decomposition of H2O2 and tert-butyl hydroperoxide as well as the low-temperature oxidation of phenol, styrene, and para-xylene have been reported [16]. The results specified that the oxidation of phenol to hydroquinone and catechol is linearly correlated with the catalytic activity of the encapsulated copper salens. Metal–salpn complexes encapsulated in Y-zeolite have been widely applied to study its catalytic activity toward the phenol oxidation [15]. These encapsulated complexes are active catalysts for the decomposition of hydrogen peroxide and also for the phenol oxidation to catechol and hydroquinone with high selectivity. The oxidation of phenol by tyrosinase enzyme produces catechol as the actual primary product. Then, the catechol transforms to its binuclear form [17]. The zeolite-Y-encapsulated complexes of iron-salen and salophen have been found to be more effective for catalytic conversion of 2-naphthol to binaphthol [18]. To reduce the coke formation from the hydroxylation of phenol, Wittayakun et al. removed aluminum from zeolite beta as a support of iron catalyst and observed that the amount of coke was less and the yields of catechol and hydroquinone were higher than aluminated catalyst [19]. Also, in another study, they changed the preparation method for iron catalysts on zeolite beta and produced light coke rather than heavy form [20].
This work deals with the encapsulation of cobalt(II) complexes of N,N′-ethylenebis (salicylideneamine) (salen), N,N′-propylenebis(salicylideneamine) (salpn), and N,N′-phenylenebis (salicylideneamine) (salophen) encapsulated in the cavities of zeolite-Y.
For this regard, the respective zeolite encapsulated cobalt(II) complexes were prepared, characterized, and their catalytic activity was evaluated for selective liquid phase hydroxylation of phenol. Furthermore, the reaction condition was optimized to obtain the highest hydroxylation efficiency.
Experimental
Materials
Commercial (analytical) reagent grade cobalt(II) chloride hexahydrate, phenol and hydrogen peroxide (30 %) were purchased from Merck. Zeolite-Y (Si/Al ratio: 10:1) obtained from Fluka. The ligands salen, salpn, and salophen were prepared according to the literature [21]. All other chemicals and solvents used were of AR grade purchased from Merck.
Preparation of catalysts
Preparation of salen, salpn, and salophen ligands
Salicylaldehyde (0.2 mol) was dissolved into 100 mL of ethyl alcohol and mixed with a solution of ethylenediamine (0.1 mol) in ethyl alcohol to prepare the N,N′-bis(salicylidene)ethylenediamine (salen) ligand. The mixed solution was refluxed for 3 h to complete the reaction. Then, the yellow precipitate was filtered, washed in cold ethyl alcohol, and dried [21]. N,N′-Bis(salicylidene)propanediamine (salpn) and N,N′-disalicylidene-1,2-phenylenediamine (salophen) were prepared through the same procedure if we replace ethylenediamine by propylenediamine and p-phenylenediamine.
Preparation of Co/zeolite-Y catalysts
About 5 g of zeolite-Y was suspended in 100 mL of distilled water containing CoCl2·6H2O (2.38 g). The suspension was stirred at room temperature for 24 h. Then, the colored solid (Co/zeolite-Y) was separated from the solution by filtration and washed with methanol to remove any chloride ions and dried at 50 °C under vacuum condition.
Preparation of Co(II) complexes/zeolite-Y catalysts
1.088 mmol of salen, salpn, or salophen and 2.176 mmol NaOH were dissolved in 100 mL methanol. About 1 g of Co/zeolite-Y was added to above solution while stirring the reaction mixture overnight. The resulting material was filtered, washed thoroughly with methanol, and then Soxhlet extracted with mixture of 1:1 methanol and chloroform in order to remove unreacted ligands (at least 72 h). Then, it was dried at 125 °C under nitrogen atmosphere for 12 h [22].
Characterization
FTIR spectra were taken using Galaxy-5000 Fourier transform IR spectrometer in the range of 4000–400 cm−1. Powder X-ray diffraction (XRD) patterns were recorded on a Shimadzu XD-D1 powder X-ray diffractometer with Cu Kα radiation.
Decomposition of H2O2
0.025 g of encapsulated catalyst was added to 5.5 g of an aqueous solution of 30 % H2O2 at room temperature and the reaction mixture was stirred for 1 and 2 h. At the end of reaction, the catalyst was filtered and the filtrate was diluted to 250 mL with deionized water. Then, 20 mL of 2 M H2SO4 and 20 mL of deionized water were added to 10 mL of this solution. This sample was then titrated against standard 0.2 M KMnO4 solution.
Hydroxylation of phenol
Catalytic phenol hydroxylation reactions were performed in a three-neekedflask fitted with a water cooled condenser. In a typical reaction, phenol (4.7 g, 0.05 mol) and catalysts (0.025 g of catalysts) were stirred at 50 °C for 30 min by a magnetic stirrer. Then, appropriate amounts of 30 wt% aqueous H2O2 (5.67 g, 0.05 mol) and 2 mL acetonitrile (MeCN) were added into the flask. The reaction mixture was refluxed for 8 h. During this process, the products were drawn from the reaction mixture at regular intervals for analysis by GC (Varian Model 3400) equipped with a flame ionization detector (FID) using OV-17 packed column. A GC–MS model QP5000 was dedicated to identify the products.
Results and discussion
Characterization of catalysts
Infrared spectral studies
IR spectra of Co/zeoliteY showed strong zeolite lattice bands in the 450–1200 cm−1 range. The strong and broad band at ~1000 cm−1 can be ascribed to the asymmetric stretching vibrations of (Si/Al)O4 units. The broad bands at 1650 and 3500 cm−1 are due to lattice water molecules and surface hydroxyl groups [23].
Table 1 reveals the selected frequencies of ligand and its encapsulated complexes. A comparison between the IR spectra of ligands and Co complexes/Y confirmed the presence of Co complexes encapsulated in zeolite-Y cages.
Although the peak intensities of the encapsulated complexes are weak due to low concentration of the complex in zeolite, the IR spectra of the encapsulated complexes are similar to the spectra of the free ligand, except for minor shifts in the positions of some of the bands and some changes in their intensities based on coordination.
The salen, salpn, and salophen ligands showed adsorption peaks associated to νC=N at 1555, 1537, and 1559 cm−1. In the spectra of the complexes, these bands are shifted toward lower frequencies and are observed at 1534, 1479, and 1539 cm−1. This shift is characteristic of the coordination of azeomethin nitrogen to cobalt as a result of complex formation [24]. The phenolic νC–O of the free ligands shifts toward higher wavenumbers in all complexes due to increased C–O band order compered to the hydrogene bonded structure in the free ligand [25]. Our findings are in agreement with the results previously reported in the literature [23, 25].
All metal complexes encapsulated zeolites exhibit band around 1100, 1035, 960, 780, and 740 cm−1 due to the zeolite framework.
No broadening or shift were observed at the positions of the zeolite vibrational bands, which indicate that no change has occurred in the zeolite framework. This clearly indicates that the metal complexes fit well within the cavities of the zeolites [8].
X-ray Diffraction (XRD)
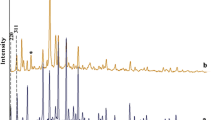
To ensure the preservation of the zeolite framework integrity, as well as encapsulation of the guest complex inside the cavity of the host, XRD patterns of zeolite-Y (a); Co/zeolite-Y (b); and the encapsulated complex [Co-(salen)]-Y (c) are recorded as shown in Fig. 1. The diffraction pattern indicates that a well crystalline zeolite encapsulated catalysts have been formed. These observations specify that the XRD patterns of [Co-(salen)]-Y are similar to those observed for zeolite-Y and Co/zeolite-Y, which indicates that they exhibit the same morphologies and crystalline structures, i.e., the framework around the guest molecule ligand is zeolite-Y (Fig. 1c) [26]. However, a slight change was observed in the intensity of the bands. This slight change in the peak intensities suggests that the supercages of the zeolite are able to store these complexes without any strain [1, 6, 23, 27, 28]. The main framework of the zeolite was not damaged. Also, no crystalline patterns were seen for the encapsulated complexes which might be ascribed to their fine distribution in the lattice [23].

XRD patterns of a zeolite-Y, b Co/zeolite-Y, and c [Co-(salen)]-Y
Fig. 1 shows that Co/zeolite-Y samples have the faujasite topological structure with high crystallinity. After complexation of the metal ions with the salen ligand and further Soxhlet extraction, the faujasite structure remained essentially unchanged. This indicates that the complexation and extraction processes have no strong influence on the structure of the parent Co/zeolite-Y materials. A noticeable difference in the XRD patterns of zeolite-Y with the encapsulated complex from that of zeolite-Y is an alteration of the relative intensities of the 220 and 311 reflections (2Θ of about 10° and 12°). For Co/zeolite-Y, I220 was slightly higher than I311, whereas after coordination to salen, I220 became lower than I311. This intensity reversal has already been identified and empirically correlated with the presence of a large transition metal complex ions within the zeolite-Y supercage. Similar observations have been reported before [18, 23, 26] and the above observation may therefore be construed as evidence for the successful encapsulation of salen complex within the supercage of Co/zeolite-Y. Our results showed good agreement with the results previously reported in the literature [18, 23].
All samples showed patterns that could be indexed to zeolite-Y and not to the complex. Therefore, it is infered that the crystallinity and morphology of the zeolite are maintained during the encapsulation [23].
These observations indicated that the zeolite framework undergoes no significant structural change during encupsulation of [Co(salen)] and thus crystallinity of zeolite-Y is mainly preserved. In Co/zeolite-Y and [Co-(salen)]-Y, no new peaks were detected in the XRD patterns which is probably due to poor loading of the metal complexes during encapsulation [15, 23].
Cataliytic activity
Decomposition of H2O2
The catalytic activity for the decomposition of H2O2 with the encapsulated complexes was investigated. The results are summarized in Table 2, which shows that the H2O2 decomposition is slow initially while increases during the reaction. This is due to the fact that the encapsulated complex used requires relatively longer time to exhibit maximum catalytic activity regarding lower number of metal centres compared to the neat complex [24]. Hence, increasing the time period of the reaction is an effective way of reaction progress with H2O2. The results show that the order of catalytic activity for decomposion of H2O2 is found to be as follows:
Hydroxylation of phenol
The liquid-phase hydroxylation of phenol using different catalysts, H2O2 as oxidant and MeCN as the solvent was scrutinized. It is known that encapsulation of metal ions in zeolites can cause abnormal oxidation states/electronic configurations resulting in catalytic activity. Hydrogen peroxide has the advantage of high mobility in the pores of zeolite systems due to its noticeably small size [23].
To determine the performance of the catalysts, the phenol conversion percentage was plotted as a function of time. As expected, only two major products (catechol and hydroquinone) were detected (Scheme 1), as –OH group of phenol is ortho and para position [24]. Other minor products, if any, presented as minor constituents which could not be detected by the gas chromatography under the conditions employed here and hence were neglected. Reaction conditions have been optimized remarking [Co-(salen)]-Y as a representative catalyst and varying different reaction parameters, i.e., the concentration of phenol and oxidant, amount of catalyst, and also the reaction mixture temperature in order to achieve maximum conversion efficiency of phenol and the formation of catechol as well as hydroquinone.

Phenol hydroxylation
Fig. 2 illustrates the effect of catalyst amount on the conversion of phenol as a function of reaction time. Four different amounts including 0.005, 0.01, 0.025, and 0.04 g were used for the constant amounts of phenol (5.67 g) and H2O2 (4.7 g) in 2 mL of MeCN at 80 °C.

Percentage phenol conversion versus time plot of phenol hydroxylation in different amount of [Co(salen)]-Y catalyst [phenol (5.47 g), H2O2 (4.7 g), MeCN (2 mL) at 80 °C at four different amounts of [Co-(salen)]-Y catalyst (0.005, 0.01, 0.025 and 0.04 g)]
It was observed that increasing the amount of catalyst from 0.005 to 0.01 g enhances the phenol conversion. Approximately 2 h resulted in 35 % conversion with 0.005 g catalyst while such conversion was achieved in 80 min using 0.01 g catalyst. The lower conversion of phenol with 0.005 g catalyst may be related to lower number of sites of catalyst. Increasing the amount of catalyst to 0.025 g enhances the conversion of phenol to 43 % while no further improvement was achieved in the conversion through increasing the catalyst amount up to 0.04 g. This has been interpreted in term of thermodynamic and mass-transfer limitations at higher reaction rate [6]. Thus, 0.025 g is the best amount for the phenol oxidation.
The concentration of hydrogen peroxide may also affect the conversion of phenol. Different molar ratios of H2O2:phenol (1:2, 1:1, 2:1) were considered while keeping the amount of phenol (5.67 g) and catalyst (0.025 g) fixed in 2 mL of MeCN. The percentage of phenol conversion after 6 h of the reaction time is compared as shown at Fig. 3. The figure reveals that 1:1 and 2:1 molar ratios score the highest percentage of conversion of about 43 and 45 within 6 h. However, the H2O2 efficiency with 1:1 molar ratio is much higher (~38 %) than 2:1 molar ratio (~24 %).

Percentage phenol conversion versus time plot of phenol hydroxylation in different molar ratios of H2O2: phenol (1:2, 1:1, 2:1) [phenol (5.67), different concentration of H2O2, MeCN (2 mL) at 80 °C and [Co-(salen)]-Y catalyst (0.025 g)]
Having compared these results, it is evident that 1:1 molar ratio is the optimum molar ratio for higher performance of catalyst with eligible percentage H2O2 efficiency and high oxidant concentration is not an essential parameter to maximize the phenol conversion. This is probably due to higher chemisorption of H2O2 and less chemisorption of phenol on the active sites and may be due to the dilution of the reaction mixture by the presence of higher amount of water molecules in H2O2 solution [24, 27, 28]. Fig. 4 shows the hydroxylation of phenol at three different temperatures 50, 65, and 80 °C as a function of time while retaining the optimized conditions of 5.67 g phenol, 4.7 g H2O2 and 0.025 g catalyst in 2 mL MeCN as concluded above. The figure demonstrates that the performance of [Co-(salen)]-Y catalyst is poor at 50 °C (36 %) and it is improved partially at 65 °C (39 %). At 80 °C, however, the performance is much higher with 43 % hydroxylation of phenol and reaches a steady state after about 6 h. Hence, 80 °C is the minimum required temperature to supply sufficient energy to reach the energy barrier of phenol transformation and maximize the conversion of phenol at minimum time [28, 29].

Percentage phenol conversion versus time plot of phenol hydroxylation in different temperatures [phenol (5.67 g), H2O2 (4.7 g), MeCN (2 mL) and [Co-(salen)]-Y catalyst (0.025 g) at four temperatures (50, 65, and 80 °C)]
Catalytic activity of other catalysts
Having obtained the optimized reaction conditions for [Co-(salen)]-Y, other catalysts viz. [Co-(salpn)]-Y and [Co-(salophen)]-Y were also studied under similar reaction conditions (i.e. 5.67 g phenol, 4.7 g H2O2 and 0.025 g catalyst and 2 mL MeCN at 80 °C) for the hydroxylation of phenol. Fig. 5 shows the outcomes of the reactions. Under the optimized reaction conditions, the performance of [Co-(salpn)]-Y, [Co-(salophen)]-Y catalysts in conversion of phenol was noticeably low at the beginning. As time passes, the former catalyst improves the conversion to 38 % in 6 h while the latter conversion was only 35 % in the above mentioned time. On the other hand, the performance of [Co-(salen)]-Y is much better compared to two above catalysts giving more than 36 % conversion of phenol within 1 h of reaction time and reaching the steady state in 6 h with a conversion of 43 %. Thus, the order of performance for three considered catalysts is as follows:

Percentage phenol conversion versus time plot of phenol hydroxylation over the different catalysts [Co-(salen)]-Y, [Co-(salpn)]-Y and [Co-(salpn)]-Y catalyst [phenol (5.67 g), H2O2 (4.7 g), MeCN (2 mL) at 80 °C over 0.025 g of different catalysts]
For the [Co-(salen)]-Y catalyst, the phenol conversion was much higher than that obtained for the other catalysts. This increase in phenol conversion can be explained by the hydrogen peroxide decomposition data as mentioned in previous section. In the presence of available hydrogen peroxide, the [Co-(salen)]-Y catalyst is able to decompose hydrogen peroxide with higher performance compared to the others. Additionally, this catalyst showed higher activities toward the oxidation of phenol compared to the others.
Running the oxidation reaction without any catalyst yielded 15 % conversion, which confirmed the necessity for the catalyst. To understand the key role of zeolite-Y-encapsulated complexes in phenol hydroxylation reaction over heterogeneous catalyst, an experiment was performed with Co/zeolite-Y as catalysts and the results shows only a maximum of 17 % conversion [30].
The metal complexes encapsulated in zeolite-Y have been reported to produce catechol in much higher proportion compared to hydroquinone. The product selectivity of these catalysts is based on the shape and size of the product molecule [13]. The predominance of catechol is not unexpected for metal-mediated hydroxylation occurring through a pathway, which involves initial weak coordination of both phenol and H2O2 to the active site. The anchoring of these two reactants in close proximity to each other leads predominantly to a cis arrangement, which in turn results in ortho-substitution of the phenol [31].
In terms of the catechol and hydroquinone formation, all three catalysts reported here are also highly selective toward the formation of catechol and least selective for hydroquinone after 6 h of reaction. Fig. 6 presents these results for phenol hydroxilation after 6 h of reaction. The figure specifies that the selectivities toward the catechole and hydroquinone are not significantly different using three catalysts (~90 % catechol formation), whereas the transformation of phenol varies from a catalyst to another.

Conversion and selectivity percentage of phenol hydroxylation over 0.025 g of [Co-(salen)]-Y, [Co-(salpn)]-Y and [Co-(salophen)]-Y catalysts [phenol (5.67 mol), H2O2 (4.7 g), MeCN (2 mL) at 80 °C] after 6 h of reaction time
Conclusions
Cobalt(II) complexes of salen, salpn, and salophen were encapsulated in zeolite-Y supercages. FTIR spectroscopy study and XRD pattern confirmed the encapsulation of salen, salpn, and salophen inside the supercage of zeolite-Y. The encapsulated complexes catalyze the oxidation of phenol efficiently using H2O2 as oxidant. The major products were catechol and hydroquinone. The effects of different factors such as the amount of oxidant, catalyst, and phenol, and also the reaction temperature were studied. The encapsulated complexes are active catalysts for the decomposition of H2O2 and also for the oxidation of phenol with high selectivity toward catechol compared to hydroquinone. Among these catalysts and under the best-suited reaction conditions, (i.e. 4.7 g phenol, 5.67 g H2O2,0.025 g catalyst in 2 mL of MeCN at 80 °C), [Co-(salen)]-Y catalyzes the hydroxylation of phenol with highest phenol conversion (~43 %) which is followed by [Co-(salpn)]-Y, which in turn is better than [Co-(salophen)]-Y. Under the optimized reaction conditions, the selectivity of catechol formation is about 90 % with all these catalysts. The performance of Co/zeolite-Y catalyst (without ligands) is of higher importance compared to [Co(ligand)]-Y catalysts, while 1.5 % coversion of phenol achieved when the hydroxylation of phenol reaction was carried out using no catalyst. The results reflect the importance of ligand and catalyst role in the hydroxylation of phenol reaction.
References
Bowers C, Dutta PK (1990) J Catal 122:271–279
Knops-Gerrits P-P, De Vos D, Thibault-Starzyk F, Jacobs PA (1994) Nature 369:543–546
Agarwal DD, Bhatnagar RP, Jain R, Srivastava S (1990) J Chem Soc Perkin Trans 2:989–992
Maurya MR, Chandrakar AK, Chand S (2007) J Mol Catal A 274:192–201
Nethravathi BP, Reddy KR, Mahendra KN (2014) J Porous Mater 21:285–291
Maurya M, Saklani H, Kumar A, Chand S (2004) Catal Lett 93:121–127
Seelan S, Sinha AK (2003) Appl Cat A 238:201–209
Nethravathi B, Mahendra K (2010) J Porous Mater 17:107–113
Nethravathi BP, Mahendra KN, Reddy KR (2011) J Porous Mater 18:389–397
Sheldon RA, van Santen RA (1995) Catalytic oxidation: principles and applications: a course of the Netherlands Institute for Catalysis Research (NIOK). World Scientific, Singapore
Arpe HJ (1999) Industrial organic chemicals: starting materials and intermediates—An Ullmann’s Encyclopedia. Wiley, New York
Maurya MR, Titinchi SJJ, Chand S (2004) J Mol Catal A 214:257–264
Maurya M, Titinchi SJ, Chand S (2003) Catal Lett 89:219–227
Maurya M, Kumar M, Titinchi SJ, Abbo H, Chand S (2003) Catal Lett 86:97–105
Maurya MR, Titinchi SJJ, Chand S, Mishra IM (2002) J Mol Catal A 180:201–209
Jacob CR, Varkey SP, Ratnasamy P (1998) Microporous Mesoporous Mater 22:465–474
Maumy M, Capdevielle P (1996) J Mol Catal A 113:159–166
Bania KK, Bharali D, Viswanathan B, Deka RC (2012) Inorg Chem 51:1657–1674
Sophiphun O, Demir D, Föttinger K, Rupprechter G, Loiha S, Neramittagapong A, Prayoonpokarach S, Wittayakun J (2016) Reac Kinet Mech Cat 117:705–713
Sophiphun O, Föttinger K, Loiha S, Neramittagapong A, Prayoonpokarach S, Rupprechter G, Wittayakun J (2015) Reac Kinet Mech Cat 116:549–561
Peyrovi MH, Mahdavi V, Salehi MA, Mahmoodian R (2005) Catal Commun 6:476–479
Alizadeh M, Farzaneh F, Ghandi M (2003) J Mol Catal A 194:283–287
Güneş A, Bayraktar O, Yılmaz S (2006) Ind Eng Chem Res 45:54–61
Maurya MR, Titinchi SJJ, Chand S (2003) J Mol Catal A 193:165–176
Joseph T, Sajanikumari CS, Deshpande SS, Gopinathan S (1999) Indian J Chem Sect A 38:792–796
Hailu SL, Nair BU, Redi-Abshiro M, Aravindhan R, Diaz I, Tessema M (2015) RSC Adv 5:88636–88645
Hosseini-Ghazvini SMB, Safari P, Mobinikhaledi A, Zendehdel M (2015) Reac Kinet Mech Cat 115:703–718
Salavati-Niasari M, Bazarganipour M (2006) Catal Commun 7:336–343
Maurya MR, Singh B, Adão P, Avecilla F, Costa Pessoa J (2007) Eur J Inorg Chem 2007:5720–5734
Mobinikhaledi A, Zendehdel M, Safari P (2014) J Porous Mater 21:565–577
Mugo JN, Mapolie SF, van Wyk JL (2010) Inorg Chim Acta 363:2643–2651
Acknowledgments
The authors are grateful to the financial support from the Islamic Azad University, South Tehran branch. This study has been done under the format of project research “Zeolite-Y encapsulated metal complexes of Cobalt(II) as catalyst for the hydroxylation of phenol”.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Khodadadi, Z., Mahmoudian, R. Zeolite-Y encapsulated metal complexes of cobalt(II) as catalyst for the hydroxylation of phenol. Reac Kinet Mech Cat 119, 685–697 (2016). https://doi.org/10.1007/s11144-016-1072-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11144-016-1072-z