
Abstract
The Food and Drug Administration (FDA) and European Medicines Agency (EMEA) are willing to consider including information on patient reported outcomes (PROs) in product labeling and advertising. Pharmaceutical industry researchers must provide sufficient evidence supporting PRO benefit before an approval may be granted. This report describes the purpose and content of a PRO Evidence Dossier, which consists of important information supporting PRO claims. The dossier should be completed by pharmaceutical industry or other researchers to document the planning of the PRO assessment strategy, psychometric evidence, desired target labeling statements, and the clinical trial evidence of PRO benefits. The systematic reporting and documentation of information on the rationale for including PROs, rationale for the selection of specific PRO instruments, evidence on the psychometric qualities of the PRO measures, and guidelines for interpreting PRO findings will facilitate achieving a PRO labeling or promotional claim. Combining all the relevant information into a single document will facilitate the review and evaluation process for clinical and regulatory reviewers. The PRO Evidence Dossier may also be helpful to industry and academic researchers in identifying further information that will need to be developed to support the clinical development program and the PRO endpoints.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the past 20 years, clinicians have recognized that understanding the patient’s perspective on the impact of disease and treatment on functioning and well-being is important for pharmaceutical, biologic, and medical device product development and evaluation. Pharmaceutical companies are increasingly incorporating health-related quality of life (HRQL) and other patient-reported outcomes (PROs) into clinical trial programs for new drugs with the expectation that these outcomes will help inform physicians and patients on the comprehensive effects of these treatments [1]. These PROs may be useful in differentiating the patient benefits among competing products with similar clinical efficacy and demonstrating and translating clinical effects into outcomes more meaningful to patients, their families and their treating physicians.
For the health products industry, the intent is to achieve labeling or promotional claims about these PRO benefits, which, if approved by the US Food and Drug Administration (FDA), can be used in marketing activities targeted at physicians and consumers [2]. In 2005, the European Medicines Agency (EMEA) released a reflection paper on the use of HRQL and PRO information in evaluating the efficacy of new medical products [3]. In February 2006 in the US, the FDA developed a draft guidance with recommendations on information on PRO measures needed to support labeling claims for medical products [2]. Although these documents differ in level of detail and substance, each provides a basis for determining PRO related evidentiary needs. Both the European and US regulatory agencies require substantial evidence supporting a PRO related claim and expect this information to be clearly summarized.
To illustrate, in the US the FDA is responsible for reviewing and evaluating the evidence on the safety and efficacy of new medical treatments or devices. Recent changes in FDA legislation have placed more focus on HRQL, patient-reported outcomes, and other endpoints [4]. The FDA views a PRO endpoint as an additional measure of ‘effectiveness– The Federal Food Drug and Cosmetic Act and related regulations require that a PRO claim, like any effectiveness claim, be backed by substantial evidence. The FDA draft guidance on PROs for labeling and promotional claims identifies information needed to evaluate the veracity of a PRO claim [2]. However, given the paucity of accumulated experiences since the inception of the draft guidance, there is uncertainty about how it will be applied.
The health outcomes research community has provided recommendations concerning good measurement science [5–8] and methodological and measurement issues related to evidence for supporting PRO claims [9–11]. Researchers should document the planning process for the PRO assessment strategy, including providing the rationale for the selection of instruments; summarizing psychometric characteristics of the PRO instruments; providing clear interpretation guidelines; and drafting labeling language regarding the PROs. In addition, there must be evidence that an a priori PRO data analysis plan was developed and filed before study unblinding; furthermore, this plan must specify methods for handling missing data and multiplicity and describe the statistical models for comparing treatment differences [10]. Currently, this information is provided across a number of different documents associated with the clinical development plan and clinical trial program, including the end of Phase II related documents, clinical trial protocols, various independent reports, statistical analysis plans, and, once the clinical trial program is completed, the clinical study reports.
This dispersion of relevant PRO related information makes the regulatory reviewer’s task much more difficult. Thus, we recommend integrating all relevant evidence into a single summary document. This is the essence of the PRO Evidence Dossier–a document intended to provide information on the PRO measurement strategy and clinical trial findings, including interpretation of clinical significance. Pharmaceutical and medical device industry personnel and other health researchers under contract to industry should develop this dossier document to illustrate the necessary evidence for substantiating a claim of PRO effectiveness. In this way, the dossier becomes a source for dialogue between the FDA or EMEA and industry researchers focused on the PRO measurement strategy and the benefits of a new treatment.
The PRO dossier is intended for use by researchers interested in including PRO endpoints for evaluating the efficacy of new pharmaceuticals, biologics or medical devices prior to review for regulatory approval. Although PRO data may also be used to document the negative consequences and side effects of a treatment or device, the focus of this paper is on the use of PRO to measure the effectiveness of a new treatment. The remainder of this paper describes the recommended content of the PRO Evidence Dossier.
Pro Evidence Dossier
The purpose of the dossier is to summarize the PRO assessment strategy, evidence on the psychometric qualities of the selected PRO instruments (i.e., reliability, content validity, construct validity, responsiveness), interpretation guidelines (i.e., minimal important difference (MID)), summary of clinical trial results, and requested PRO labeling language. The intent is to inform the reviewer about the planned PRO assessment strategy associated with a clinical development program. The seven components of the PRO Evidence Dossier are (1) rationale for measuring PRO endpoints; (2) rationale and selection of PRO instruments; (3) background on the development of the PRO Instruments; (4) summary of psychometric characteristics of the PRO instruments; (5) interpretation guidelines and minimal important differences; (6) summary of clinical trial results; and (7) requested PRO labeling statements. Each of these seven sections of the dossier will be briefly described below.
Rationale for measuring PRO endpoints
PRO endpoints are often incorporated into clinical trials and into the clinical development programs for new medical treatments and rationale for including these endpoints should be provided by researchers. PRO measures should be considered for all clinical development programs, and, if not included, some justification should be provided. However, preparation of the PRO Evidence Dossier assumes that the investigators have decided to incorporate PRO endpoints into the clinical trial program. This section of the dossier should provide the rationale for including PRO endpoints in the clinical development program. The rationale should be based on the medical and health outcomes literature and other available information as to why PROs are important for understanding the outcomes of treatment for the targeted disease indication. The researcher should identify those PRO domains that are salient for the disease.
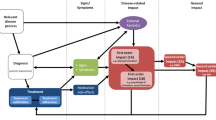
The FDA guidance recommends including an endpoint model and conceptual framework for the PROs as part of any claims submission [2]. The endpoint model provides insight into the proximal and distal nature of the relationships between the clinical and PRO measures within treatment and disease progression context [6, 12]. The conceptual framework links the individual items and PRO domains, and if included, the domains with overall summary scores. Evidence supporting the PRO rationale should be gathered based on the epidemiologic data on the disease, clinical understanding of the condition, and previous medical and health outcomes research. Documentation from qualitative research on involved stakeholder groups (i.e., patients and their families, clinicians, others) may be used as a basis for this rationale. This information needs to be summarized to provide the rationale and justification for measuring PROs for the targeted disease. This section of the dossier should also provide some insight into the important domains of health outcomes for the targeted disease population.
Rationale and selection of PRO instruments
In many cases, multiple potentially acceptable PRO instruments are available for assessing the domains identified during the planning of the PRO strategy for a clinical development program. In this section, the researcher should document the rationale for the selection of specific PRO measures. The rationale should consider to what extent the objectives of the clinical trial program and other important domains that need to be measured are reflected in the content of the PRO measures [9, 10]. Consideration needs to be taken in selecting instruments that assess the relevant part of the health outcome continuum in the targeted patient population. In addition, the researcher should review the psychometric characteristics (i.e., reliability, validity, responsiveness), language translations for international studies, relevant recall period for the population and disease, and the timing of PRO assessment in the clinical trials. The support provided should illustrate the rationale for use of PROs in general and justify the selection of specific PRO instrument(s) in a way that is convincing to a health outcomes or clinical researcher.
Background on the development of the PRO instruments
Based on the instrument manual, publications on the development of the PRO instrument, and if needed, contact with the instrument developer(s), a brief summary should be developed documenting the instrument development methods and procedures. Evidence supporting a systematic development approach is especially critical for recently developed PRO measures. This section should cover the methods used to identify the key outcome domains, item generation and reduction procedures, and scoring subscale and total scores. Since the FDA view is that PRO instruments should be based on significant patient input [2], there should be evidence that the content, domains and items in the PRO instrument are derived from patient focus groups and/or interviews (see section on content validity below). For well established and accepted measures, this section can be fairly brief, but still needs to provide the needed information.
Summary of psychometric characteristics of the PRO instruments
The measurement qualities of the PRO instruments for the PRO claim need to be summarized in sufficient detail to allow an experienced researcher to fully understand the instrument’s psychometric characteristics. This section of the PRO Evidence Dossier should include a summary of the available evidence on the content validity, reliability, construct validity, and responsiveness of the PRO measures. This information should be derived from the medical literature and targeted independent studies. In addition, blinded analysis of clinical trial data may be used to further confirm the psychometric characteristics of the PRO instruments in the targeted patient population. The relevant information on psychometric qualities of the measure(s) should be based on patient samples that are comparable to those in the clinical trials, or at least similar enough so that the psychometric evidence is generalizable to the targeted population.
Content validity
For content validity, information should be included on the original development of the PRO instruments and the involvement of patients and clinicians in identifying and confirming the content of the measure. This evidence should be generated from physician focus groups, interviews, or surveys and from patient focus groups, interviews or surveys performed during the instrument development process or through independent studies. The FDA views the patient perspective as most critical for PROs [2]; thus, patient-derived information for generating or confirming the content of the PRO measure is of primary importance.
There is little guidance as to how much information is enough, and most data from focus groups and cognitive interviewing related to PRO instruments are based on relatively small samples. However, confirmation of relevant content and domain coverage can be obtained from larger patient surveys. Evaluation of content validity is more qualitative, and the evidence supplied needs to be sufficient enough to demonstrate that the content of the construct or domain is covered adequately. In addition, content validity–the coverage of the construct or domain–differs from face validity [13, 14] and must be examined for the PRO measures.
Reliability
Reliability for PRO measures is demonstrated through internal consistency reliability and test–retest reliability data. All the available information on reliability should be summarized. A table can be used to summarize the reliability evidence, especially when extensive data are available, such as for the St George’s Respiratory Questionnaire [15] or SF-36 Health Survey [16]. Although reliability evidence across populations is of interest, the focus should be on showing reliability in the targeted patient population.
Construct validity
The construct validity of the PRO instruments should be summarized for the target patient population. Validity refers to the degree to which the measure reflects what it is supposed to measure rather than something else [7]. Validity varies by patient population and application, and validation is an ongoing process of accumulating evidence supporting the PRO measure over multiple studies. For example, the SF-36 may have demonstrated validity for applications in the general and chronic disease population in community settings but may lack evidence of validity for applications in nursing home patients. There are few guidelines as to how much validity evidence is enough [5]; however, current recommendations indicate that the more evidence the better [7, 10].
The evidence supporting construct validity of the PRO measure should include attention to convergent, divergent, discriminant, and known groups validity. The information presented should be sufficient to convince reviewers that the PRO instrument is operating as expected. This includes demonstrating that the instrument is related to clinical and other PRO measures in meaningful ways and that these associations are of the direction and magnitude hypothesized. The researcher should provide a text summary of the validity evidence and highlight relevant validity findings in tables or figures. If the PRO measure has been used previously in clinical trials, this information is important to provide as supportive evidence of validity and responsiveness (see below).
Responsiveness
Responsiveness is a component of validity and represents the PRO measure’s capability to detect changes in clinical status or other relevant outcome measures. This section of the document should summarize the evidence that the PRO scores are responsive or sensitive to changes in clinical status. Evidence from previous clinical trials should be reviewed and summarized in this part of the dossier. The responsiveness of PROs is evaluated using multiple approaches, such as the application of a treatment of known efficacy, relating changes in the PRO to changes in clinical status, and in relating changes in the PRO to patient- or clinician-rated changes in clinical status. Responsiveness is critical for supporting any claim in randomized clinical trials and is tied to demonstrations of clinical significance and MID.
The psychometric evidence section of the dossier should be assembled early in the planning of the clinical trial program. An assessment of the completeness of the psychometric information on the selected PRO measures will help in planning studies to provide additional measurement evidence. For example, new disease-specific instruments may have little data available as to responsiveness and MID. Therefore, the sponsor may need to plan and complete a study designed to provide responsiveness and MID information as well as additional data on reliability and validity. Blinded secondary analyses of the PRO data collected in Phase II studies can also be used to examine psychometric characteristics.
Interpretation guidelines and MID
This section of the dossier provides guidance to those reviewing the clinical trial results as to whether statistically significant group differences or changes are meaningful [17–19]. The MID is the smallest change that patients perceive as beneficial or which would require a change in clinical management [17]. To assist in understanding the importance of statistically significant PRO results, information should be provided as to the MID for the primary PRO endpoints that will be used to make labeling or promotional claims [2]. The MID information should be determined primarily using anchor-based methods, with the results from distribution-based methods used as supportive data [17–19].
The MIDs for each of the primary PRO endpoints should be summarized based on the medical and health outcomes research literature or independently conducted studies designed to estimate MIDs. MIDs can also be based on blinded analyses of Phase II clinical trial data. However, MID data from sources independent of the clinical trial program may be more meaningful and more likely to be accepted by regulatory agencies. In the absence of good evidence on MID, the more conservative 1/2 standard deviation approach might be specified [18].
The MID section of the dossier should specify a numeric value for the MID and summarize the rationale and evidence supporting this MID value. Guidance on MIDs should be provided for all the pre-specified primary PRO endpoints.
Summary of clinical trial results
This section is focused on summarizing the PRO findings from all clinical trials submitted to regulatory agencies. The emphasis should be on information and results that may assist the reviewer in evaluating whether or not the PRO results are scientifically adequate (i.e., substantive evidence) in supporting a labeling or promotional claim. This summary is not expected to substitute for the full clinical study reports or peer-reviewed publications; rather, it is intended to complement the study reports and bring attention on the primary PRO endpoints. This section should include a brief summary of the clinical trial protocols, the clinical efficacy findings, primary PRO endpoints, and any secondary PRO endpoints that may be useful to support results of the primary PRO endpoints. The clinical trial summaries should provide information on the research protocols, the timing of PRO assessments, statistical power, and other relevant information [21, 22]. Attention should be given to whether the clinical trials were designed to evaluate superiority or non-inferiority between treatments.
The researcher should provide information demonstrating consistency of results across the clinical and PRO endpoints, as contradictory results may lead regulatory agency reviewers to have difficulty accepting PRO findings. Appropriate references should be made to the more detailed data and results contained in the clinical study reports. The PRO clinical trial results should be displayed in well-conceived and clear tables and figures [21]. The primary focus of this summary is to demonstrate the evidence from the clinical trials supporting the treatment’s impact on the PRO endpoints. This information supplies the critical foundation for supporting the PRO labeling claim.
Requested PRO labeling statements
The final section of the PRO Evidence Dossier should contain the desired labeling or promotional claim statements related to the PRO endpoints. The FDA has suggested that it is important to identify the intended targeted claim early in the clinical trial program to allow the agency to understand and evaluate the sponsor’s plans [2]. Clearly, the final claim statement should not be drafted until the PRO results are known. The claim language should be based on the content of the pre-specified primary PRO endpoints in the clinical trial protocol and statistical analysis plan [2].
For regulatory agencies, the claim statements are driven by the PRO results, based on statistical significance and meeting the specified MID criteria as well as the content of the PRO measures. The claim statements should be developed to clearly communicate the PRO findings to physicians and patients. General statements, such as ‘quality of life– ‘overall well-being– etc., are difficult to substantiate, and for many reviewers these types of statements are too ambiguous. Achieving a health-related quality of life claim will likely require consistent and positive findings across multiple domains of a health outcome measure (or at least mostly positive results with some no difference results).
In general, simple and straightforward statements are included in product labels, such as those related to relief from pain or improving physical function in labels for products for treating rheumatoid arthritis. PRO-related findings may be included in the indication, such as this example for remicade: “Remicade, in combination with methotrexate, is indicated for reducing signs and symptoms, inhibiting the progression of structural damage, and improving physical function in patients with moderately to severely active rheumatoid arthritis.–Most often, however, PRO evidence is included in the clinical studies section of the label [1] or in Summary of Product Characteristics (SPC) [23].
When developing the PRO label statements, we recommend constructing simple and clear statements of fact. For example, ‘In two randomized clinical trials, Product X improved physical functioning compared to placebo in patients with rheumatoid arthritis.–Review of previously accepted label statements for similar indications is useful to provide insight into potentially acceptable language on PRO results. Regulatory agencies evaluate statements for instances of puffery (i.e., statements that expand and generalize beyond the results) for PROs as well as other effectiveness endpoints. These kinds of claim statements will be denied for incorporation into product labels or SPCs. Care should be taken to ensure that PRO statements are clear and based on the clinical trial evidence.
Summary
This paper describes the purpose and content of a PRO Evidence Dossier supporting PRO claims to regulatory agencies. This dossier can provide documentation related to the planning of the PRO assessment strategy, desired labeling statements, and summaries of the clinical trial evidence of PRO benefits. Achievement of a PRO labeling or promotional claim is facilitated through the systematic reporting and documentation of information on the rationale for including PROs, the rationale for the selection of specific PRO instruments, the evidence on the psychometric qualities of the PRO measures, and guidelines for interpreting PRO findings.
Combining all the relevant information and evidence into a single document may make the review and evaluation of the PRO evidence easier for the clinical and regulatory reviewers within the FDA, EMEA and other agencies. The PRO Evidence Dossier should also be considered a living document in that it will function as a record of key elements in the PRO assessment and development strategy. The dossier can be updated and revised by researchers within the industry as new information is developed or located in the published literature. The PRO Evidence Dossier may also help industry and contracted researchers in identifying information that will need to be further developed to support the clinical development program and targeted PRO claims.
The PRO Evidence Dossier is typically developed for reviewers in the health products industry and regulatory agencies; however, this document may also be helpful to health insurers, health care organizations and others involved in evidence-based medicine. PROs provide a valuable indicator of treatment benefit for health care organizations, and the PRO dossier can provide a useful framework for assessing the efficacy and effectiveness of health care interventions for health care organizations. Although some of the PRO information is included in product dossiers for formulary and technology related decision making [24, 25], these data are usually only briefly summarized and the focus is most often on clinical efficacy, safety and economic outcomes. Health care decision makers may be able to use the information summarized in the dossier to make more informed decisions about the health outcome benefits of different interventions.
References
Wilke, R. J., Burke, L. B., & Erickson, P. (2004). Measuring treatment impact: A review of patient-reported outcomes and other efficacy endpoints in approved labels. Control Clinical Trials, 25, 535–52.
Food and Drug Administration. (February 2006). Guidance for industry –patient-reported outcome measures: Use in medical product development to support labeling claims. Silver Spring, MD: FDA.
Committee for Medicinal Products for Human Use. (July 2005). Reflection Paper on the regulatory guidance for the use of Health-Related Quality of Life (HRQL) measures in the evaluation of medicinal products. London: EMEA.
Food and Drug Administration Modernization Act, 1997. Available at http://www.fda.gov/cder/guidance. Accessed September 9, 2004.
Lohr, K. (2002). Assessing health status and quality-of-life instruments: Attributes and review criteria. Quality of Life Research, 11, 193–05.
Patrick, D. L., & Chiang, Y. P. (2000). Measurement of health outcomes in treatment effectiveness evaluations: Conceptual and methodological challenges. Medical Care, 38, (9 Suppl): II14–5.
Hays, R., & Revicki, D. A. (2005). Reliability and validity, including responsiveness. In P. Fayers, & R. Hays (Eds.), Assessing quality of life in clinical trials (2nd ed.). New York: Oxford University Press.
Fayers, P., & Hays, R. (Eds.), (2005). Assessing quality of life in clinical trials (2nd ed.). New York: Oxford University Press.
Leidy, N. K., Revicki, D. A., & Geneste, B. (1999). Recommendations for evaluating the validity of quality of life claims for labeling and promotion. Value in Health, 2, 113–27.
Revicki, D. A., Osoba, D., Fairclough, D., Barofsky, I., Berzon, R., Leidy, N. K., & Rothman M. (2000). Recommendations on health related quality of life research to support labeling and promotional claims in the United States. Quality of Life Research, 9, 887–00.
Acquadro, C., Berzon, R., Dubois, D., Kline Leidy, N., Marquis, P., Revicki, D., & Rothman, M. (2003). Incorporating the patient’s perspective into drug development and communication: An ad hoc task force report of the patient-reported outcomes (PRO) harmonization group meeting at the Food and Drug Administration, February 16, 2001. Value in Health, 6, 522–31.
Wilson, I. B. & Cleary, P. D. (1995). Linking clinical variables with health-related quality of life A conceptual model of patient outcomes. JAMA, 273, (1):59–5.
Lynn, M. R. (1986). Determination and quantification of content validity. Nursing Research, 35, 382–85.
Guilford, J. P. (1954). Psychometric methods New York: McGraw-Hill.
Jones, P. W., Quirk, F. H., Baveystock, C. M., & Littlejohns, P. A. (1992). Self-completed measure for chronic airflow limitation –the St George’s Respiratory Questionnaire. American Review of Respiratory Disease, 145, 1321–327.
Ware, J. E., Snow, K. K., Kosinski, M., & Gandek, B. (1993). SF-36 Health survey: Manual and interpretation guide. Boston, Massachusetts: The Health Institute, New England Medical Center.
Guyatt, G., Osoba, D., Wu, A., Wyrwich, K., & Norman, G. (2002) Methods to explain the clinical significance of health status measures. Mayo Clinic Proceedings, 77, 371–83.
Sloan, J. A., Cella, D., & Hays, R. D. (2005). Clinical significance of patient-reported questionnaire data: Another step toward consensus. Journal of Clinical Epidemiology, 58, 1217–219.
Revicki, D. A., Cella, D., Hays, R. D., Sloan, J. A., Lenderking, W. R., & Aaronson, N. K. (2006). Responsiveness and minimal important differences for patient reported outcomes. Health Quality of Life Outcomes, 4, 70.
Wyrwich, K. W., Bullinger, M., Aaronson, N., Hays, R. D., Patrick, D. L., Symonds, T., & Sloan, J. A. (2005). Estimating clinically significant differences in quality of life outcomes. Quality of Life Research, 14, 285–95.
Revicki, D. A. (2000). Reporting analyses for clinical trials. In P. Fayers, & R. Hays, (Eds.), Assessing quality of life in clinical trials 2nd ed.). New York: Oxford University Press.
Efficace, F., Bottomly, A., Osoba, D., Gotay, C., Flechtner, H., D’Haese, S., & Zurlo, A. (2003). Beyond the development of health-related quality-of-life (HRQOL) measures: A checklist for evaluating HRQOL outcomes in cancer clinical trials–does HRQOL evaluation in prostate cancer research inform clinical decision making? Journal of Clinical Oncology, 21, 3502–511.
Szende, A., Leidy, N. K., & Revicki, D. A. (2005). Health-related quality of life and other patient-reported outcomes in the European centralized drug regulatory process: A review of guidance documents and performed authorizations of medicinal products 1995 to 2003. Value in Health, 8, 534–48.
Academy of Managed Care Pharmacy. (2005). The AMCP format for formulary submissions version 2.1. Alexandria, VA: Academy of managed Care Pharmacy.
National Institute for Clinical Excellence (2004). A guide for manufacturers and sponsors contributing to a technology appraisal London: National Institute for Clinical Excellence.
Acknowledgements
The authors would like to thank the anonymous reviewers and Kathy Lohr, PhD for their helpful comments toward improving this paper. We also appreciate the careful review and editing by Chris Sexton, PhD.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Revicki, D.A., Gnanasakthy, A. & Weinfurt, K. Documenting the rationale and psychometric characteristics of patient reported outcomes for labeling and promotional claims: the PRO Evidence Dossier. Qual Life Res 16, 717–723 (2007). https://doi.org/10.1007/s11136-006-9153-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11136-006-9153-5