
Abstract
High-light-inducible proteins (Hlips) are single-helix transmembrane proteins that are essential for the survival of cyanobacteria under stress conditions. The model cyanobacterium Synechocystis sp. PCC 6803 contains four Hlip isoforms (HliA-D) that associate with Photosystem II (PSII) during its assembly. HliC and HliD are known to form pigmented (hetero)dimers that associate with the newly synthesized PSII reaction center protein D1 in a configuration that allows thermal dissipation of excitation energy. Thus, it is expected that they photoprotect the early steps of PSII biogenesis. HliA and HliB, on the other hand, bind the PSII inner antenna protein CP47, but the mode of interaction and pigment binding have not been resolved. Here, we isolated His-tagged HliA and HliB from Synechocystis and show that these two very similar Hlips do not interact with each other as anticipated, rather they form HliAC and HliBC heterodimers. Both dimers bind Chl and β-carotene in a quenching conformation and associate with the CP47 assembly module as well as later PSII assembly intermediates containing CP47. In the absence of HliC, the cellular levels of HliA and HliB were reduced, and both bound atypically to HliD. We postulate a model in which HliAC-, HliBC-, and HliDC-dimers are the functional Hlip units in Synechocystis. The smallest Hlip, HliC, acts as a ‘generalist’ that prevents unspecific dimerization of PSII assembly intermediates, while the N-termini of ‘specialists’ (HliA, B or D) dictate interactions with proteins other than Hlips.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The structure and function of photosynthetic reaction centers are highly conserved between all oxygenic phototrophs. Photosystem I (PSI) consists of two large pigment-binding reaction center proteins (PsaA and PsaB) and ten to fourteen smaller subunits depending on the species (Jordan et al. 2001; Ben-Shem et al. 2003; Mazor et al. 2017; Malavath et al. 2018). Photosystem II (PSII) reaction centers, on the other hand, contain four major pigment-binding proteins (D1, D2, CP43, CP47) in addition to sixteen smaller membrane spanning subunits and three extrinsic lumenal subunits of the oxygen-evolving complex (Umena et al. 2011; Wei et al. 2016). The PSII reaction center proteins D1 and D2 contain the chlorophylls (Chls) and pheophytins responsible for primary charge separation, while the two inner antenna proteins, CP43 and CP47, transfer captured light energy to the reaction center. Both photosystems utilize additional light-harvesting antenna systems to increase their light-harvesting capacity. Cyanobacteria contain the peripheral phycobilisome antennae, whereas in plants and different types of algae, the outer light-harvesting antennae of PSI and PSII are composed of membrane embedded light-harvesting complex proteins (LHCs). LHCs belong to the LHC superfamily, which is characterized by a highly conserved Chl-binding motif (ExxN/HxR; Engelken et al. 2010). LHCs contain this motif in two of their three transmembrane helices. The conserved Glu and Arg residues from one helix form salt bridges with the Arg and Glu residues of the other helix, respectively, locking the helices in a cross formation that is further stabilized by associated carotenoid (Car) and Chl molecules (Kühlbrandt et al. 1994; Bassi et al. 1999; Liu et al. 2004).
LHC superfamily proteins are present in all oxygenic phototrophs, but they vary in the number of transmembrane helices and Chl-binding motifs, and not all of them function in light harvesting (Engelken et al. 2010). The most ancient LHC proteins are cyanobacterial high-light-inducible proteins (Hlips), which contain a single transmembrane helix with the ExxNxR motif. Homologues of Hlips called one-helix proteins (OHPs; an alternative name Ycf17 is used in some algae) also seem to be ubiquitous in oxygenic eukaryotes (Engelken et al. 2010). The model cyanobacterium Synechocystis sp. PCC 6803 (hereafter Synechocystis) contains four Hlip isoforms (HliA-D) and one Hlip-like domain fused to the ferrochelatase enzyme (Pazderník et al. 2019). Based on known LHC structures, it has been proposed that Hlips and OHPs form pigment-binding dimers (Staleva et al. 2015; Hey and Grimm 2018). Indeed, pigment binding has been demonstrated experimentally for purified Synechocystis HliC- and HliD-dimers (Staleva et al. 2015; Shukla et al. 2018) and in vitro refolded plant OHPs (Hey and Grimm 2020).
Expression of Hlip genes is highly induced by stress conditions (Dolganov et al. 1995; He et al. 2001) and, as shown by studies made on ΔHlip strains, Hlips are essential for cell survival already under moderately high light intensities (He et al. 2001; Havaux et al. 2003). The exact mechanism of how Hlips ensure cell survival under adverse conditions remains unclear, but all four Synechocystis Hlips have been indicated in one way or another in PSII assembly (Komenda and Sobotka 2016). PSII assembly is a complicated and highly regulated process that is initiated by the formation of four separate PSII assembly modules. Each module contains one major Chl-binding PSII subunit (D1, D2, CP43 or CP47) preloaded with pigments and subjoined by small PSII subunits. The modules then assemble in a stepwise manner forming several distinct PSII assembly intermediates (Komenda et al. 2012b). First, the D1 and D2 modules dimerize to form the PSII reaction center core (RCII). Next, the CP47 module (CP47m) attaches to RCII, yielding the RC47 assembly intermediate (Dobáková et al. 2009; Boehm et al. 2012). The addition of the CP43 module to RC47 results in the monomeric (non-oxygen-evolving) PSII reaction center core complex (RCCII). Finally, the lumenal oxygen-evolving complex is assembled and the resulting PSII-monomers dimerize to the fully active PSII-dimer (Becker et al. 2011).
In addition to de novo biosynthesis, PSII also undergoes rapid repair cycles in the cell due to its susceptibility to photo-oxidative damage (Vass 2012). It is likely that PSII assembly and repair utilize, at least partially, the same accessory proteins and involve similar intermediary complexes (Komenda et al. 2012b; Järvi et al. 2015). Both PSII assembly and repair rely on the assistance of various proteins (assembly factors) that are not part of the active PSII complex. These factors bind to the assembly or repair intermediates during different stages: for instance, the RCII complex is associated with lumenal proteins Ycf48 and CyanoP, whereas the later PSII intermediates contain Psb28 and Psb27 factors connected to CP47 and CP43 antenna subunits, respectively. Synechocystis HliC and HliD form a heterodimer that binds tightly to the PSII assembly factor Ycf39; the resulting HliCD-Ycf39 subcomplex interacts with RCII, giving rise to a complex called RCII* (Knoppová et al. 2014). The HliCD-dimer is capable of quenching light energy via energy transfer from Chl to β-carotene (β-Car) (Staleva et al. 2015), and thus very likely protects the newly forming reaction center against photo-oxidation. Moreover, it has been proposed that HliCD-Ycf39 aids in the delivery of Chl to the nascent D1 reaction center protein, as another protein partner of HliCD is the Chl synthase enzyme (ChlG) (Chidgey et al. 2014). In the course of PSII biosynthesis, the HliCD-Ycf39 subcomplex is detached prior to the formation of RC47 (Knoppová et al. 2014). Similar to HliC and HliD, plant OHPs bind the plant homologue of Ycf39 (HCF244) and associate with RCII (Myouga et al. 2018; Hey and Grimm 2018; Li et al. 2019).
In contrast to HliC and HliD, much less is known about the HliA and HliB proteins. In Synechocystis, HliA, HliB, and HliC associate with the CP47 antenna, but the stage(s) of PSII biogenesis at which this occurs remain to be clarified (Promnares et al. 2006; Yao et al. 2007; Pascual-Aznar et al. 2021). In addition, whether HliA and HliB bind pigments or interact with other Hlips is not known. In this study, we characterized HliA- and HliB-associated protein complexes purified from Synechocystis. We found that both HliA and HliB form heterodimers with HliC and that the resulting HliAC and HliBC pairs bind Chl and β-Car in a quenched configuration. Both dimers associate with the CP47m as well as later PSII assembly intermediates, RC47 and RCCII. Using a detailed mass spectrometry analysis of the isolated HliA/B complexes, we identified several previously uncharacterized proteins that potentially participate in PSII biogenesis. The results presented here make a complete picture of the Synechocystis Hlip ‘interactome.’
Results
HliAC and HliBC heterodimers bind pigments in an energy-dissipating configuration
To study the biochemical properties of HliA and HliB, we expressed N-terminally His-tagged recombinant proteins (9.0 kDa and 8.7 kDa, respectively) in Synechocystis cells under the strong, high-light-inducible psbA2 promoter and deleted the respective native genes to create the strains his-hliA/ΔhliA and his-hliB/ΔhliB. We tested the accumulation of recombinant proteins by shifting the cells from normal light (40 µmol m−2 s−1) to high light (500 µmol m−2 s−1) for up to 5 h. Cellular levels of His-HliA and His-HliB proteins were comparable with their native counterparts in wild-type cells (WT) (Supplemental Fig. 1) confirming that the ectopically expressed His-HliA/B were present in membranes at physiologically relevant concentrations. For all of the following experiments, we used a membrane fraction isolated from cells treated by high light for 3 h.
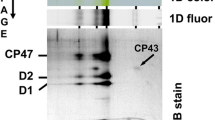
In order to establish whether HliA and HliB bind pigments, we first purified the His-tagged Hlips from n-dodecyl β-d-maltoside (DDM)-solubilized membranes in phosphate buffer (pH 7.8) containing no Mg2+ and Ca2+ ions; these conditions, according to our previous results, destabilize the interaction between Hlips and PSII subunits allowing isolation of free Hlip-dimers (Shukla et al. 2018). His-HliA/B eluates showed a similar yellow-green color and, as revealed by 2D clear native (CN)/SDS-electrophoresis, they contained mostly free Hlip-dimers composed of the His-tagged bait proteins in complex with HliC (5.2 kDa; Fig. 1). Quantification of the proteins from the Coomassie-stained 2D gel yielded a ratio of 1:0.6 for His-HliA and HliC (Fig. 1a), and 1:0.7 for His-HliB and HliC (Fig. 1b). We did not detect the native HliA in His-HliB pull-down nor the native HliB in His-HliA pull-down. HliD was not present in either eluate. Based on these results, we concluded that HliC is the preferred Hlip interacting with HliA and HliB in vivo. The eluates also contained low amounts of the RCCII and RC47 assembly complexes, as revealed by Chl fluorescence imaging of the CN strips and the pattern of PSII reaction center proteins on the 2D gels. Importantly, free Hlips co-migrated with pigments but did not show any Chl fluorescence in the CN gel, indicating that they were present in a quenched conformation.

Two-dimensional CN/SDS-PAGE separation of Ni-NTA pull-downs from his-hliA/ΔhliA (a) and his-hliB/ΔhliB (b) strains Hlip complexes were purified in phosphate buffer from the membrane fractions of high-light -treated cells and separated by 2D CN/SDS-PAGE. The pigment–protein bands containing (His-)HliAC and (His-)HliBC heterodimers that were cut for absorption spectrum and pigment measurements (Fig. 2; Table 1) are indicated by dashed-line boxes. His-HliA, His-HliB, and HliC abundances were quantified from the Coomassie-stained gels and the raw intensities were normalized to the molecular weights of the proteins. The molar ratios of the proteins are indicated in brackets. CBB coomassie stain, Fluor Chl fluorescence, u.p. unbound pigments
To characterize the (His-)HliAC and (His-)HliBC-dimers further, we cut the Hlip-bands from the CN gel strips as indicated (Fig. 1) and measured their in-gel absorption spectra (Fig. 2a). The spectra were almost identical with Chl absorption maxima at 437 nm and 677 nm and a Car peak around 488 nm with a shoulder at 520 nm (Fig. 2a). Next, we eluted the Hlips from the gel, extracted pigments into methanol and quantified individual pigment species by HPLC (Fig. 2b, Table 1). In addition to Chl and β-Car as the main pigments, both Hlip-dimers contained small amounts of echinenone and β-cryptoxanthin. The Chl to total Car ratio in (His-)HliAC- and (His-)HliBC-dimers was 1:0.6.

Absorption spectra (a) and pigment analysis (b) of purified Hlips. a (His-)HliAC- and (His-)HliBC-dimers were cut from CN-PAGE (see Fig. 1) and in-gel absorption spectra recorded. Spectra were normalized to Chl QY band (677 nm). Absorption maxima of Chl a (437 nm and 677 nm) and Cars (488 nm and 520 nm) are indicated above the spectra. Spectra represent an average from three independent purifications. b Pigments were eluted from the CN-PAGE separated Hlips (see Fig. 1) and analyzed with HPLC. The eluted pigments were quantified (see Table 1) and the relative amounts compared to Chl calculated (indicated in brackets). Ech echinenone, a.u. arbitrary units
HliA and HliB are specifically associated with the assembling PSII complexes
To clarify the association of HliA and HliB with different PSII (sub)complexes, we separated solubilized cellular membranes by 2D CN/SDS-PAGE and immunodetected both Hlips and their expected interaction partner, CP47 (Supplemental Fig. 2). However, this time we used a mildly acidic MES buffer (pH 6.5) supplemented with Mg2+ and Ca2+ for membrane isolation and solubilized the thylakoids using a combination of DDM together with the very mild detergent glycodiosgenin (GDN) in order to stabilize protein–protein interactions (Chae et al. 2012). The majority of Hlips in WT co-migrated with the RCCII complex and CP47m (Supplemental Fig. 2a) in agreement with previous findings (Promnares et al. 2006). Notably, CP47m was observed as at least three distinct spots with Hlips present as a diffuse signal around the two higher molecular weight forms. According to published results (Boehm et al. 2011; Pascual-Aznar et al. 2021), the fastest migrating CP47m spot corresponds to a ‘standard’ CP47m complex containing CP47 subjoined with the small PSII subunits PsbH, PsbL, and PsbT. A fraction of CP47m further associates with the small Psb35 protein and the mass difference between the RC47m and RC47m-Psb35 complexes is clearly resolved by CN gel (Pascual-Aznar et al. 2021). Thus, it seems likely that the higher MW CP47m forms detected here represent CP47m + Hlips with and without Psb35. Minor HliA/B signals were also observed around the RC47 complex and in the high molecular weight region. Notably, almost no Hlips were detected as free dimers, indicating that our solubilization conditions preserved the native interactions of Hlips.
To differentiate between the highly similar HliA and HliB, which react with the same antibody, we also utilized the single-mutant strains ΔhliA and ΔhliB (Supplemental Fig. 2b, c). Both Hlips had a very similar distribution in the gel, with the majority of the protein found in RCCII and CP47m. Both Hlips were also observed in the two higher molecular weight spots of CP47m as in WT. Interestingly, a significant amount of HliA was also observed in the high molecular weight region at the top of the gel, whereas HliB was more enriched around CP47m. Neither Hlip was present in high quantities in the dimeric PSII, suggesting a preferential binding to assembly and/or repair complexes. To study the effect of HliC on HliA/B localization, we created ΔhliA/ΔhliC and ΔhliB/ΔhliC strains and repeated the experiment (Supplemental Fig. 2d, e). The signal of immunodetected Hlips was very faint, indicating an important role of HliC for the stability of HliA/B (see later). The Hlips also dispersed to multiple (unidentified) protein complexes that seemed to be distinct from their native localization, suggesting that HliC is important not only for HliA/B accumulation, but also for their proper targeting in the cell.
To obtain more detailed insight on the HliA/B interactomes and their dependency on HliC, we isolated thylakoid membranes and performed pull-downs from high-light-treated WT, his-hliA/ΔhliA, his-hliB/ΔhliB, his-hliA/ΔhliA/ΔhliC, and his-hliB/ΔhliB/ΔhliC strains. In order to preserve a maximal amount of native interactions, we performed the pull-downs in similar mild conditions as was used for the 2D-analysis of thylakoids (solubilization with DDM/GDN in MES buffer). The membranes were first analyzed by 1D SDS-PAGE followed by immunodetection to compare the levels of (His-)HliA/B proteins in the strains (Fig. 3a). Notably, the presence of His-HliA and native HliA in the membranes lowered dramatically after deletion of the hliC gene. Amounts of His-HliB and native HliB were also significantly reduced in ΔhliC, but to a lesser extent than (His-)HliA. We then analyzed the Ni-NTA pull-downs by CN-PAGE (Fig. 3b). Four green bands co-eluted specifically with His-HliA and His-HliB proteins and an additional, fast migrating green-yellow band was visible in the His-HliB eluate. In accordance with the dramatic reduction in His-HliA/B levels upon deletion of hliC, the eluates from ΔhliC background contained significantly less pigment–protein complexes; while some complexes were still present in the his-hliB/ΔhliB/ΔhliC pull-down, no visible bands were detected in the CN-separated his-hliA/ΔhliA/ΔhliC eluate (Fig. 3b).

Accumulation of Hlips in the thylakoid membranes (a) and CN-PAGE separation of Ni-NTA pull-downs (b) from WT, his-hliA/ΔhliA, his-hliB/ΔhliB, his-hliA/ΔhliA/ΔhliC, and his-hliB/ΔhliB/ΔhliC strains. a Membranes from high-light-treated cells were isolated in MES buffer, separated on SDS-PAGE and immunoblotted using an antibody against HliA/B. The different Hlip-forms are indicated with arrows. Total staining of the gel with SYPRO® Orange prior to blotting is shown as loading control. b Hlip complexes were purified in MES buffer using a combination of DDM and GDN detergents. The eluates were separated on CN-PAGE and the gel was scanned. Protein complexes were identified based on 2D-analysis of the gel strips (see Fig. 4). PSI[3] trimeric PSI, u.p. unbound pigments
The eluates obtained from his-hliA/ΔhliA and his-hliB/ΔhliB were further separated by 2D CN/SDS-PAGE (Fig. 4). The green bands co-eluting with the Hlips were identified as RCCII, RC47, and CP47m assembly intermediates; the small green-yellow band in his-hliB/ΔhliB was identified as free Hlip-dimers. In both eluates, the CP47m was present as two distinct bands: with and without Hlips. This implied that a fraction of Hlips was stripped out from the CP47m during CN-PAGE; indeed free (His-)HliAC- and (His-)HliBC-dimers were detectable on the Coomassie-stained 2D gels (Fig. 4). Free (His-)HliBC was more abundant, and the pigments associated with the dimer were readily visible on the CN gel (Fig. 3b). In agreement with preferential binding of HliBC to CP47m (Supplemental Fig. 2), the CP47m to RCCII ratio in the His-HliB preparation was much higher than in the His-HliA pull-down (Fig. 4). Even though the RC47 assembly intermediate was below/close to the detection limit in the 2D-blots from thylakoid membranes (Supplemental Fig. 2), it was clearly visible in the His-HliA/B pull-downs (Fig. 4) demonstrating the presence of Hlips in this complex as well.

Two-dimensional CN/SDS-PAGE separation of His-HliA and His-HliB pull-downs CN-PAGE strips from his-hliA/ΔhliA and his-hliB/ΔhliB pull-downs (see Fig. 3b) were separated in the second dimension with SDS-PAGE and the gels were stained with Coomassie blue (CBB). Protein complexes co-eluted with Hlips are marked by dashed line boxes. Chl-binding PSII subunits and Hlips are indicated with arrows
To get a holistic view of all the HliA/B interactors, we repeated the pull-downs three times and analyzed the eluates with mass spectrometry (Fig. 5, Supplemental Data 2). PSII core subunits were clearly enriched in the pull-downs from his-hliA/ΔhliA and his-hliB/ΔhliB (Fig. 5a and b), while PSI was not significantly enriched, consistent with the CN-PAGE analysis where PSI[3] was a contamination present also in the WT control elution (Fig. 3b). In addition to the PSII subunits (D1, D2, CP43, CP47, cytochrome b559 α and β, and PsbH), the assembly factors Psb27, Psb28-1, Psb28-2, Psb35, Ycf48, and CyanoP were significantly enriched. Moreover, no oxygen-evolving complex proteins were detected, confirming that the eluates consisted only of PSII assembly intermediates or PSII repair complexes and not of fully assembled PSII. As was already observed in the phosphate buffer preparations (Fig. 1), native HliB was not present in the his-hliA/ΔhliA pull-down and vice versa. HliD was not detected in either eluate, whereas HliC was enriched in both, confirming that HliA and HliB dimerize preferentially with HliC in vivo. A set of previously unknown interactors were also detected. Most notably, the export proteins SecD and SecF, and the uncharacterized proteins Slr2105 and Sll1071 were enriched in both eluates. The his-hliB/ΔhliB eluates contained an additional set of eighteen high confidence interactors, most of which are uncharacterized proteins (Supplemental Data 2).

Volcano plots of his-hliA/ΔhliA (a), his-hliB/ΔhliB (b), his-hliA/ΔhliA/ΔhliC (c), and his-hliB/ΔhliB/ΔhliC (d) pull-downs analyzed by MS Pull-downs from WT thylakoids were used as a negative control. The differences between the sample and control (log2-transformed intensities) are plotted on the x-axis and the negative logarithms of p-values on the y-axis. All pull-downs were performed three times. Cut-offs for high confidence (FDR = 0.1%) and low-confidence (FDR = 1%) interactors are marked by solid and dashed lines, respectively (lines in panels b and c are overlapping). Individual proteins are indicated with gray squares. All proteins that were enriched in both his-hliA/ΔhliA and his-hliB/ΔhliB, all Hlips and the HliD-interactors Ycf39 and ChlG have been indicated with different colors and symbols in each plot: Hlips are indicated with orange circles, PSII subunits with green triangles, known PSII assembly factors with magenta diamonds, and novel HliA/B interactors with blue crosses. Hlips and their direct interactors (ChlG, CP47, PsbH, and Ycf39) as well as the novel co-eluted proteins (SecD, SecF, Sll1071, and Slr2105) are additionally marked with corresponding font colors
MS analysis of his-hliA/ΔhliA/ΔhliC and his-hliB/ΔhliB/ΔhliC pull-downs (Fig. 5c, d; Supplemental Data 2) was in line with the reduction of Hlips (Fig. 3a) and their aberrant localization in the thylakoid membranes (Supplemental Fig. 2d, e) upon deletion of hliC. Due to low accumulation of the bait proteins, we obtained significantly less interacting proteins compared to the preparations containing HliC. Intriguingly, both Hlips co-purified with a significant amount of HliD and HliD-specific partners, Ycf39 and ChlG. Deletion of HliC resulted in the presence of small amounts of native HliB in the His-HliA elution and, analogously, HliA was co-purified with His-HliB. Overall, the MS results confirmed our observations from the CN-PAGE analysis (Fig. 3b): the His-HliA eluate did not contain significant amounts of PSII core subunits nor the novel interactors (Fig. 5c), whereas some PSII assembly complexes were still enriched with His-HliB, even in the absence of HliC (Fig. 5d). Moreover, some interaction with SecD and SecF as well as Sll1071 was retained in his-hliB/ΔhliB/ΔhliC.
Lysine 35 destabilizes HliA in the absence of HliC
HliC and HliD proteins form a heterodimer, but homodimers of these Hlips can also accumulate in large quantities, particularly if one of the two is eliminated (Staleva et al. 2015; Shukla et al. 2018). The amino-acid sequences of HliA and HliB are very similar (87% amino-acid identity). However, in contrast to HliB, which is still rather stable in the absence of HliC, the accumulation of HliA depends strictly upon HliC (Fig. 3a). The most plausible explanation for this result is a fast degradation of monomeric, pigment-less HliA produced in the absence of HliC. As we discuss later, HliB is most likely able to form homodimers, whereas HliA seems to occur in the cell almost exclusively in heterodimers with HliC.
To clarify the mechanism of selective Hlip pairing, we created point-mutated His-HliA constructs at residues E22 and K35. These two sites are the most divergent between HliA and HliB and therefore might be important for determining the difference between these proteins (Fig. 6). The mutants His-HliA(E22P) and His-HliA(K35E) were created to mimic HliB and the mutant His-HliA(K35A) to mimic HliC (Fig. 6). All mutated His-HliA variants were expressed in the ΔhliA/ΔhliC background and membranes from high-light-treated cells were analyzed with 1D-SDS-PAGE (Fig. 7a). An immunoblot of the gel showed that the level of His-HliA(E22P) remained low in the membranes, while both K35 variants were able to accumulate to a higher extent – up to a comparable amount with His-HliB expressed in the ΔhliC background. We also investigated the effect of the point mutations on protein–protein interactions by performing pull-downs in MES buffer (Fig. 7b). With the increased accumulation of His-HliA(K35A) and His-HliA(K35E) in the thylakoid membranes, the proteins also co-purified with more CP47. Interestingly, the amount of HliD was also increased in both eluates.

Sequence comparisons of Synechocystis Hlips and Psb34. a Alignment of Synechocystis Hlips. The conserved LHC-motif is underlined. b Alignment of Synechocystis HliA, HliB, and Psb34 proteins. Identical amino-acid residues are highlighted with dark gray and highly similar residues with light gray. Amino-acid residues mutated in this work in His-HliA are indicated in bold and red

Stability of His-tagged Hlips including their mutant variants and the level of co-eluted proteins Cells expressing His-HliA, His-HliB, or His-HliA variants were treated with 500 µmol m−2 s−1 light for 3 h prior to thylakoid isolation and affinity purification. a Thylakoid membranes were loaded based on Chl content (2–5 μg, depending on the antibody; 200% of WT thylakoids was loaded on the HliA/B blot to increase the visibility of the native proteins). b Pull-downs of His-tagged Hlips were performed with equal amounts of starting material and equal volumes of the final eluates were loaded on the gel. Proteins were separated on SDS-PAGE and blotted. Blots were probed with specific antibodies against the indicated proteins; HliA and HliB can be detected with the same antibody raised against HliA
Discussion
The universal and specific features of pigment binding to Hlips
In this study, we isolated and characterized highly pure (His-)HliAC- and (His-)HliBC-dimers by a combination of nickel-affinity chromatography and CN-PAGE separation (Fig. 1). The dimers were pigmented with a Chl:Car ratio of 1:0.6, very close to the 1:0.57 reported previously for homo-oligomeric HliC isolated from the genetic background accumulating HliC as the only Hlip (Shukla et al. 2018). The pigment configuration of dimeric HliC, purified by a combination of nickel-affinity chromatography and anion-exchange chromatography in DDM, was accordingly suggested to correspond to 4 Chls and 2 Cars (Shukla et al. 2018). Here, we utilized A8-35 amphipol for separation of Hlip-dimers during CN-PAGE and analyzed the pigments from gel-extracted proteins. This method prevented the commonly observed (unspecific) oligomerization of Hlips during the gel run (see, e.g., Shukla et al. 2018, Fig. 1) and yielded extremely pure and probably very well-preserved native conformations of Hlips. The observed Chl:Car ratio of 1:0.6 suggests the alternative possibility that dimeric HliC as well as HliAC and HliBC bind 5 Chls and 3 Cars, with the third Car binding site being weaker. In contrast to other Hlip-dimers, HliCD contains 5–6 Chls per 2 Cars (ratio 1:0.36; Niedzwiedzki et al. 2016) but, as demonstrated recently, another (weak) xanthophyll-binding site in HliCD facilitates its interaction with Chl synthase (Proctor et al. 2020). Indeed, a small fraction of echinenone and β-cryptoxanthin (~ 15% of total Cars), found in both HliAC and HliBC, might partially replace β-Car in a less specific, more polar and peripheral binding site.
Similar to HliC (Hontani et al. 2018; Shukla et al. 2018) and HliD (Staleva et al. 2015), the HliAC- and HliBC-dimers were clearly in a quenching conformation, as shown by the absence of Chl fluorescence from the purified dimers (Fig. 1). This result further corroborates that constitutive quenching is a universal feature of Hlips. The absorption spectra of HliAC and HliBC exhibit a typical red-shift (> 520 nm) in Car absorbance, which appears to be characteristic for all LHC-like proteins able of thermal dissipation via Car S1 state (Llansola-Portoles et al. 2017; Hontani et al. 2018; Skotnicová et al. 2021). As shown by Knoppová et al. (2014), the HliCD pair is able to dissipate excitation energy from the RCII complex. It is tempting to speculate that Hlips can also dissipate energy from the CP47 antenna. However, one has to keep in mind that the RCII complex contains only six Chl molecules, whereas the CP47m binds 16 Chls. Effectively quenching such a complicated pigment system would require a fast energy-transfer channel from Chls in CP47 to a Car in the associated Hlips. Whether CP47m can be photoprotected by Hlips, or HliAC and HliBC serve some different function (e.g., Chl delivery) needs to be addressed in future work.
The Synechocystis Hlip-interactome
HliA and HliB were previously found to co-elute after separation of thylakoid proteins by gel-filtration (He et al. 2001), which led to the proposition that these proteins form a complex. Based on 2D gel analysis of purified Hlips (Figs. 1 and 4) and MS analysis of His-HliA and His-HliB pull-downs (Fig. 5), we demonstrated that HliA and HliB form heterodimers specifically with HliC. HliC seems to be crucial for the accumulation of HliA/B (Fig. 3a) and for the correct localization of these proteins in thylakoids (Supplemental Fig. 2). These results imply a very limited stability of monomeric Hlips and we speculate that the only ‘surviving’ Hlips in ΔhliC mutant cells are those forming either homodimers or heterodimers with other Hlips. Indeed, only in the absence of HliC does HliA/B interact mutually or with HliD (Fig. 5c, d). It should be noted that in contrast to the aberrant interaction with HliD, which could be detected by immunoblotting (Fig. 7b), the formation of HliAB heterodimers was detectable only by the very sensitive MS approach (Fig. 5c, d) and thus must be extremely rare even in the ΔhliC mutant. Together, these results also explain the localization of HliA/B in ΔhliC thylakoids (Supplemental Fig. 2d, e): the unspecific pairing between HliA/B and HliD might lead to some HliA/B binding to HliD-associated complexes and vice versa.
The mechanism behind the strict selectivity of Hlip pairing is enigmatic. To clarify the dimerization ‘rules’, we mutated Lys35 of HliA into Glu (opposite charge) or Ala (small, neutral residue). Importantly, we found that the affinity of the K35E mutant protein to HliD and CP47 was significantly increased, while the K35A mutation also slightly increased the interactions (Fig. 7b). It seems plausible that K35 mutation strengthens the interaction with HliD. Additionally, it might facilitate the formation of homodimers, thereby increasing the stability of His-HliA in the ΔhliC background. As HliB contains a Glu residue in the ‘-3’ position before the Chl-binding motif, these results are consistent with the higher stability of this Hlip in the absence of HliC. It is also noteworthy that the N-terminal extensions of HliA and HliB are relatively long in comparison to HliC and HliD (Fig. 6a). Indeed, both HliC and HliD are able to accumulate as homodimers (Staleva et al. 2015; Shukla et al. 2018). Thus, it is possible that the long N-terminal extensions of HliA and HliB could further hinder their mutual dimerization. Based on these observations, we speculate that the very short stromal-exposed N-terminus and a non-polar residue in position ‘-3’ are some of the key determinants for the ability of HliC to bind any other Hlip.
HliB was previously shown to bind near CP47 and PsbH (Promnares et al. 2006). Recently solved Thermosynechococcus RCCII structures revealed the presence of a novel Psb34 assembly factor next to PsbH and CP47 (Zabret et al. 2021; Xiao et al. 2021). Interestingly, the N-terminal sequence of this assembly factor shares significant similarity with HliA and HliB N-termini (Fig. 6b). The N-terminus of Psb34 forms a loop along the surface of CP47 with several hydrogen bonds with PsbH, PsbL, and CP47 stabilizing the interaction (Supplemental Fig. 3). As we did not observe Psb34 in our MS data (Supplemental Table 1), it is likely that both HliA and HliB compete with Psb34 for the same binding site of CP47. Furthermore, Hlips were not detected in FLAG-Psb34 affinity pull-downs even from high-light-treated cells (Rahimzadeh Karvansara et al. accepted). Thus, the N-terminal sequence of HliA/B most likely mediates the interaction between Hlip-dimers and CP47 in a manner similar to the Psb34 N-terminus. Because HliD interacts with Ycf39 even in the absence of HliC (Staleva et al. 2015), this interaction is also most probably mediated via the N-terminal sequence of HliD. Based on these data, we propose a model for Hlip-dimerization where HliC functions as a truncated ‘generalist’ helix that prevents aggregation of PSII assembly complexes, while the role of the other ‘specialist’ Hlip partner is to target Hlips to the desired complexes.
HliA and HliB in PSII biogenesis
All Chl-binding PSII core subunits as well as several known PSII assembly factors co-purified with His-HliA and His-HliB (Fig. 5, Supplemental Data 2), whereas no oxygen-evolving complex proteins were detected. The pattern of the eluted complexes on CN- and 2D-PAGE (Figs. 3b and 4) in combination with the MS data gave strong evidence that HliA and HliB associate with PSII assembly intermediates and not with the active complex. Based on the Hlip-interactome data presented in this publication (Figs. 4 and 5) and previous data describing the roles of HliCD-dimers during early steps of PSII biogenesis (Chidgey et al. 2014; Knoppová et al. 2014), we prepared a model depicting the participation of Hlips in PSII assembly/repair under stress conditions (Fig. 8). Hlips are involved in PSII biogenesis from the earliest steps: HliC and HliD form a complex with Chl synthase and Ycf39, which also associates with the YidC insertase (Chidgey et al. 2014). Together, this complex might assist Chl delivery to the newly formed D1 reaction center protein (Chidgey et al. 2014). The HliCD-Ycf39 complex remains attached to the D1 module and the RCII* complex, which they protect against photo-oxidative damage (Knoppová et al. 2014; Staleva et al. 2015). Upon CP47m binding, the HliCD-dimer and Ycf39 likely detach, as they are not seen in the RC47 complex (Boehm et al. 2012; this study Fig. 5a, b). HliAC- and HliBC-dimers pre-assemble to the CP47m under stress conditions, remain bound until formation of the monomeric PSII reaction center complex (RCCII), and detach before/during oxygen-evolving complex (OEC) assembly. We suggest that the protection of CP47m and later assembly steps against photo-oxidation is the main role of HliA/B proteins. As shown by Rahimzadeh Karvansara et al. (accepted), these Hlips have different dynamics after light stress, with HliB accumulating more transiently (maximal expression after 2 h of high light stress) and HliA showing more stable accumulation even after prolonged (24 h) high light stress. The different affinity of HliA and HliB to ‘free’ CP47m reported here supports the fine-tuning and specialized roles of these Hlips during stress conditions.

Scheme of PSII assembly intermediates involving Hlips The PSII complex is assembled from four building blocks called modules (D1m, D2m, CP43m, CP47m), each of which is preloaded with Chl and Car cofactors before association to larger PSII assembly intermediates (Komenda et al. 2012b). During stress conditions, Hlips attach to distinct assembly/repair intermediates, potentially protecting the whole pathway. HliAC- and HliBC-dimers likely occupy the same binding site as the Psb34 assembly factor (Zabret et al. 2021; Xiao et al. 2021). Although both heterodimers bind to complexes containing CP47, HliBC appears to have a higher affinity to ‘free’ CP47m while HliAC might be more specific for later assembly steps. Small PSII subunits PsbE (Cyt b559 α), F (Cyt b559 β), I, K, L, M, T, X, Z and Ycf12 have been omitted for clarity. Known assembly factors Ycf48 (Komenda et al. 2008), CyanoP (Knoppová et al. 2016), Psb28 (Dobáková et al. 2009), Psb27 (Komenda et al. 2012a) and Psb35 (Pascual-Aznar et al. 2021) that were also identified in our His-HliA/B pull-downs are indicated. D1m D1 module, CP47m CP47 module, CP43m CP43 module, P CyanoP, H PsbH, 28 Psb28, 27 Psb27
MS analysis of His-HliA and His-HliB pull-downs (Fig. 5a and b) revealed as yet unknown interactors for both Hlip isoforms. The discovery of SecD and SecF translocon proteins was especially interesting. In heterotrophic bacteria, the SecDF-dimer utilizes the transmembrane proton gradient to promote protein export across the plasma membrane (Gardel et al. 1990; Pogliano and Beckwith 1994; Arkowitz and Wickner 1994). Together with YidC, SecDF is also a part of the holo-translocon complex, which is required for efficient membrane insertion of polytopic membrane proteins (Komar et al. 2016; Botte et al. 2016). Our results suggest that the SecDF-dimer is also a component of the thylakoid holo-translocon and might participate in PSII biogenesis. The other identified Hlip-interactors, Sll1071 and Slr2105, are proteins of unknown function and thus candidates for novel (possibly high-light-specific) PSII assembly factors.
Materials and methods
Synechocystis strains
All mutant strains described in this work are constructed using the Synechocystis WT-P substrain (Tichý et al. 2016). To prepare a ΔhliA strain, we fully segregated the deletion construct described in (Xu et al. 2002) in WT-P; the ΔhliB and his-hliB/ΔhliB mutants were described in Promnares et al. (2006). To construct his-hliA/ΔhliA, we transformed the ΔhliA strain with the plasmid (pPD-his8-hliA) and segregated transformants on plates with increasing kanamycin concentration (starting with 10 μg ml−1 and doubling the amount until full segregation was achieved). To create the pPD-his8-hliA plasmid, the hliA gene was amplified from the Synechocystis chromosomal DNA using primers his8-hliA-fw and hliA-rev. The resulting insert was cloned into the pPD-FLAG plasmid (Hollingshead et al. 2012) using NdeI and BglII restriction sites. The hliC gene was deleted by chloramphenicol resistance cassette from the plasmid pACYC184. The deletion construct was created by overlap extension PCR (Lee et al. 2004) using the scpB1, scpB4, ScpBCm5, and scpBCm6 primers. The deletion construct was transformed into Synechocystis and segregated with increasing chloramphenicol concentration. Genomic DNA from this strain was used to transform the his-hliA/ΔhliA and his-hliB/ΔhliB strains to obtain his-hliA/ΔhliA/ΔhliC and his-hliB/ΔhliB/ΔhliC, respectively. The ΔhliA/ΔhliC strain was created by transforming genomic DNA isolated from ΔhliA into the ΔhliC strain and segregating transformants on plates with increasing erythromycin concentration (starting with 3 μg ml−1 and doubling the amount until full segregation was achieved). The ΔhliB/ΔhliC strain was created by transforming genomic DNA isolated from ΔhliC into the ΔhliB strain and segregating transformants on plates with increasing chloramphenicol concentration (starting with 10 μg ml−1 and doubling the amount until full segregation was achieved). His-HliA point mutants were constructed by mutating the pPD-his8-hliA plasmid using the QuikChange II XL kit (Agilent) with primers his-hliA(E22P), his-hliA(K35A), and his-hliA(K35E). The mutated plasmids were sequenced and transformed to the ΔhliA/ΔhliC background. All primer sequences are listed in Supplemental Table 1.
Growth conditions
Synechocystis cells were inoculated by suspending one loop of cells to 50 ml of BG11 medium in 250 ml Erlenmeyer flasks. Cells were grown under 40 µmol m−2 s−1 in a shaker at + 28 °C. The starting cultures were grown until OD750 > 1.0 (WPA S1200 Spectrawave, Biochrom) and used to inoculate experimental cultures at OD750 0.1–0.2. For the expression analysis of the His-constructs, the cells were grown under the same conditions as above until logarithmic growth phase and then transferred to high light (500 µmol m−2 s−1) for 1, 3, and 5 h; the control flask (0 h) was kept in 40 µmol m−2 s−1. Cells were harvested at OD750 0.8–1.0 by centrifuging at 10,000 rpm and + 4 °C for 10 min (Sigma 3K30, rotor 12,155) and resuspended to MES buffer [25 mM MES-NaOH (2-(N-morpholino)ethanesulfonic acid) pH 6.5, 10 mM CaCl2, 10 mM MgCl2, 25% glycerol] for storage. For localization of Hlips in thylakoid membranes, the cells were grown as above and treated with 3 h of high light before harvesting. For pull-down assays, cells were grown in 1 l cylinders with 600 ml of BG11, air bubbling and mixing by magnetic stirrer with 40 µmol m−2 s−1 illumination from the side at + 28 °C. Cells were grown until OD750 1.0–1.5 and treated with 500 µmol m−2 s−1 light for 3 h before harvesting. Cells were harvested by centrifuging at 6600 rpm and + 4 °C for 20 min (Sigma 8KS, rotor 12505-H) and resuspended either to phosphate buffer (25 mM Na-phosphate pH 7.8, 50 mM NaCl, 10% glycerol) or to MES buffer and pelleted again by centrifuging at 10,000 rpm and + 4 °C for 10 min (Sigma 3K30, rotor 12155). Cells were resuspended to the respective buffer for storage. Cells were frozen in N2(l) and stored in − 80 °C.
Thylakoid membrane extraction
Thylakoid membranes for expression analysis were extracted by breaking the cells in MES buffer. Pelleted cells were resuspended in 400 µl of MES buffer and mixed with 200 µl of glass beads (100–200 µm) and cOmplete™ protease inhibitor cocktail (Roche) in 2-ml lysis tubes. This suspension was vortexed 4 × 20 s and samples were cooled on ice for 3 min between each cycle. After breaking, the membranes were extracted by washing the glass beads with 200 µl MES buffer 4–5 times. The obtained suspension was pelleted by centrifuging at 36,000 g and + 4 °C for 20 min and resuspended to 100 µl of MES buffer. Thylakoids for localization studies were broken with Precellys® Evolution (Bertin Technologies) in 2-ml lysis tubes in MES buffer. Cells were pelleted in the lysis tubes and supplemented with 200 μl of buffer, 200 μl of glass beads (100–200 µm) and 10 μl of 50 × cOmplete™ protease inhibitor cocktail (Roche) (one tablet dissolved to 1 ml of H2O). Lysis program was set to 3 × 20 s at 5 500 rpm with 120 s pause between the cycles; cooling was set to 0 °C. The glass beads were washed repeatedly with 200 μl of buffer and thylakoids collected in 2-ml eppendorf tubes. Thylakoids were pelleted by centrifuging at 18,000 rpm and + 4 °C for 30 min (Sigma 3K30, rotor 12131) and resuspended to ~ 100 μl of buffer for storage. For pull-down assays, the cells were broken in 7-ml lysis tubes either in phosphate or MES buffer. Cells were pelleted in the lysis tubes and supplemented with 1 ml of buffer, 2 ml of glass beads (100–200 µm) and 100 μl of 50× protease inhibitor. The same lysis program was used as above, but the program was run twice with 5 min incubation of samples on ice between the cycles. The glass beads were washed repeatedly with 1–2 ml of buffer and thylakoids collected in 30-ml centrifugation tubes. Thylakoids were pelleted by centrifuging at 20,000 rpm and + 4 °C for 30 min (Sigma 3K30, rotor 12158) and suspended in a few ml of buffer for storage. Membranes were frozen in N2(l) and stored in − 80 °C until use. After cell lysis, the samples were kept constantly cooled and protected from light. Thylakoid Chl concentration was determined in methanol according to Lichtenthaler and Wellburn 1983.
Pull-down assays
Thylakoid membranes were solubilized using 1% DDM (AppliChem). Thylakoid concentration was set to 0.5 µg µl−1 Chl with phosphate or MES buffer and the membranes were incubated with the detergent and SIGMAFAST™ EDTA-free protease inhibitor (Sigma-Aldrich) for 10 min in + 10 °C with gentle rotation. Insoluble membrane debris was removed by centrifuging at 20,000 rpm and + 4 °C for 30 min (Sigma 3K30, rotor 12131). For MES buffer pull-downs, 1% glycodiosgenin (GDN, Anatrace) was added to the solubilized membranes to stabilize protein complexes (Chae et al. 2012). Solubilized membranes were mixed with ~ 100 µl of pre-equilibrated Protino® Ni-NTA agarose (Macherey–Nagel) per 1 mg of Chl in the starting material (typically 1.5 mg Chl was used). 500 mM NaCl and 10 mM imidazole were added to reduce unspecific binding. Proteins were bound to the Ni-NTA agarose by incubating for 1 h at + 10 °C with gentle rotation. All buffers used during the purification were supplemented with 0.04% DDM and MES buffer pull-downs with additional 0.04% GDN. Purification was performed in 10 ml Poly-Prep® chromatography columns (Bio-Rad). The nickel resin was washed with 20 column volumes (CV) of equilibration buffer (10 mM imidazole, 500 mM NaCl in phosphate or MES buffer) and 20 CV of washing buffer (20 mM imidazole in phosphate or MES buffer). Elution was performed with 5 or 10 CV of elution buffer (200 mM imidazole in phosphate or MES buffer, respectively). The final eluates were concentrated using 50 kDa cut-off centrifugal columns (Vivaspin® 6, Sartorius or Amicon® Ultra, Merck). All genotypes were purified in parallel for the MS analysis, and the experiment was replicated three times in total. First and second replicates were purified with new Ni-NTA resin and the second replicate with one-time-used and washed resin.
Electrophoretic methods
Clear native gel electrophoresis was performed as in (Komenda et al. 2019) with minor modifications. CN samples were prepared by solubilizing thylakoid membranes in 1% DDM + 1% GDN at 0.5 μg μl−1 Chl concentration for 10 min on ice. We also omitted deoxycholate from the cathode running buffer and added 1% amphipol A8-35 (Tribet et al. 1996; Kameo et al. 2021) to the CN-PAGE samples before loading. CN-run was performed at constant current of 11 mA. 2D- and 1D-SDS-PAGE were performed on 16–20% acryl amide, 0–10% sucrose gradient gels prepared in 0.65 M Tris–HCl (pH8.6) (acryl amide:bis acryl amide 60:1). Proteins were transferred to PVDF membranes (Immobilon-P, Merck) in carbonate buffer (3 mM Na2CO3, 10 mM NaHCO3) using Trans-Blot® Cell (Bio-Rad). Blotting was performed at 850 mA for 3 h with water cooling at + 4 °C and mixing with magnetic stirrer. Antibodies against HliA/B (AS10 1603), HliD (AS10 1615), and CP47 (AS04 038) were purchased from Agrisera and used as 1:4000 dilutions. Antibody against HliC (Moravian Biotechnology) was generated against a synthetic N-terminal peptide (amino acids 1–17) in rabbit and used as 1:500 dilution. All antibodies were diluted in TTBS (10 mM Tris–HCl pH 7.6, 150 mM NaCl, 0.05% TWEEN® 20) and incubated on the membranes overnight at + 10 °C with gentle shaking. For detection, proteins were labeled with Peroxidase-conjugated goat anti-rabbit secondary antibody (A6154, Sigma-Aldrich, 1:10,000 dilution) for 1 h at room temperature and incubated for 1 min in Immobilon® Crescendo Western HRP Substrate (Merck). Chemiluminescence was detected with LAS-4000 (Fujifilm).
Pigment analysis
To identify pigments associated with purified His-Hlips, we cut a piece of CN gel (~ 3 × 2 mm) containing separated Hlips and cut it further into several smaller pieces. Gel pieces were incubated in 200 μl of MES with 0.04% DDM buffer overnight. After centrifugation, 150 μl of the supernatant was injected into the Agilent-1260 HPLC system equipped with a diode-array detector. Pigments were separated on a reverse-phase column (Zorbax Eclipse C18, 5 μm particle size, 3.9 × 150 mm; Agilent) with 35% (v/v) methanol and 15% (v/v) acetonitrile in 0.25 M pyridine (solvent A) and 20% (v/v) methanol, 20% (v/v) acetone, 60% (v/v) acetonitrile as solvent B. Pigments were eluted with a linear gradient of solvent B (30–95% (v/v) in 25 min) in solvent A followed by 95% of solvent B in solvent A at a flow rate of 0.8 ml min−1 at 40 °C. Chl a and Cars were detected at 440 nm; the obtained peaks were integrated and the molar stoichiometries calculated from calibration curves prepared using authentic standards.
Absorption spectra
Absorption spectra of purified Hlips were measured by cutting a piece from the CN gel containing the pigmented Hlip complexes and setting it on a plastic slip attached to a custom made adaptor to fit into a standard 1-ml cuvette slot in a spectrophotometer. Spectra were recorded from 750 to 350 nm in 0.5 nm intervals with a Shimadzu UV-3000 spectrophotometer (slit width 5 nm); an empty gel was used as blank. The spectra were normalized to the absorption maximum of the red region of Chl a (QY band). Three independent pull-downs were analyzed per strain and the spectra averaged.
Mass spectrometry
Concentrated Ni-NTA eluates (5 μl) were diluted with 45 µl of 50 mM ammonium bicarbonate supplemented with 0.1% (v/v) Rapigest (Waters) surfactant and incubated at + 60 °C. After 45 min, proteomic grade trypsin (Sigma) was added to a final concentration of 10 ng µl−1 and incubated at + 37 °C. After 12 h, samples were acidified and peptides were isolated by the StageTip procedure (Rappsilber et al. 2007) to produce 30 µl of sample. LC–MS/MS analysis was performed on an UltiMate 3000 UHPLC (Thermo Fisher Scientific) on-line coupled to a TimsTOF pro (Bruker) mass spectrometer. Samples (2 μl) were trapped for 1 min on a ThermoFisher trap (0.3 × 5 mm, C18, 5 µm) column, then separated by reverse phase liquid chromatography on a Acclaim PepMap RSLC column (75 µm × 15 cm, C18, 2 µm, 100 Å; Thermo Fisher Scientific). During 30 min, peptides were eluted by increasing ratio of acetonitrile (solvent B) in 0.1% formic acid (solvent A) from 3 to 50% directly into the CaptiveSpray nano ion source of the mass spectrometer. Spectra were acquired in PASEF (Parallel Accumulation/SErial Fragmentation) data dependent mode with an accuracy of 0.2 ppm for precursors and 0.5 ppm for peptides. Each sample was run twice (two technical repetitions). Raw data were processed by the MaxQuant/Andromeda software (Cox and Mann 2008; Cox et al. 2011; Tyanova et al. 2016a) and compared to a species specific Synechocystis protein database downloaded from Uniprot and Cyanobase.
Statistical analysis of protein groups obtained from MaxQuant was performed in Perseus 1.6.14.0 (Tyanova et al. 2016b). Proteins identified only by site or in the reverse dataset and potential contaminants were removed. Data were log2-transformed and inspected by hierarchical clustering and one technical replicate was discarded due to low quality of the MS run (WT replicate 1.2). Other technical replicates were averaged with their respective pairs and data filtered to include only proteins that were detected in all three pull-downs at least in one genetic background. Missing data was imputed from normal distribution for statistical analysis. Data was inspected for significant background interactions (i.e., batch effect) by hierarchical clustering and checking the correlations between samples. A significant batch effect was detected between the different pull-down batches and subsequently removed using the Limma package (Ritchie et al. 2015) via the PerseusR-plugin (Rudolph and Cox 2019). Significant interactors were determined using the Hawaii plot function in Perseus, which uses a global permutation-based false-discovery rate (FDR) to detect significant hits (Rudolph and Cox 2019). High confidence (Class A) FDR rate was set to 0.1%, low-confidence (Class B) FDR rate was set to 1%, and s0 was set to 2.
In silico analyses
Coding sequences of Synechocystis Hlips (hliA, ssl2542; hliB, ssr2595; hliC, ssl1633 and hliD, ssr1789) and psb34 (ssl1498) were obtained from the genomic sequence (GenBank assembly GCA_000009725.1) and translated. Sequences were aligned using the EMBL-EBI T-coffee multiple sequence alignment tool (Madeira et al. 2019).
Image manipulation
Histograms of gel images were adjusted with Photoshop CS2 (Adobe) to improve visual clarity. Volcano plots were exported from Perseus v. 1.6.15.0 (Tyanova et al. 2016b). Vector art was added and images were assembled in CorelDRAW X6.
Data Availabilty
The raw MS data has been submitted to the MassIVE repository (CCMS, University of California, San Diego) and can be accessed via the link ftp://massive.ucsd.edu/MSV000088753/.
Change history
23 March 2023
A Correction to this paper has been published: https://doi.org/10.1007/s11120-023-01016-y
References
Arkowitz RA, Wickner W (1994) SecD and SecF are required for the proton electrochemical gradient stimulation of preprotein translocation. EMBO J 13:954–963. https://doi.org/10.1002/j.1460-2075.1994.tb06340.x
Bassi R, Croce R, Cugini D, Sandona D (1999) Mutational analysis of a higher plant antenna protein provides identification of chromophores bound into multiple sites. Proc Natl Acad Sci 96:10056–10061. https://doi.org/10.1073/pnas.96.18.10056
Becker K, Cormann KU, Nowaczyk MM (2011) Assembly of the water-oxidizing complex in photosystem II. J Photochem Photobiol B 104:204–211. https://doi.org/10.1016/j.jphotobiol.2011.02.005
Ben-Shem A, Frolow F, Nelson N (2003) Crystal structure of plant photosystem I. Nature 426:630–635. https://doi.org/10.1038/nature02200
Boehm M, Romero E, Reisinger V, Yu J, Komenda J, Eichacker LA, Dekker JP, Nixon PJ (2011) Investigating the early stages of photosystem II assembly in Synechocystis sp. PCC 6803. J Biol Chem 286:14812–14819. https://doi.org/10.1074/jbc.M110.207944
Boehm M, Yu J, Reisinger V, Bečková M, Eichacker LA, Schlodder E, Komenda J, Nixon PJ (2012) Subunit composition of CP43-less photosystem II complexes of Synechocystis sp. PCC 6803: implications for the assembly and repair of photosystem II. Philos Trans R Soc B 367:3444–3454. https://doi.org/10.1098/rstb.2012.0066
Botte M, Zaccai NR, Lycklama à J, Nijeholt JL, Martin R, Knoops K, Papai G, Zou J, Deniaud A, Karuppasamy M, Jiang Q, Roy AS, Schulten K, Schultz P, Rappsilber J, Zaccai G, Berger I, Collinson I, Schaffitzel C (2016) A central cavity within the holo-translocon suggests a mechanism for membrane protein insertion. Sci Rep 6:38399. https://doi.org/10.1038/srep38399
Chae PS, Rasmussen SGF, Rana RR, Gotfryd K, Kruse AC, Manglik A, Cho KH, Nurva S, Gether U, Guan L, Loland CJ, Byrne B, Kobilka BK, Gellman SH (2012) A new class of amphiphiles bearing rigid hydrophobic groups for solubilization and stabilization of membrane proteins. Chem Eur J 18:9485–9490. https://doi.org/10.1002/chem.201200069
Chidgey J, Linhartová M, Komenda J, Jackson PJ, Dickman MJ, Canniffe DP, Koník P, Pilný J, Hunter CN, Sobotka R (2014) A cyanobacterial chlorophyll synthase-HliD complex associates with the Ycf39 protein and the YidC/Alb3 insertase. Plant Cell 26:1267–1279. https://doi.org/10.1105/tpc.114.124495
Cox J, Mann M (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26(12):1367–1372. https://doi.org/10.1038/NBT.1511
Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res 10:1794–1805. https://doi.org/10.1021/pr101065j
Dobáková M, Sobotka R, Tichý M, Komenda J (2009) Psb28 protein is involved in the biogenesis of the photosystem II inner antenna CP47 (PsbB) in the cyanobacterium Synechocystis sp. PCC 6803. Plant Physiol 149:1076–1086. https://doi.org/10.1104/pp.108.130039
Dolganov NA, Bhayat D, Grossman AR (1995) Cyanobacterial protein with similarity to the chlorophyll a/b binding proteins of higher plants: Evolution and regulation. Proc Natl Acad Sci USA 92:636–640. https://doi.org/10.1073/pnas.92.2.636
Engelken J, Brinkmann H, Adamska I (2010) Taxonomic distribution and origins of the extended LHC (light-harvesting complex) antenna protein superfamily. BMC Evol Biol 10:233. https://doi.org/10.1186/1471-2148-10-233
Gardel C, Johnson K, Jacq A, Beckwith J (1990) The secD locus of E. coli codes for two membrane proteins required for protein export. EMBO J 9:3209–3216. https://doi.org/10.1002/j.1460-2075.1990.tb07519.x
Havaux M, Guedeney G, He Q, Grossman AR (2003) Elimination of high-light-inducible polypeptides related to eukaryotic chlorophyll a/b-binding proteins results in aberrant photoacclimation in Synechocystis PCC6803. Biochim Biophys Acta Bioenerget 1557:21–33. https://doi.org/10.1016/S0005-2728(02)00391-2
He Q, Dolganov N, Björkman O, Grossman AR (2001) The high light-inducible polypeptides in Synechocystis PCC6803. Expression and function in high light. J Biol Chem 276:306–314. https://doi.org/10.1074/jbc.M008686200
Hey D, Grimm B (2018) ONE-HELIX PROTEIN2 (OHP2) is required for the stability of OHP1 and assembly factor HCF244 and is functionally linked to PSII biogenesis. Plant Physiol 177:1453–1472. https://doi.org/10.1104/pp.18.00540
Hey D, Grimm B (2020) ONE-HELIX PROTEIN1 and 2 form heterodimers to bind chlorophyll in photosystem II biogenesis. Plant Physiol 183:179–193. https://doi.org/10.1104/pp.19.01304
Hollingshead S, Kopečná J, Jackson PJ, Canniffe DP, Davison PA, Dickman MJ, Sobotka R, Hunter CN (2012) Conserved chloroplast open-reading frame ycf54 is required for activity of the magnesium protoporphyrin monomethylester oxidative cyclase in Synechocystis PCC 6803. J Biol Chem 287:27823–27833. https://doi.org/10.1074/jbc.M112.352526
Hontani Y, Kloz M, Polívka T, Shukla MK, Sobotka R, Kennis JTM (2018) Molecular origin of photoprotection in cyanobacteria probed by watermarked femtosecond stimulated Raman spectroscopy. J Phys Chem Lett 9:1788–1792. https://doi.org/10.1021/acs.jpclett.8b00663
Järvi S, Suorsa M, Aro EM (2015) Photosystem II repair in plant chloroplasts—regulation, assisting proteins and shared components with photosystem II biogenesis. Biochim Biophys Acta (BBA) - Bioenerg 1847:900–909. https://doi.org/10.1016/J.BBABIO.2015.01.006
Jordan P, Fromme P, Witt HT, Klukas O, Saenger W, Krauß N (2001) Three-dimensional structure of cyanobacterial photosystem I at 2.5 Å resolution. Nature 411:909–917. https://doi.org/10.1038/35082000
Kameo S, Aso M, Furukawa R, Matsumae R, Yokono M, Fujita T, Tanaka A, Tanaka R, Takabayashi A (2021) Substitution of deoxycholate with the amphiphilic polymer amphipol A8–35 improves the stability of large protein complexes during native electrophoresis. Plant Cell Physiol 62:348–355. https://doi.org/10.1093/pcp/pcaa165
Knoppová J, Sobotka R, Tichý M, Yu J, Konik P, Halada P, Nixon PJ, Komenda J (2014) Discovery of a chlorophyll binding protein complex involved in the early steps of photosystem II assembly in Synechocystis. Plant Cell 26:1200–1212. https://doi.org/10.1105/tpc.114.123919
Knoppová J, Yu J, Konik P, Nixon PJ, Komenda J (2016) CyanoP is involved in the early steps of photosystem II assembly in the cyanobacterium Synechocystis sp. PCC 6803. Plant Cell Physiol 57:1921–1931. https://doi.org/10.1093/pcp/pcw115
Komar J, Alvira S, Schulze RJ, Martin R, Nijeholt JL, Lee SC, Dafforn TR, Deckers-Hebestreit G, Berger I, Schaffitzel C, Collinson I (2016) Membrane protein insertion and assembly by the bacterial holo-translocon SecYEG–SecDF–YajC–YidC. Biochem J 473:3341–3354. https://doi.org/10.1042/BCJ20160545
Komenda J, Sobotka R (2016) Cyanobacterial high-light-inducible proteins - Protectors of chlorophyll-protein synthesis and assembly. Biochim Biophys Acta - Bioenerg 1857:288–295. https://doi.org/10.1016/j.bbabio.2015.08.011
Komenda J, Nickelsen J, Tichý M, Prášil O, Eichacker LA, Nixon PJ (2008) The cyanobacterial homologue of HCF136/YCF48 is a component of an early photosystem II assembly complex and is important for both the efficient assembly and repair of photosystem II in Synechocystis sp. PCC 6803. J Biol Chem 283:22390–22399. https://doi.org/10.1074/jbc.M801917200
Komenda J, Knoppová J, Kopečná J, Sobotka R, Halada P, Yu J, Nickelsen J, Boehm M, Nixon PJ (2012a) The Psb27 assembly factor binds to the CP43 complex of photosystem II in the cyanobacterium Synechocystis sp. PCC 6803. Plant Physiol 158:476–486. https://doi.org/10.1104/pp.111.184184
Komenda J, Sobotka R, Nixon PJ (2012b) Assembling and maintaining the photosystem II complex in chloroplasts and cyanobacteria. Curr Opin Plant Biol 15:245–251. https://doi.org/10.1016/j.pbi.2012.01.017
Komenda J, Krynická V, Zakar T (2019) Isolation of thylakoid membranes from the cyanobacterium Synechocystis sp. PCC 6803 and analysis of their photosynthetic pigment-protein complexes by clear native-PAGE. Bio-Protoc 9:3126. https://doi.org/10.21769/BIOPROTOC.3126
Kühlbrandt W, Wang DN, Fujiyoshi Y (1994) Atomic model of plant light-harvesting complex by electron crystallography. Nature 367:614–621. https://doi.org/10.1038/367614a0
Lee J, Lee HJ, Shin MK, Ryu WS (2004) Versatile PCR-mediated insertion or deletion mutagenesis. Biotechniques 36:398–400. https://doi.org/10.2144/04363BM04
Lichtenthaler HK, Wellburn AR (1983) Determinations of total carotenoids and chlorophylls a and b of leaf extracts in different solvents. Biochem Soc Trans 11:591–592
Liu Z, Yan H, Wang K, Kuang T, Gui L, An X, Chang W (2004) Crystal structure of spinach major light-harvesting complex at 2.72 Å resolution. Nature 428:287–292. https://doi.org/10.1038/nature02373
Li Y, Liu B, Zhang J, Kong F, Meng H, Li W, Rochaix JD, Li D, Peng L (2019) OHP1, OHP2, and HCF244 form a transient functional complex with the photosystem II reaction center. Plant Physiol 179:195–208. https://doi.org/10.1104/pp.18.01231
Llansola-Portoles MJ, Sobotka R, Kish E, Shukla MK, Pascal AA, Polívka T, Robert B (2017) Twisting a β-carotene, an adaptive trick from nature for dissipating energy during photoprotection. J Biol Chem 292:1396. https://doi.org/10.1074/JBC.M116.753723
Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, Lopez R (2019) The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47:W636–W641. https://doi.org/10.1093/nar/gkz268
Malavath T, Caspy I, Netzer-El SY, Klaiman D, Nelson N (2018) Structure and function of wild-type and subunit-depleted photosystem I in Synechocystis. Biochim Biophys Acta - Bioenerg 1859:645–654. https://doi.org/10.1016/j.bbabio.2018.02.002
Mazor Y, Borovikova A, Caspy I, Nelson N (2017) Structure of the plant photosystem I supercomplex at 2.6 Å resolution. Nat Plants 3:17014. https://doi.org/10.1038/nplants.2017.14
Myouga F, Takahashi K, Tanaka R, Nagata N, Kiss AS, Funk C, Nomura Y, Nakagami H, Jansson S, Shinozaki K (2018) Stable accumulation of photosystem II requires ONE-HELIX PROTEIN1 (OHP1) of the light harvesting-like family. Plant Physiol 176:2277–2291. https://doi.org/10.1104/pp.17.01782
Niedzwiedzki DM, Tronina T, Liu H, Staleva H, Komenda J, Sobotka R, Blankenship RE, Polívka T (2016) Carotenoid-induced non-photochemical quenching in the cyanobacterial chlorophyll synthase-HliC/D complex. Biochim Biophys Acta - Bioenerg 1857:1430–1439. https://doi.org/10.1016/j.bbabio.2016.04.280
Pascual-Aznar G, Konert G, Bečková M, Kotabová E, Gardian Z, Knoppová J, Bučinská L, Kaňa T, Sobotka R, Komenda J (2021) Psb35 protein stabilizes the CP47 assembly module and associated high-light inducible proteins during the biogenesis of photosystem ii in the cyanobacterium Synechocystis sp. PCC6803. Plant Cell Physiol 62:178–190. https://doi.org/10.1093/pcp/pcaa148
Pazderník M, Mareš J, Pilný J, Sobotka R (2019) The antenna-like domain of the cyanobacterial ferrochelatase can bind chlorophyll and carotenoids in an energy-dissipative configuration. J Biol Chem 294:11131–11143. https://doi.org/10.1074/jbc.ra119.008434
Pogliano JA, Beckwith J (1994) SecD and SecF facilitate protein export in Escherichia coli. EMBO J 13:554–561. https://doi.org/10.1002/j.1460-2075.1994.tb06293.x
Proctor MS, Pazderník M, Jackson PJ, Pilný J, Martin EC, Dickman MJ, Canniffe DP, Johnson MP, Hunter CN, Sobotka R, Hitckcock A (2020) Xanthophyll carotenoids stabilise the association of cyanobacterial chlorophyll synthase with the LHC-like protein HliD. Biochem J 477:4021–4036. https://doi.org/10.1042/BCJ20200561
Promnares K, Komenda J, Bumba L, Nebesarova J, Vácha F, Tichý M (2006) Cyanobacterial small chlorophyll-binding protein ScpD (HliB) is located on the periphery of photosystem II in the vicinity of PsbH and CP47 subunits. J Biol Chem 281:32705–32713. https://doi.org/10.1074/jbc.M606360200
Rahimzadeh Karvansara P, Pascual Aznar G, Bečková M, Komenda J (2022) The Psb34 protein modulates binding of high-light-inducible proteins to CP47 containing photosystem II assembly intermediates in the cyanobacterium Synechocystis sp. PCC 6803. Photosynth Res, accepted
Rappsilber J, Mann M, Ishihama Y (2007) Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2(8):1896–1906. https://doi.org/10.1038/NPROT.2007.261
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK (2015) Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43:e47. https://doi.org/10.1093/nar/gkv007
Rudolph JD, Cox J (2019) A network module for the Perseus software for computational proteomics facilitates proteome interaction graph analysis. J Proteome Res 18:2052–2064. https://doi.org/10.1021/acs.jproteome.8b00927
Shukla MK, Llansola-Portoles MJ, Tichý M, Pascal AA, Robert B, Sobotka R (2018) Binding of pigments to the cyanobacterial high-light-inducible protein HliC. Photosynth Res 137:29–39. https://doi.org/10.1007/s11120-017-0475-7
Skotnicová P, Staleva H, Kuznetsova V, Bína D, Konert M, Lu S, Polivka T, Sobotka R (2021) Plant LHC-like proteins show robust folding and static non-photochemical quenching. Nat Commun 12:6890. https://doi.org/10.1038/s41467-021-27155-1
Staleva H, Komenda J, Shukla MK, Šlouf V, Kaňa R, Polívka T, Sobotka R (2015) Mechanism of photoprotection in the cyanobacterial ancestor of plant antenna proteins. Nat Chem Biol 11:287–291. https://doi.org/10.1038/nchembio.1755
Tichý M, Bečková M, Kopečná J, Noda J, Sobotka R, Komenda J (2016) Strain of Synechocystis PCC 6803 with aberrant assembly of photosystem II contains tandem duplication of a large chromosomal region. Front Plant Sci 7:648. https://doi.org/10.3389/FPLS.2016.00648
Tribet C, Audebert R, Popot J-L (1996) Amphipols: polymers that keep membrane proteins soluble in aqueous solutions. Proc Natl Acad Sci 93:15047–15050. https://doi.org/10.1073/pnas.93.26.15047
Tyanova S, Temu T, Cox J (2016a) The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc 11:2301–2319. https://doi.org/10.1038/NPROT.2016.136
Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016b) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13:731–740. https://doi.org/10.1038/nmeth.3901
Umena Y, Kawakami K, Shen JR, Kamiya N (2011) Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9Å. Nature 473:55–60. https://doi.org/10.1038/nature09913
Vass I (2012) Molecular mechanisms of photodamage in the photosystem II complex. Biochim Biophys Acta (BBA) - Bioenerg 1817:209–217. https://doi.org/10.1016/J.BBABIO.2011.04.014
Wei X, Su X, Cao P, Liu X, Chang W, Li M, Zhang X, Liu Z (2016) Structure of spinach photosystem II-LHCII supercomplex at 3.2 Å resolution. Nature 534:69–74. https://doi.org/10.1038/nature18020
Xiao Y, Huang G, You X, Wang W, Kuang T, Han G, Sui SF, Shen JR (2021) Structural insights into cyanobacterial photosystem II intermediates associated with Psb28 and Tsl0063. Nat Plants 7:1132–1142. https://doi.org/10.1038/s41477-021-00961-7
Xu H, Vavilin D, Funk C, Vermaas WFJ (2002) Small Cab-like proteins regulating tetrapyrrole biosynthesis in the cyanobacterium Synechocystis sp. PCC 6803 Plant Mol Biol 49:149–160. https://doi.org/10.1023/A:1014900806905
Yao D, Kieselbach T, Komenda J, Promnares K, Hernández Prieto MA, Tichy M, Vermaas W, Funk C (2007) Localization of the small CAB-like proteins in photosystem II. J Biol Chem 282:267–276. https://doi.org/10.1074/jbc.M605463200
Zabret J, Bohn S, Schuller S, Arnolds O, Möller M, Meier-Credo J, Liauw P, Chan A, Tajkhorshid E, Langer J, Stoll R, Krieger-Liszkay A, Engel B, Rudack T, Schuller J, Nowaczyk M (2021) Structural insights into photosystem II assembly. Nat Plants 7:524–538. https://doi.org/10.1038/s41477-021-00895-0
Acknowledgements
European Research Council Synergy Award 854126 and the Czech Science Foundation, grant No. 19-29225X funded this work. RS also acknowledges institutional support RVO 61388971. PK acknowledges support from the Czech Ministry of Education, Youth and Sports project ‘Mechanisms and dynamics of macromolecular complexes: from single molecules to cells’, CZ.02.1.01/0.0/0.0/15_003/0000441. Jan Pilný is thanked for HPLC analysis of pigments.
Author information
Authors and Affiliations
Contributions
MMK and RS designed the study; AW, MMK, and PK performed the experiments; MMK, PK, and RS analyzed the data; MMK and RS wrote the paper. All authors read and accepted the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Konert, M.M., Wysocka, A., Koník, P. et al. High-light-inducible proteins HliA and HliB: pigment binding and protein–protein interactions. Photosynth Res 152, 317–332 (2022). https://doi.org/10.1007/s11120-022-00904-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-022-00904-z