
Abstract
The OJDIP rise in chlorophyll fluorescence during induction at different light intensities was mathematically modeled using 24 master equations describing electron transport through photosystem II (PSII) plus ordinary differential equations for electron budgets in plastoquinone, cytochrome f, plastocyanin, photosystem I, and ferredoxin. A novel feature of the model is consideration of electron in- and outflow budgets resulting in changes in redox states of Tyrosine Z, P680, and QA as sole bases for changes in fluorescence yield during the transient. Ad hoc contributions by transmembrane electric fields, protein conformational changes, or other putative quenching species were unnecessary to account for primary features of the phenomenon, except a peculiar slowdown of intra-PSII electron transport during induction at low light intensities. The lower than F m post-flash fluorescence yield F f was related to oxidized tyrosine Z. The transient J peak was associated with equal rates of electron arrival to and departure from QA and requires that electron transfer from QA − to QB be slower than that from QA − to QB −. Strong quenching by oxidized P680 caused the dip D. Reduced plastoquinone, a competitive product inhibitor of PSII, blocked electron transport proportionally with its concentration. Electron transport rate indicated by fluorescence quenching was faster than the rate indicated by O2 evolution, because oxidized donor side carriers quench fluorescence but do not transport electrons. The thermal phase of the fluorescence rise beyond the J phase was caused by a progressive increase in the fraction of PSII with reduced QA and reduced donor side.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Chlorophyll fluorescence is an easily measurable signal representing excitations not used for photosynthetic electron transport. A classical understanding is that fluorescence is in its maximum, F m, when in PSII the photosynthetic electron transfer is blocked by reduction of the first acceptor quinone QA (Duysens and Sweers 1963). In many cases, this understanding works well, except when QA is reduced very rapidly by single-turnover flashes (STF). During illumination with intensities (PFD) of up to 10,000 µmol m−2 s−1, fluorescence begins at the low F o level and approaches the maximum F m level after about 300 ms. The slow saturation is explained by fast electron transfer away from QA, which stops only after the entire electron transport chain becomes reduced. When PFD is increased to the level of xenon or laser flashes exceeding 1 mol m−2 s−1, QA becomes reduced faster than oxidized by the electron transfer, so that the F m-level fluorescence is expected after a few microseconds. In experiments, this does not happen and instead a lower F f level of fluorescence is recorded at 50 µs after the flash, followed by quenching as QA − is re-oxidized by electron transfer to QB (de Wijn and van Gorkom 2001). A persistent enigma of the “QA model” of fluorescence is that the “maximum” fluorescence yield F f, measured 50 µs after a ST flash, is significantly below the F m level determined from a second-long multiple-turnover pulse (Joliot and Joliot 1964, 1977, 1981; Neubauer and Schreiber 1987; Samson and Bruce 1996). This has opened possibilities for alternative interpretations, such that the flash-level fluorescence F f is indeed the actual maximum fluorescence yield, corresponding to blocked charge separation within PSII, but the following “thermal phase” is caused by the release of other, hypothetical quenchers (Delosme 1967; Schreiber and Neubauer 1987; Neubauer and Schreiber 1987). These potential quenchers have been listed (Vredenberg 2008b; Stirbet and Govindjee 2012; Koblížek et al. 2001): (1) P680+ can quench Chl a fluorescence as efficiently as QA reduction; (2) P680 triplet, 3P680, most likely 3ChlD1 quenches in equilibrium with 3PD1; (3) carotenoid triplet 3Car is an efficient quencher in the antenna; (4) non-photochemical quenching by oxidized PQ molecules; (5) reduced PheoD1 may be a quencher due to charge separation equilibrium P680* ↔ P680+Pheo−, shifted by transmembrane electric field photo-electrochemical quenching (Vredenberg et al. 2009); (6) quenching by charge recombination from QB (Schreiber 2002); and (7) quenching by conformational changes in Chl proteins (Schansker et al. 2011).
In this work, we focus on this paradox by mathematical modeling of the fluorescence rise kinetics in terms of gradual reduction of all electron carriers, without a need for excitation quenchers other than photochemical quenching by electron transfer and non-photochemical quenching by oxidized donor side carriers. Models have been created to describe fluorescence induction, differing mainly in mathematical details (Laisk et al. 2009; Lazár and Schansker 2009). A widely applied approach has been to synthesize the complex FI curve with a series of exponentials, each representing a partial reaction approximated by the first-order kinetics (Strasser et al. 2004; Vredenberg 2015). In a more detailed approach, a system of mass action-based ordinary differential equations considered kinetic properties of individual reactions (Baake and Strasser 1990; Baake and Schlöder 1992; Zhu et al. 2005). The most sophisticated approaches apply large systems of first-order master equations to describe transformations among the numerous “forms” of a protein complex, incorporating several redox-related electron carriers (Lazár 2003, 2009, 2013; Lebedeva et al. 2002; Belyaeva et al. 2011, 2015). The model by Lebedeva et al. (2002) involved electron transfer redox reactions, accompanied by the generation of transmembrane field and a proton gradient. The detailed approach by Lazár (2003) involved PSII and the water-splitting complex, covering 12 orders of time magnitude from the excitation of PSII antenna to the reduction of plastoquinone. The model was extended involving Q-cycle and cyclic electron transport, up to a reduction of ferredoxin and NADP+ at the PSI acceptor side (Lazár 2009).
Despite various mathematical techniques, no consensus has yet been established concerning a central question: how are the PSII donor and acceptor side electron transfer processes combined to determine the fluorescence rise from F o to F m (Schansker et al. 2011, 2014; Vredenberg 2008b)? There are at least three states (or forms) of the PSII complex co-determining the fluorescence yield: QA reduction on the acceptor side and oxidized TyrZ and oxidized P680 on the donor side. In this work, we have designed a mathematical model where these three states, considered together, can determine the whole fluorescence induction with its OJDIP inflections, at least at high light intensities. The experimental observations which were difficult to reconcile with the simple QA model (Schansker et al. 2014) can still be reconciled. The principle contradiction—O2 evolution is much slower than electron transport determined from fluorescence quenching (Laisk and Oja 2013)—is explained by the inability of the donor-oxidized states to transport electrons, though they are still quenching excitation. Competitive rebinding of reduced plastoquinone to the QB site on the acceptor side explains the closely proportional dependence of fluorescence on the redox state of this electron carrier.
The model
Applying the mathematical methods used by Lebedeva et al. (2002), Lazár (2003), and Belyaeva et al. (2006, 2011), but grouping PSII internal structure in partial reactions differently from the predecessors, we have constructed a computer model that adequately simulates PSII electron transport and Chl fluorescence in leaves. The model explains the low ST flash-induced fluorescence yield and the OJDIP inflection in the dark-light fluorescence rise on the basis of kinetic restrictions and different quenching properties of electron carriers on the donor and acceptor sides of PSII, without a need to postulate changes in transmembrane electric field, protein conformation, or other potential excitation quenchers. The internal structure of the PSII core is simplified to the sequence YPAB of four electron carriers, where Y is tyrosine Z, P is P680, A is QA, and B is QB. In this denotation, P680 is the complex of PD1·PD2·ChlD1·ChlD2·PheoD1·PheoD2 (Renger 2010). The basic assumption of the model is that the whole coherence complex covering the two PSII branches, but with an exciton mainly concentrating on ChlD1, PD1·PD2, PheoD1 (Novoderezhkin et al. 2015) and involving the PheoD1 − ·PD1 + intermediate charge-separated state, is equilibrated with antenna excitation. The charge-stabilized state with QA − and P680+ is formed as a result of a charge transfer from excited P680* to QA via the PheoD1 − ·PD1 + intermediate state, but is immediately (within less than 1 µs) reduced by electron transfer to P680+ from TyrZ (Christen et al. 1998), resulting in the TyrZox·P680 donor complex YoPrArBoo. Double oxidation of the TyrZox·P680 pair is rare, but may happen at high PFDs wherein P680+ (YoPoArBro form) accumulates in an appreciable amount.
On the acceptor side, QA (denoted A) is the bound quinone primary electron acceptor, from which electrons are transferred to the mobile secondary quinone acceptor QB (denoted B). The latter accepts sequentially two electrons, after which reduced plastoquinone (PQH2) formed in the QB site (denoted Brr) equilibrates diffusionally with the free PQH2 pool. Both quinone forms, PQH2 and PQ, may leave the QB site with a first-order rate constant, but the empty QB site may absorb either PQ or PQH2 competitively, depending on the second-order rate constant and concentration of PQ and PQH2—as is typical for the active site of an enzyme. Fluorescence transients could be reproduced assuming relatively high affinity of PQH2 for the QB site, which makes this product a strong inhibitor of PSII as the “water–plastoquinone oxidoreductase” enzyme. The fraction of PSII closed due to the doubly reduced QB increases in reversible equilibrium about proportionally with the free PQH2 pool.
A direct output of the model is mechanistic explanation of the kinetic features of the rising fluorescence induction (FI) traces for leaves. For easier comparison with experiment, inputs of the model are incident photon flux density PFD (mol m−2 s−1) and Chl content (mol m−2). The area densities of PSII (PS2T) and PSI (PSIT) are calculated as
and
where a II is partitioning of absorbed excitation to PSII and PSU2 is the antenna size (Chl per center). Equation 2 is the analogous expression for PSI. Excitation rates per center n 2 and n 1 (s−1) are calculated considering the incident photon flux density (PFD, mol m−2 s−1), absorption coefficient (Labs), and antenna size:
and
The redox states of the YPAB system components or forms (Lazár 2003) are conveniently expressed with subscripts denoting reduction (r) or oxidation (o). For example, after dark adaptation the most likely PSII form is YrPrAoBoo, meaning reduced TyrZ and P680, but oxidized QA and QB.
Emphasis is placed on the donor side oxidation states, that is the forms containing YoPr and YoPo. After an electron is transferred, the YrPrAoBoo form becomes YoPrArBoo. We assume an immediate electron transfer from TyrZ to P680+, precluding the accumulation of YrPo form. The oxidized Yo receives an electron from an S-state with first-order kinetics, rate constant j d (for rate constants, the subscript d means “direct” and r means “reverse,” but in this analysis we neglect the reversibility of electron donation from TyrZ back to the S-state). The j d value actually depends on the advancement of the S-state, but for the sake of simplicity we did not include different S-states in the model. Inductions involving the transfer of several electrons are characterized by a single effective j d value.
The heart of the model is the system of 24 master equations, each describing the budget (derivative, rate of change of the fraction) of the particular YPAB form. The sum of the forms is normalized to 1. In contrast to prior studies, the rate constants of excitation energy capture were normalized to the sum of the physical excitation capture constants (dissipation + triplet formation + fluorescence) = k f = 1. Photosynthetically important rate constants are expressed in relation to the physical constant, e.g., k p = 7.5 (Table 1). As a result, the calculated fluorescence is normalized to F m = 1. The following differential equations describe the transformation of each of the forms, where d(YxPxAxBxx)/dt is the rate of change of the fraction (s−1) of each of these forms.
0 e− forms:
1 e− forms:
2 e− forms:
3 e− forms:
4 e− forms:
5 e− forms:
Equations (5–28) contain forms YxPxAxBrr and YxPxAxBoo with doubly reduced and doubly oxidized QB, which reversibly dissociates, joining the diffusible PQH2 and PQ pools, whose level is determined by the following budget equations:
Equation (31) describes electron transport through the Q-cycle in the Cyt b6f complex. Although the Q-cycle is a complex reaction system (Lebedeva et al. 2002), its overall kinetics are well represented by two first-order reactions in series (Laisk et al. 2016). Here we neglect the two-step nature of the process, but apply single first-order reaction kinetics with respect to PQH2. Although being first-order, with increasing PQH2 level the reaction rate still becomes limited by the availability of oxidized Cyt f as an electron acceptor.
Electron transfer between Cyt f, PC, and P700 is very fast, being close to redox equilibrium (Oja et al. 2010). In the Cyt b6f complex, the Q-cycle reduces 2 Cyt f at the expense of oxidation of one PQH2. From Cytfr electrons rapidly leave, reducing oxidized PC with the first-order kinetics:
Plastocyanin is oxidized by electron transfer to P700+:
In the latter equations (31–36), the oxidized form is found as the difference (total pool − reduced form). In Eqs. (34, 35), n 1 is the PSI excitation rate (Eq. 4) and only those PSI are photochemically 100% efficient whose P700 is reduced and the primary acceptor (denoted FX) is oxidized. Equilibrium constants kE were adjusted to kE cytf = 3, kE pc = 35, and kE fx = 100 (Oja et al. 2010; Laisk et al. 2016). The large equilibrium constants kE cytf and kE pc ensure free electron flow through the Cyt b6f complex, while electrons accumulate on the PSI donor side and the large kE fx avoids the blockade on the acceptor side. As PSII room-temperature Chl fluorescence is only indirectly influenced by PSI electron transport via the redox sequence of electron carriers, we used ordinary differential equations for this part of the electron transport chain.
The amount of electrons transported through PSII donor side, measurable as 4·O2 evolution, e− per PSII, is expressed as follows:
Equation (37) actually presents the electron transport rate, so for better comparison with experiments based on light pulsing an integrated amount of electrons is calculated for each time moment.
Different Chl fluorescence yield was ascribed to each distinct PSII form. The forms with reduced donor and primary acceptor, YrPrArBxx (independent of whether xx is oo, ro, or rr), emit fluorescence at the maximum yield F m, normalized to unity in the absence of non-photochemical quenching. The forms with reduced donor and oxidized primary acceptor, YrPrAoBxx, emit at the F o yield. Forms with oxidized TyrZ, but reduced P680, YoPrArBxx, are assumed to emit at an intermediate “flash” level F f, which is about a half to two-thirds of the F m value, dependent on the constant k r = 1 or 0.5. In units with Yo and Ao, fluorescence is below F o, rather close to zero (k q >2 k p) (Steffen et al. 2005). Thus, the PSII fluorescence is calculated as follows:
where the last term represents forms emitting at F m, but the yield decreases progressively in the preceding terms. In principle, Eq. (38) states that “closed” centers emit fluorescence (Duysens and Sweers 1963), but differently dependent on the reduction state of their donor and acceptor side carriers. In the calculations, the rate constant for non-photochemical quenching k n = 0, i.e., F m, is at its maximum “predawn” value and normalized to unity. For comparison with O2 evolution, electron transport was calculated from fluorescence as
Equation (39) also presents the electron transport rate, as does Eq. (37), but the sum of transferred electrons per PSII is calculated for each time moment during an induction.
Reduced ferredoxin FDr is the end of the dark-adapted electron transport sequence. Although the absolute PSII density PS2T was calculated for every preset Chl content and antenna size, for calculations the sum of all PSII partial states was normalized to unity. For convenience, pool sizes of electron carriers were expressed in relation to PSII density, e.g., PQT = 5, but antenna sizes and Chl partitioning were chosen such that PSIIT = PSIT.
These equations were solved by computer using the parameters shown in Table 1, modified as shown in the figure legends when necessary. Euler’s method was used by automatically adjusting the time step dt so as not to exceed a change of 0.1% of the amplitude of the fastest changing variable. This stabilized the system even during very steep changes in the rate (e.g., ST flash light of 10 mol photons m−2 s−1).
Results
The model was tuned on the basis of experimental FI curves presented in Figs. 1 and 3 of Schansker et al. (2011). Although the Chl content of leaves typically approaches 400–600 µmol m−2, in calculations Chl was set at 300 µmol m−2. Such a choice was an attempt to minimize distortions arising from the gradient in photon density in the leaf cross-section during measurements: most of the photons are absorbed in the upper leaf layer, and most of the fluorescence is detected from the upper layer, accommodating only a part of the whole leaf chlorophyll (Sušila et al. 2004). Partitioning of Chl to PSII was set at a II = 0.5, and antenna sizes were chosen PSU2 = PSU1 = 170 Chl. This resulted in equal PSII and PSI densities PS2T = PS1T = 0.88 µmol m−2, actively participating in the modeled process. With these parameters, PSI electron transport slightly exceeded that of PSII—due to the losses in PSII caused by fluorescence emission—resulting in about 10% oxidation of P700 during steady-state exposure to a low PFD of 30 µmol m−2 s−1.

Simulating a DCMU-poisoned leaf. A very high light (PFD as shown in the panel, mol m−2 s−1) was turned on at Time = 0. Upper line shows the time course of total fluorescence, and lower line of the same color shows fluorescence emitted by the donor-oxidized form YoPrAr(Brr). Electron transfer from QA to QB was blocked by setting b1d = b2d = 0 (equivalent to the Brr state). Electron donation rate j d = 18,200 s−1, characteristic of the S1 state
The DCMU case
Responses of the model were systematically investigated from simpler to more complex situations, adjusting the parameters towards better fit to the experimental examples, mainly as reported in Schansker et al. (2011). Best-fit coincidence of the calculated and measured traces was not attempted, rather qualitative reproduction of characteristic features—such as timing and shape of the JIP inflections—was the objective.
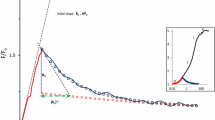
The DCMU-inhibited case was modeled by blocking the electron flow between Ar and Boo (QA and QB) by setting the rate constants b1d (for the first electron) and b2d (for the second electron transfer) to zero. The initial state YrPrAo(Brr) is converted to the state YoPrAr(Brr) upon the onset of light (electron transfer from Yr to Po is immediate), which is re-reduced to YrPrAr(Brr) by transfer from an S-state. A very high xenon flash illumination of 10 mol m−2 s−1 was necessary to accomplish electron transfer in all PSII centers within 5 µs (an approximate Xe flash length, Fig. 1). Toward the end of the flash fluorescence yield becomes equal to F f = 0.5 F m, as defined by the rate constant k r = 1, competing for excitation quenching in the YoPrAr(Brr) form (parentheses indicate that the QB site is occupied by DCMU). This low yield rapidly increases thanks to fast electron donation from the S1 state (j d = 18,200 s−1). After 50 µs, the yield is already 0.8 F m, approaching F m beyond 100 µs (normalized F m = 1, corresponding to QA and donor side carriers reduced in all PSII). The fast electron donation from the S1 state makes it difficult to measure the time course of fluorescence approaching F m in the presence of DCMU. Flash power is also critical—1 M (million µmol m−2 s−1) is still a sufficient intensity, but with 0.1 M the characteristic fluorescence yield at 50 µs past the flash is significantly unsaturated.
Electron transfer time from an S-state to TyrZ depends on the number of the S-states, being 250 µs from S0 to S1, 55 µs from S1 to S2, 290 µs from S2 to S3, and 1200 µs from S3 to S0 (Rappaport et al. 1994). Fluorescence traces of Fig. 1 were calculated with the fast rate constant of the S1→S2 transfer (j d = 18,180 s−1), dominating in the dark-adapted state. Slower electron donation from other S-states causes slower substitution of the flash-generated YoPrAr(Brr) form with YrPrAr(Brr): S2→S3 (j d = 3448) and S3→S4→S0 (j d = 833 s−1, Fig. 2).

Dependence on the S-state. Electron donation rate, j d, was varied in accordance with the dominating S-state number: 18,180 s−1 (S1 to S2 red), 3448 s−1 (S2 to S3 green), and 833 s−1 (S3 to S4 to S0 blue). At PFD of 1 mol m−2 s−1, the upper line shows the time course of accumulation of the YrAr(Brr) form, and lower line of the same color shows re-reduction of the YoPrAr(Brr) form
In longer transients below, where more than one electron is transferred, we applied some effective averaged j d values. Extremely low j d values of 200 and 20 s−1 (Fig. 3) closely reproduced the DCMU-inhibited fluorescence inductions recorded at the negative temperatures of −20 °C and lower (Fig. 3 in Schansker et al. 2011).

Extremely slow electron donation. Electron donation rate j d was slowed down in a DCMU-poisoned leaf in order to simulate the low-temperature measurements of Fig. 3 in Schansker et al. (2011). PFD = 3000 µmol m−2 s−1. Upper line shows total fluorescence, and lower line of the same color shows fluorescence emitted by the YoPrAr(Brr) form
\({{\text{Q}}_{\text{A}}} \to {{\text{Q}}_{\text{B}}}{\text{ electron transfer}}\)
In this section, the two electrons accumulate at QB, but electron transfer from QB to PQ is blocked by zeroing the rate constants for dissociation from and association with the QB site of the reduced and oxidized plastoquinone species: rq d = oq d = rq r = oq r = 0. The initial condition (dominating form) was set YrPrAoBoo = 1 (PSII donor side reduced, acceptor side oxidized) and three additional electrons were accumulated until the form YrPrArBrr approached unity.
At the high PFD of 15,000 µmol m−2 s−1, the modeled fluorescence approaches F m at about 3 ms. The characteristic J inflection is present in this transient, though donor side limitation was set absent (Fig. 4a). Importantly, the J inflection is present when the first QA → QB electron transfer is slower (b1d = 2000) than the second electron transfer (b2d = 5000 s−1). This is a critical requirement because the J inflection disappears when the rate constant values are reversed, and the first electron is transferred faster than the second (b1d = 5000, b2d = 2000 s−1, Fig. 4b). The level of the J inflection decreases and a dip appears when electron donation from the S-states becomes rate limiting (j d = 3000 s−1, Fig. 4c). With this j d value, the depth of the dip was still smaller than in experiments (Fig. 1 in Schansker et al. 2011), but j d of 2000 s−1 (donation time 0.5 ms) gave a deeper dip. The strong donor side limitation, however, lowered the height of the J inflection significantly under the experimental value of 0.6 F m (Fig. 4d).
A limitation of the current model is negligence of the different electron donation times from S-states advancing during the induction. A qualitative exploration of this process appears in panels A, C, and D of Fig. 4. During the transfer of the first electron (S1 state), fluorescence follows the trace in panel A (actually to the somewhat lower J level, but the overly fast j d = 100,000 s−1 was chosen to ensure the presence of the J inflection independent of donor side processes). When the second electron is transferred with j d = 3000 s−1, the fluorescence curve shifts from that in panel A to that in C, describing a dip actually deeper than visible in C. The latter dip could be even deeper due to slower donation from the S3 state, but a mixture of S-states in the leaf may dampen the transition.

In these panels, QB reduction by QA − was activated, but QB exchange with free PQH2 and PQ pools was forbidden by setting the rate constants rq d = oq d = rq r = oq r = 0. Plotted are fluorescence yield (red line) and groups of forms with Y, P, and A reduction state as indicated. Each of these represents the sum of four forms—with empty QB site, with Boo, Bro, and Brr. PFD = 15,000 µmol m−2 s−1 in all panels. Panel a j d = 100,000 s−1 (no donor side limitation), b1d = 2000, b2d = 5000 s−1. The J inflection appears independent of donor side limitation when b2d > b1d. Panel b j d = 100,000 s−1 (no donor side limitation), b1d = 5000, b2d = 2000 s−1. The J inflection disappears when b2d < b1d. Panel c j d = 3000 s−1, b1d = 2000, b2d = 5000 s−1; the dip after the J inflection is related to donor side limitation. Panel d j d = 2000 s−1. The dip deepens with stronger donor side limitation, but the height of the J inflection decreases too
Summarizing this section, the J inflection is not a straightforwardly interpretable phenomenon, but its basis is a complicated balance between donor and acceptor side electron transfer rates within PSII. Even without donor side limitation, at the J level QA is reduced only in 70% of PSII units, and in a still smaller fraction when the donor side is rate limiting. Fluorescence remains briefly constant at the J level, while the fraction of forms with QA reduced remains constant as well. This occurs when the rate of QA −→QB transfer is equal to the rate of QA reduction by photochemistry. At lower PFDs, this balance occurs with a smaller fraction of QA reduced, corresponding to a lower J-level fluorescence. The J level increases with increasing PFD provided that electron availability from the Mn complex is non-limiting. In the contrary case, i.e., light saturation due to the donor side limitation, the latter determines the maximum J level which may be lowered due to quenching by accumulation of TyrZox and P680+. In this case, application of a STF does not increase J-level fluorescence (Schreiber et al. 2012). The J inflection is pronounced when the transfer of the second electron is faster than that of the first electron, blending into a more or less constant balance between electron arrival to and departure from QA. On the other hand, when the first electron is transferred faster than the second electron, equilibration of QA − with the QB pool by the first electron occurs at a lower J level than that by the second electron, generating a continuously increasing fluorescence transient. The dip (D level) after the J inflection is generated when the second electron is transferred much faster than the first, and is enhanced when the forms with oxidized donor side carriers YoPoAo, YoPoAr, and YoPrAo strongly quench fluorescence. Their combined fraction is small, but enough to cause the temporary dip in fluorescence. These fractions not only deepen the dip, but also quench the height of the J inflection below the experimental level of Schansker et al. (2011). A compromise parameter value for fitting to the experiment would be k r = 0.5, for which the flash fluorescence F f = 0.66 F m. The following calculations use this k r value and b1d = 2000 and b2d = 5000 s−1.
Plastoquinone reduction
Plastoquinone reduction is a kinetically complex process involving exchange between free PQ and PQH2 pools and an empty QB site. To investigate the process, the initial (dark) condition was set to YrPrAoBoo = 1 (all PSII bound with PQ in the QB site) and the additional free pool PQ = 4 per PSII. Thus, there are altogether 5 PQ per PSII, initially all oxidized. Light intensity was set high, PFD = 15,000 µmol m−2 s−1, to match the fastest induction curve in Fig. 1 of Schansker et al. (2011). Oxidation of PQH2 by Cyt b6f was blocked by setting the rate constant k b6f = 0. In accordance with the common understanding that “reduced quinone is released from, but oxidized quinone replaces it in the QB site,” the rate constants were set at rq d = oq r = 2000, while the reversal rates were set at rq r = oq d = 100 s−1 (note that the “direct” rate is defined as away from PSII). The resulting induction curve did not resemble the experimental curve at all (Fig. 5a, red line). The reason was the large equilibrium constant between QA and PQH2, preventing QA reduction before PQH2 becomes highly reduced (Fig. 5b). To prevent the redox equilibration between PQH2 and QA, the binding and unbinding rate constants were set such that about equal occupancy of the QB site by both ligands was ensured at their medium reduction level (Crofts and Wraight 1983; Robinson and Crofts 1983). The absolute values of 2000 s−1 ensured fast exchange of quinone between PSII and Cyt b6f within 1 ms (Laisk et al. 2016), but the second-order rebinding rate constant of 2000 s−1 per PQH2 ensured sufficient competitive rebinding of PQH2, particularly at its higher reduction levels, when PQH2 = 2 to 4, but PQ is low. With these rate constants, the modeled approach of fluorescence to F m was rather similar to the experimental trace in the log time axis (Fig. 5a, blue line). As expected, fluorescence yield increased about proportionally with increasing PQ reduction (Fig. 5b, blue line). This is caused by product inhibition by PQH2 of PSII as the water–plastoquinone reductase, due to fast re-association of PQH2 with the QB site. Naturally, the I inflection is still absent in this transient, since PQH2 oxidation is blocked.

The exchange of PQH2 and PQ with the QB site was activated, but oxidation of PQH2 by Cyt b6f was blocked by setting k b6f = 0. PFD = 15,000 µmol m−2 s−1 j d = 3000 s−1, b1d = 2000, and b2d = 5000 s−1. Red lines rq d = 2000 s−1, rq r = 100 s−1 per PQH2 per PSII, oq r = 2000 s−1 per 1 PQ per PSII, oq d = 100 s−1 (“direct” means away from PSII). Blue lines rq d = 2000 s−1, rq r = 2000 s−1 per PQH2 per PSII, oq r = 2000 s−1 per 1 PQ per PSII, oq d = 2000 s−1
Full induction
Mathematical description of the full induction requires modeling of electron transport through the Q-cycle and through PSI. The last processes are not reflected by Chl fluorescence in such detail as the PSII kinetics. Therefore, here we neglect the master-equation approach (see Lebedeva et al. 2002), but apply ordinary differential equations to describe the reduction–oxidation budgets of individual carriers, despite the fact that some of them occur within a common protein complex.
Fluorescence induction from the dark-adapted state is a dead-end process, where electron transport is blocked beyond ferredoxin—most likely caused by reversal of GAP-dehydrogenase in the dark, leading to over-reduction of NADPH. As light is turned on, electrons are transported from the PSI donor side to its acceptor side, accumulating in FD, but leaving the donor side carriers ready to accept electrons generated by PSII. Ferredoxin becomes reduced first, followed by P700, PC, Cyt f, and PQH2 (Fig. 6). The induction lasts until all electron carriers become reduced, ending with F m-level fluorescence.

Reduction state of electron carriers during a full induction. PFD = 3000 µmol m−2 s−1, other parameters as in Table 1
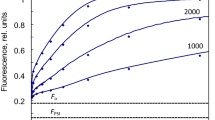
The set of induction curves modeled with the rate constant of PQH2 oxidation k b6f of 50 s−1 exhibits the I inflection at 20–50 ms (Fig. 7). In order to slow down the whole transient in accordance with the experiment (Schansker et al. 2011), the product inhibition of PSII by PQH2 was increased by setting rq r = 5000 s−1 per 1 PQH2 per PSII. The characteristic light dependence of the I level is caused by the first-order kinetics of the Q-cycle reactions—the level of PQH2 increases proportionally with the electron transport rate, but fluorescence in its turn increases in proportion with the PQH2 level. At very high PFDs, the PSII donor side becomes rate limiting, determining the highest I level saturated with respect to the light intensity. At PFDs down from 15,000 to 3000 µmol m−2 s−1, the shape of the calculated FI curves is in accordance with those in Fig. 1a of Schansker et al. (2011) measured in pea leaves, but at lower PFDs the calculated curves did not match the measurements of Fig. 1b of the same study carried out with tobacco leaves. The calculated example at a PFD of 600 µmol m−2 s−1 stays at the low fluorescence <0.3 F m until 50 ms, but the measured curve increased to about 0.6 F m at the same time.

Full inductions with different light intensities. j d = 2000 s−1, rq d = 2000 s−1, rq r = 5000 s−1 per 1 PQH2 per PSII, oq r = 2000 s−1 per 1 PQ per PSII, oq d = 2000 s−1, k b6f = 50 s−1, other constants as in Table 1. The “slow PSII” curve was calculated with b1d = 1000 and b2d = 500 s−1
Literature reports indicate that considerable differences can occur among different plant species in antenna size, PQ pool size, etc. But PSII core structure is highly conserved. Nevertheless, the too fast initial rise of fluorescence between 0.1 and 10 ms at low light intensity suggests decreased or downregulated electron transport within PSII, but not beyond it. This assumption was confirmed by the trace calculated for the PFD of 600 µmol m−2 s−1 with b1d = 1000 and b2d = 500 s−1, fitting the corresponding curve in Fig. 1b of Schansker et al. (2011).
The K peak in heat-treated leaves
Even a brief exposure to >45 °C destroys the Mn complex, resulting in a drastic change in FI curves: the J and I steps disappear, but a novel K peak develops at 300–400 μs at high PFDs (Tóth et al. 2007a). This K peak was suggested to be related to oxidation of TyrZ and is thus a suitable candidate for modeling donor side oxidation.
The heat-inhibited FI transient was modeled with the same set of constants as in Fig. 7, except that the donor rate j d was decreased from 2000 to 200 s−1 (Fig. 8). The slow residual water-splitting activity was retained to simulate electron donation from stromal reductants. The K peak is indeed produced, but like the J inflection it is not directly related to oxidized TyrZ. The forms containing Yo together approach 50% at 0.6–0.8 ms, the rest remaining in the initial state YrPrAo. Soon after the K peak, fully oxidized YoAoPo dominates, decreasing fluorescence to the minimum due to quenching by P680+.

Full induction reproducing the K peak when the water-splitting complex was inhibited by setting j d = 200 s−1. PFD = 3000 µmol m−2 s−1, other parameters as in Fig. 7
This model does not reproduce the P peak as recorded in Tóth et al. (2007a), probably because ferredoxin-NADP reductase is not activated in the model, but could become activated in experiments, where the recording is extended to 1 s. If this happened, fluorescence decreased, because the ET chain became re-oxidized. Re-activation of ferredoxin-NADP reductase could be facilitated by ascorbate, as observed in the experiments of Tóth et al. (2007a).
Fluorescence and electron transport rate
Occupancy (presence) of the donor-oxidized forms of PSII is significant during the initial phase of induction, when total electron transport rate is fast. Each of these YPA forms (Fig. 9) is the sum of four forms—YPA without B (empty QB site), YPABoo, YPABro, and YPABrr. We reiterate that fluorescence yield was defined differently depending on the reduction state of YPA, but independent of B (Eq. 38). Fluorescence is initially at the F o level in YrPrAo forms; it is at the flash F f level in YoPrAr forms after one electron is transferred and approaches the F m level when all the three become reduced (YrPrAr). Transient double oxidation of the donor side is assumed possible, occupying YoPoAr and even YoPoAo forms—each up to 10% at a PFD of 7500 µmol m−2 s−1—emitting very low fluorescence (Eq. 38). During the first milliseconds of illumination, the occupancy of the YrPrAo form rapidly decreases (note the rare diamonds), being substituted mainly by YoPrAr, but other electron transfers occur simultaneously: S-state→Yo transfer creates YrPrAr emitting at F m, QA→QB transfer creates YoPrAo emitting at <F o, but double oxidation of the donor side inhibits YoPoAr and even YoPoAo forms emitting at very low level due to strong quenching by P680+. Accumulation of these strongly quenching forms causes the dip past the J inflection at around 1–2 ms of induction. All these donor-oxidized forms remain present during the whole induction, though their occupancy gradually decreases as the electron transport rate decreases. This explains why the true F m can be reached only after the complete reduction of the whole electron transport chain—because the donor (TyrZox and P680+) and acceptor side (QA) quenching disappears only when electron transport rate decreases to zero.

Time courses of different PSII forms during full dark–light inductions of fluorescence. Each of the indicated forms is a sum of four “subforms”—with empty QB site, with Boo, Bro, and Brr. PFD = 7500 µmol m−2 s−1, other parameters as in Fig. 7
Accumulation of the fluorescence-quenching but not electron-transporting donor-oxidized forms leads to a discrepancy between integrated electron transport calculated from fluorescence and O2 evolution (Eqs. 37 and 39). During an induction at PFD = 3000 µmol m−2 s−1, 20 electrons per PSII were transferred during the whole induction as detected from O2 evolution (an integral of the S-state to Yo electron transfer rate in the model, Eq. 37), but 25.3 electrons were calculated from fluorescence quenching (Fig. 10). This overestimation is small at low PFDs, but increases with increasing light intensity (Fig. 11a). Such a dependence is analogous to a counter of photons operating with a dead time, like a Geiger–Müller counter of nuclear particles:


Overestimated electron transport from fluorescence quenching. Panel a SumF/SumO2 dependent on the PSII excitation rate (parameters as in Figs. 7, 9, 10), Panel b The corresponding dead time τ. Blue lines indicate the values calculated assuming the possibility for double oxidation of the donor side (P680+ may accumulate), and red lines indicate the values calculated assuming no electron transfer in PSII with oxidized TyrZ (no accumulation of P680+)
where n is the number of counted particles, n 0 is the actual number of particles hitting the counter, and τ is the dead time—the time interval after each successful hit during which the detector does not respond. During photosynthesis, photons exciting PSII are the incident particles and transferred electrons are the actual “counted” ones, but τ is the time interval after each electron transfer, during which the exciting photons are missed, which can be expressed using Eq. (40) as
where n 0 is SumF—the number of transferred electrons calculated from fluorescence quenching—and n is SumO2—the number of transferred electrons donated from the S-states. The ratio n 0/n is unity at low PFDs, but non-linearly increases with increasing PSII excitation rate (Fig. 11a). The non-linearity translates into increasing τ, beginning with 0.1 ms at low PFDs and approaching 0.25 ms at the highest PFD of 10,000 µmol m−2 s−1 (n 2 = 4.86 ms−1). This non-linearity is caused by the “buffering effect” due to the dual oxidation on the donor side. Electron donation rate to TyrZ was constantly set at 2000 s−1, but due to the allowed dual oxidation on the donor side two electrons could be transferred before PSII became closed on the donor side. This was more likely to happen at high than at low PFDs. This condition was tested by forbidding light-induced electron transfer from all YoPrAo forms in the model, though P680 was reduced and QA was oxidized—resulting in constant τ of 0.28 ms instead of 0.5 ms as defined by j d = 2000 (the reason for this apparent discrepancy is the preset fluorescence yield of 0.66 F m for the tyrosine-oxidized PSII units, which only partially reflected the donor-blocked electron transfer in fluorescence quenching).
Discussion
Scientific output of modeling
The model program was created in accordance with our best notion about the photosynthetic electron transport. The computation results are thus expected to mimic the dynamic features of photosynthesis. If successful, the work would confirm that the program is close to the notion and the latter is close to Nature—how the process actually occurs. Thus, more interesting are cases when the computed results differ from the actual data. A valid caution is vigilance for limitations in the program. An evident deficiency of our program is the neglected dependence of electron donation rate on the number of S-states, which we attempted to ease by mixing computing and thinking (Fig. 4). But could it be that the present notion taken for the basis of the program is wrong or insufficient itself? We reject the case that the Z-scheme is wrong in its general structure, but how about details—like values of some rate constants? For the sake of better coincidence with experiments, we had to use a partially quenched fluorescence value F f < F m for PSII with reduced QA but oxidized Tyr Z—not recognized in the common notion. In order to reproduce the J inflection, we needed to use an electron transfer rate constant slower for the QA −→QB and faster for the QA −→QB − transfer—in contradiction to some literature reports. In order to reproduce the linear (on a logarithmic scale) rise of fluorescence between the J and I inflections, we needed to use a higher empty QB site affinity for PQH2 than for PQ—suggesting a strong product inhibition of PSII by reduced plastoquinone not appreciated before. These novel, yet testable, observations invite confirmatory efforts using alternate methodologies.
Usually, model rate constants are based on experiments with isolated chloroplasts, thylakoids, or enzymes, believed to be more direct and reliable than fittings of kinetics with intact leaves. On the other hand, a fluorescence induction trace measured on an intact leaf is also an informative experiment, despite the necessity of computation to evaluate rate constants. Calculations are needed to express the characteristic constants even in in vitro studies. A relevant example is the measurement of electron transport rate from QA − to QB and QA − to QB − in a frequently cited work (de Wijn and van Gorkom 2001). The study involved the measurement of Chl fluorescence yield decay after laser flashes in a series of advancing S-states and altered QB reduction state. In thylakoids isolated from spinach leaves (de Wijn and van Gorkom 2001), as well as in chlorella cells (Kolber et al. 1998) and leaves of different plant species (Osmond et al. 2017), the post-flash decay of chlorophyll fluorescence yield appeared to be highly polyphasic, with only slight differences between the flashes. Deconvolution of the data from the mixture of S-states and correction for the non-linear dependence of fluorescence yield on the concentration of QA − for excitonic connectivity between PSII units were required. As a result of such sophisticated mathematical modeling and application of advanced statistical procedures, two kinetic components could be distinguished in the oxidation of QA − with time constants in the range of 0.2–0.8 and 2–3 ms, respectively, in addition to a minor, much slower component. An original report (Bowes and Crofts 1980) that QB is reduced faster than QB − could be confirmed only conditionally: the fastest time constant of 0.2 ms covered only a part of the QB reduction transient, but the overall fluorescence decay was actually slower than that for the QB − reduction—due to the presence of a slow decay phase that occurs on QB, but not on QB −, reduction. In our work, the J inflection could be reproduced when QB was reduced within 0.5 and QB − within 0.2 ms, slower values being clearly insufficient to cope with the fast electron transport at light intensities above 3000 µmol m−2 s−1. Such a result is even expected in the sense of the quinone stability: while one electron is shared between the QA and QB semiquinones, its stabilization on QB − with an equilibrium constant (kE1) of ~20 is determined by protein environment. As soon as the second electron arrives, the very stable QB quinol rapidly and strongly binds both electrons (de Wijn and van Gorkom 2001). Since the J inflection is a strong experimental fact, we used the rate constants b1d = 2000 for the first and b2d = 5000 s−1 for the second electron. The dip after the J inflection was due to strong quenching by forms with oxidized P680 and/or oxidized TyrZ; YoPoAo, YoPoAr, and YoPrAo.
Thus, the application of computing to the experimental curves is a logically correct—and actually widely used—way of obtaining scientifically novel results. Usually, a limited number of parameters are related to a particular kinetic feature, so that the fear of multiple equivalent solutions is exaggerated.
Thermal phase of fluorescence induction—the notion
We have shown that the “thermal phase” of fluorescence rise kinetics is explained by gradual relaxation of two types of quenching—photochemical quenching due to oxidized QA and donor side quenching due to photo-oxidized TyrZ and P680. The observed fluorescence is a sum of emission from individual PSII units: some are at state F o, some at F m, some at F f, and some quenched to about zero. Total electron transport rate through PSII gradually decreases during the fluorescence rise, but this smooth decline is an average, not reflected in each PSII unit—electron transport is still maximal in units with oxidized QA and reduced donor side, but becomes zero in PSII where QA is reduced or the donor side is oxidized. At the beginning of FI, the forms emitting at F o dominate. The first electron transfer to QA generates TyrZox, but in our time-scale leaves P680 unoxidized, the form assumed to be emitting at F f. Secondary donation from the S-states reduces the TyrZox, generating a fraction of PSII units emitting at F m. Simultaneous electron transfer to QB re-oxidizes QA −, restoring emission at the F o level. Forms of PSII emitting at the F o level and at the F f level gradually disappear, while the forms emitting at the F m level become dominant. At very high PFDs, the model allows double oxidation of the donor side, transiently accumulating a form with TyrZox and P680+, emitting at a level about zero. This complex understanding was facilitated by prior mathematical modeling applying techniques of master equations (Paillotin et al. 1983; Lebedeva et al. 2002; Lazár 2003), but developed further in this work by differently grouping intrinsic electron transfer steps within PSII. It is worth noting that the first-order reactions describing conversions between the PSII forms are actually exponential functions with the given time (or rate) constants. Due to this, the initially synchronized forms rapidly become mixed, e.g., if the S-state donation time constant is 0.2 ms, then at this time only 0.74 of all flash-oxidized TyrZ have become re-reduced.
Our effort is offered with an eye to general scientific progress, i.e., no model is above being revisited. Contemporary information about PSII structure and function discerns too many kinetically complex electron transfer steps to be realistically described with a system of master equations. This brings us to the problem of optimal simplification, as different approaches lead to different simulation results (Lazár and Jablonsky 2009). In the present model, we combined the reversible charge-separated states, Pheo− and PD1 + (or ChlD1 +), into one vibrationally coherent complex P680. In this denotation, P680 is the “six-pack” complex of PD1·PD2·ChlD1·ChlD2·PheoD1·PheoD2 (Renger 2010). Our basic assumption is that exciton localization on PD1·PD2·ChlD1·PheoD1 of this complex, specifically the PheoD1 − ·PD1 + intermediate charge-separated state, is in equilibrium with excitation on the antenna pigment complex (Novoderezhkin et al. 2015). Excitation is photochemically quenched only after an electron is stabilized on QA. The earlier assumption that charge recombination between Pheo− and PD1 + dissipates excitation caused problems in the study of Lazár (2003), which did not reproduce period-four oscillations in flash O2 evolution, and for Belyaeva et al. (2011), who reported a significant influence of membrane potential and lumen acidification on PSII charge transfer efficiency during induction. If P680 would have been considered as the above “six-pack” or “four-pack” (Novoderezhkin et al. 2015) and the state with TyrZox as a normal long-lived form of PSII, then the mysterious “intermediate S-states” (Jablonsky and Lazar 2008) would have been unnecessary.
As reported in this work, earlier models sought to reproduce the fluorescence rise with its OJDIP inflections. The mass action linear approach—equivalent to expression as a sum of exponentials (Strasser and Govindjee 1991; Strasser et al. 2004; Vredenberg 2008a, 2015)—has qualitatively resolved the characteristic sub-processes, laying the base for a series of reports culminating in computer-calculated parameters by a commercial photosynthesis efficiency analyzer (PEA). The master-equation approach was developed to its apotheosis in Lebedeva et al. (2002), Belyaeva et al. (2006), and Lazár (2003). How was it possible that these models reproduced the light-dependent induction curves with their J and I inflections without incorporation of the novel features we introduced in this study? The low flash fluorescence was actually ignored by these models and the J inflection was generated and explained as follows. “Phase J is clearly discerned only under intense enough light, and is associated with accumulation of fluorescing states in which the acceptor is not fully reduced (not more than one electron on QB)” (Lebedeva et al. 2002). “J step is caused by the superposition of the accumulation of excited states that are formed when only QA is reduced, or when QA is reduced together with singly reduced QB” (Lazár 2003). These statements demonstrate a frequent problem: a program may succeed in mimicking the natural process, but the verbal explanation of its internal relationships—notion—may be different. Above the PSII form with QA reduced was stated to emit fluorescence at the J level, but why not at the F m level? Also how can an electron on QB partially quench fluorescence to the J level? In this work, we aimed to promote the understanding of fluorescence emission in terms of PSII electron transport kinetics.
During the J inflection, fluorescence remains briefly static as electron arrival to and departure from QA remain equal, resulting in constant occupation of the QA − form at an average reduction level of about 0.7 at the high light intensity (Fig. 4). The arrival rate depends on light intensity, but it approaches a ceiling limited by donation from the S-states. The departure rate depends on the QA→QB transfer. As discussed above, de Wijn and van Gorkom (2001) revealed the complex nature of this process—the first electron reducing QB initially faster, but the whole process being slower than the single exponential reduction of QB − by the second electron. Our modeling confirms that with the time constants of 0.2 for the first and 0.5 ms for the second electron the J inflection did not appear (Fig. 4b), because the equilibrium QA − occupancy was lower for the first than for the following second electron, continuously rising with time due to the mixing of forms. Contrary to this, the J inflection was pronounced when the transfer of the second electron was set faster than that of the first electron, blending the two rates into a more or less constant balance between the arrival and departure of the two electrons (Fig. 4a). Notably, these kinetics were expressed while QA and QB became reduced within PSII, without exchange with free PQ. Such a test computation was not shown by Lazár (2003, 2009), Lebedeva et al. (2002), or Belyaeva et al. (2011). Therefore, in these models the temporarily balanced QA − state during the J inflection involved the QA→QB as well as the QB→PQ electron transfer. In our hands, a similar computation resulted in Fig. 5a—an over-pronounced J inflection and D dip lasting while the PQ pool became almost fully reduced. This happened because the gradual accumulation of PQH2 had no feedback to QA reduction, due to the extremely large equilibrium constant between the semiquinone and quinol. In order to eliminate such an evidently incorrect response, PQH2 was assumed to be able to reduce QA to some extent and fluorescence quenching by oxidized plastoquinone was assumed (Lazár 2003, 2009). Although possible in thylakoids, this process is not present in leaves (Tóth et al. 2005b). In this work, we suggest a mechanistic process instead of “quenching by oxidized plastoquinone”—competitive binding of reduced plastoquinone to the QB site.
An important electron transfer step is equilibration of the QB site with diffusible PQH2 and PQ species, explicitly modeled in this work compared to Lazár (2009). Although sometimes diffusion of the quinone species is considered rate limiting for photosynthesis (Lavergne and Joliot 1991; Joliot et al. 1992; Kirchhoff et al. 2000; Haferkamp et al. 2010), our recent work on intact leaves showed very fast equilibration (within less than 1 ms) of the diffusible quinone species with the QB site on one hand and with the primary quinone binding p-site of Cyt b6f on the other hand (Laisk et al. 2016). In accordance with this, the rate constants for dissociation of PQH2 from the QB site (rq d = 2000 s−1) and association of PQ with the QB site (oq r = 2000 s−1 per PQ = 1 per PSII) were set fast. The common understanding that PQH2 is always rapidly exchanged for PQ in the QB site did not work, since the calculated induction trace was strongly sigmoidal, expressing a very long J inflection in the log scale, significantly different from the experimental one (Fig. 5). A better result was obtained when fast re-association of PQH2 with and dissociation of PQ from the QB site were allowed, in accordance with the notion that neither quinone nor quinol bind tightly (Crofts and Wraight 1983): QBH2 + PQ ↔ QB + PQH2, K 3 = 1. In order to fit the calculated induction trace to the measured one, the PQH2 re-association rate constant rq r had to be set at least equal to or even faster (rq r = 5000 s−1 per PQH2 = 1 per PSII) than its dissociation rate constant (rq d = 2000 s−1). Initially, the high affinity of the QB site for reduced PQH2 was detected from light pulsing experiments in intact sunflower leaves. When PQ was reduced illuminating a leaf with gradually longer multiple-turnover light pulses, the rate of PSII electron transport decreased proportionally with the decreasing fraction of oxidized PQ. Such apparently linear kinetics with respect to PQ, earlier detected from fluorescence measurements (Tóth et al. 2007b), were suggested to be caused by strong product inhibition by PQH2, blocking the QB site for electron transfer in proportion with its pool size (Oja et al. 2011; Laisk et al. 2015). As a result, fluorescence yield is rather proportional to PQH2 reduction state during induction (Tóth et al. 2007b; Fig. 5b).
This kinetic property explains the peculiar discrepancy in temporal courses of fluorescence induction (Tóth et al. 2005a; Laisk et al. 2015; Schansker et al. 2011). When light is turned on for the first time, fluorescence rise is slow, approaching F m during a light pulse of about 200 ms. In the dark after the pulse fluorescence decreases initially faster, but approaches a low value of about 2 F o within a few seconds. When the light is turned on again, fluorescence approaches F m within a millisecond, quite like in DCMU-treated leaves. In kinetic terms, the result indicates that after the pulse PQH2, and in equilibrium with it QB, remains reduced for a longer time, but QA becomes oxidized, probably due to charge recombination—despite the fact that QB is occupied by PQH2. When light is turned on again, the re-induction of fluorescence is fast, as with DCMU.
PQH2 oxidation by the Q-cycle in the Cyt b6f complex is the major rate-limiting event in photosynthetic electron transport. Although a complex process involving two oxidations of PQH2 and one reduction of PQ, its overall kinetics are equivalent to two first-order reactions in series, the first being faster and the second slower (Laisk et al. 2016). In the present approach, complete PQH2 oxidation was modeled with a single first-order rate constant of 50 s−1—the sum of the two component reaction times. It must be emphasized that the frequently cited rate of post-illumination reduction of P700 and PC of about 100 to 200 s−1 is not the rate constant of PQH2 double oxidation, but involves the pool sizes of P700 and PC, but each possessing one redox electron. During induction, the Q-cycle rate gradually decreases, being limited by the availability of the electron acceptor, oxidized Cyt f. Thylakoid membrane potential may also participate in this process (Lebedeva et al. 2002; Belyaeva et al. 2011), but during the relatively fast fluorescence rise its role is indistinguishable from that of the gradually reducing electron acceptors. Neglecting master equations for the Cyt b6f complex and PSI complex (see Lebedeva et al. 2002) could cause inaccuracies in calculating the true reduction states of Cyt f, P700, and the PSI acceptor FX (a formal denotation, not necessarily the corresponding FeS complex), but this could hardly be reflected in the corresponding PSII fluorescence traces.
Similarity of the calculated traces to the measured curves (Schansker et al. 2011) confirmed that the kinetic characteristics of electron transport employed and their assumed relationship to Chl fluorescence are realistic. This proves that electrochemical quenching due to membrane potential difference (Vredenberg et al. 2009) and control of transmembrane charge transfer by membrane potential (Lebedeva et al. 2002; Belyaeva et al. 2006, 2011), which were omitted from our model, do not significantly control PSII electron transport and fluorescence emission during the dark–light fluorescence rise, at least at high light intensities. Most kinetic phenomena considered unexplainable in the framework of the QA model and requiring involvement of protein conformational changes (Schansker et al. 2011, 2014) are explainable as follows:
-
A saturating single-turnover flash cannot generate more than 60–65% of the maximum fluorescence intensity and the J step is only 60–65% of the Fm value.
We propose that oxidized TyrZ is related to quenching of the flash fluorescence F f. The fast (within a microsecond) onset of this quenching during the ST flash cannot involve conformational changes. At the J step QA is partially reduced indeed.
-
Electron flow through PSI has a kinetic effect on the Chl a fluorescence rise, i.e., a reduced electron transport chain is a pre-condition for reaching Fm.
A reduced chain involves reduced plastoquinone. We propose that PQH2 is a product inhibitor of PSII, binding competitively to the QB site. With highly reduced PQH2, electron transport kinetically simulates inhibition by DCMU.
-
Even high light of 15,000 μmol photons m−2 s−1 is not sufficient to eliminate the IP-phase.
The I inflection level is lower than F m because the balanced level of PQH2 adjusts below its complete reduction. The rate of PQ reduction increases with light intensity, but the ceiling is donation by water splitting. This high PFD exceeds the maximum donation rate, and thus the I level becomes independent of light.
-
The J step does not represent a single charge separation but 2–3 charge separations, and the J and I steps do not change position in response to changes in the light intensity.
The J step appears as a temporarily stabilized QA reduction state, while electron donation by photochemistry and departure by QA→QB transition remain equal. The J step occurs while the first and second electrons are reducing QB, but may be extended if QBH2 exchange with QB is fast enough. The J inflection does not change position because at low light the lower J level compensates for the shift to the right due to slow excitation. The I step is mainly determined by electron transfer kinetics from PSII to PSI, not photochemistry.
-
The relaxation kinetics of the thermal phase, occurring within 100 ms, cannot be explained by any known redox reaction involving the re-oxidation of QA.
If a process cannot be explained by any known redox reduction, then (a) a novel redox reaction has to be suggested or the rate constant of a known reaction modified or (b) a conformational change may be suggested. We propose the modification of rate constants of charge recombination between QA − and some S-states, rather than a conformational change relaxing faster than within a millisecond.
-
The kinetics of the fluorescence rise at low temperatures do not agree with a photochemical reaction leading to the reduction of QA.
Fluorescence rise kinetics rather similar to the experimental observations were modeled by assuming slow electron donation from an S-state at low temperature (Fig. 3).
The above arguments however do not mean that conformational changes accompanying PSII electron transport are excluded. The faster increase of the measured compared to the calculated fluorescence at low light intensities (Fig. 7) required light-dependent adjustment of kinetic parameters within PSII: the QA-to-QB transfer rates had to be decreased and QB reduction set faster than QB − reduction. Notably, for the low-light FI curves the fitting QA→QB rate constants were 1000 for the first and 500 s−1 for the second electron, as appeared to be necessary to eliminate the low-level J inflection and ensure the continuous rise of fluorescence up to the I inflection at about 80 ms. Conditions and details of this phenomenon need further investigation and modeling, but it may just be the experimental indication of conformational changes controlling QA to QB electron transfer within PSII, as once suggested for bacterial photosynthetic centers (Xu et al. 2002). Another peculiar feature—the temporary decay upon darkening and fast recovery of fluorescence upon re-illumination—should promote further investigations of charge recombination from QA − in the dark and QA re-reduction upon repeated illumination.
The present modeling approach was motivated by measurements showing significantly smaller rates of PSII electron transport based on O2 evolution compared to fluorescence quenching (Oja et al. 2011; Laisk et al. 2012, 2015; Laisk and Oja 2013). According to these results, some of the fluorescence quenching must be of non-photochemical origin or related to non-oxygenic cyclic electron transport within PSII (Laisk et al. 1994). In most of the cited models, P680+ was assumed to be the non-photochemically quenching species (for alternative suggestions see Introduction). Strasser (1997) explained the K step observable with damaged water-splitting complex (Fig. 8) in terms of accumulation of oxidized TyrZ, causing subsequent accumulation of P680+ as the actual quenching form. Vredenberg (2008b) considered the possibility of fluorescence quenching by oxidized TyrZ, but could not find convincing arguments from experiments with DCMU-inhibited Chenopodium chloroplasts. Nevertheless, the amply documented period-four oscillation of the STF-induced variable fluorescence (Schreiber and Neubauer 1987; Kolber et al. 1998; Koblížek et al. 2001; Shinkarev 2004) was a strong argument for the S-state-related mechanism of diminished F f fluorescence yield (Vredenberg 2008b).
The water-splitting complex becomes inhibited in leaves when exposed to temperatures exceeding 45 °C. Indeed, when temperature was gradually increased with a darkened leaf, fluorescence, initially at the F o level, gradually increased to 0.5 F m in the dark at temperatures >45 °C (Laisk et al. 1998). When saturation pulses were imposed on the same leaf, F m was initially high but gradually decreased to a half at >45 °C—variable fluorescence disappeared. In this experiment, the yield of 0.5 F m was adjusted in the absence of electron transport, when the q E-type non-photochemical quenching was certainly absent. Therefore, it is very tempting to interpret this fluorescence yield as that of PSII with TyrZ being oxidized by the saturation pulse or by back-reaction to the damaged S-state in the dark.
Fluorescence yield, F f, of flash-excited PSII units is about 3 to 3.5F o, while the F m values are about 5 to 7F o (Neubauer and Schreiber 1987; Samson and Bruce 1996; Vredenberg et al. 2007). In our calculations, the F f yield was initially set at 0.5F m (the DCMU calculations), but later increased to 0.66F m based on the fit of the calculated J inflection level to the experimental curves. Instead of F o, we used F m as a natural fluorescence unit, because it is represented by a basic physical rate constant—the sum of excitation conversion rates to heat, triplet, and fluorescence—normalized to unity. F o involves also the rate constant of photochemistry, which may vary depending on conditions. In well-conditioned leaves F m = 9F o for the PSII fluorescence (Peterson et al. 2014), and therefore 0.5F m = 4.5, but 0.66F m = 6 F o. The fast response measurements of (Steffen et al. 2005) did not detect TyrZox as a quencher, but focused on QA, P680+, and triplet 3Chl (or 3Car). But these authors overlooked that their maximum fluorescence F m approached only 2F o at 50 µs after the flash—when triplets had decayed and P680+ was re-reduced. Knowing the actual high F m/F o ratio, the peak denoted F m in Steffen et al. (2005) was significantly underestimated—most likely because TyrZ was simultaneously oxidized but not considered as a quencher.
The partial quenching of excitation to the level F f < F m by oxidized TyrZ is difficult to explain. Earlier (Laisk et al. 2012), we suggested a photochemical quenching hypothesis, based on cycling of electrons within PSII bypassing QA. Since tyrosine is to some extent similar to quinone functioning as an electron carrier, Laisk et al. (2012) and Laisk and Oja (2013) suggested electron cycling in a triangular pathway.
In the state of oxidized TyrZ and reduced P680 and QA, the excited electron leaves P680* quenching excitation, but arrives at TyrZox, from where it rapidly cycles to oxidized P680. This cycle would need only 1 µs of electron transfer time from TyrZ to P680+, plus about 2 ns for electron transfer from excited P680* to TyrZox. Such a fast charge transfer from the excited ChlD1 to oxidized TyrZ would be allowed by the tunneling distance of about 8 Å between these electron carriers (Umena et al. 2011), but the problem is the too large free energy gap (Moser et al. 2005). During laser flashes of many excitations per µs, this model would still predict the accumulation of P680+ with reduced TyrZ, which would quench fluorescence below F o right after the flash—as actually reported by Belyaeva et al. (2015), but suggested to be the quenching by 3Car. Although not conclusive, these arguments still allow oxidized TyrZ to quench excitation non-photochemically.
The obvious discrepancy between the fluorescence-based and O2-based electron transport (Oja et al. 2011; Laisk et al. 2012; Laisk and Oja 2013) increased with light intensity, suggesting a mechanism similar to the Geiger–Müller counter of nuclear particles: after every photon transporting an electron, a dead time follows, during which photons absorbed by the antenna are lost for charge transfer (Laisk and Oja 2013). Due to the assumed possibility of double oxidation (accumulation of TyrZox and P680+) generating a strong quencher, the discrepancy—expressed as the dead time τ after each successful electron transfer—is low (dead time short) at low PFDs and increases non-linearly with rising PFD. This is caused by the buffering effect of the double oxidation: in the single oxidation case, the second photon is lost in pairs separated by time interval < τ, but in the double oxidation case only the third photon is lost in triples separated by time interval < τ. In the single oxidation case, the dead time is constant, but in the double oxidation case the effective dead time is shorter than that of the single oxidation case at low light intensities, approaching the single oxidation value with increasing light intensity (Fig. 11b). The measured dependence of the type of Fig. 11a was not concave as predicted by the double oxidation, but rather was convex, transforming into decreasing τ with increasing light intensity (Laisk and Oja 2013). This experiment however was not definite due to the limited amplitude of PSII excitation rate. More significant may be the very low 810-nm signal from P680+ detected in sunflower leaves at a PFD of 7000 µmol m−2 s−1, equivalent to 2% of the full signal from P700+ (Laisk et al. 2012). At this PFD, the YoPo forms were calculated to accumulate up to 20% of all PSII forms (Fig. 4d). Experimentally, P680+ has been detected only when the water-splitting complex was inhibited (Steffen et al. 2005; Belyaeva et al. 2014, 2015). Thus, it may be possible that the state with TyrZ oxidized but P680 unoxidized is unable for photochemical charge transfer.
In conclusion, the post-flash fluorescence yield F f is observed when TyrZ is oxidized. Therefore, we relate the quenching to TyrZ, but cannot say that oxidized TyrZ is the quencher. Our model explains the S-state-dependent rate of relaxation of the F f quenching, but not the S-state dependency of the F f level. These facts still leave room for transmembrane electric field and proton concentration, as well as for protein conformational changes as factors controlling excitation quenching.
Abbreviations
- F o :
-
Photochemically quenched fluorescence yield
- F f :
-
Post-flash fluorescence yield
- F m :
-
Maximum unquenched fluorescence yield
- FI:
-
Fluorescence induction rise from F o to F m
- PFD:
-
Photon flux density
- TyrZ:
-
Tyrosine Z
- STF:
-
Single-turnover flash
References
Baake E, Schlöder JP (1992) Modeling the fast fluorescence rise of photosynthesis. Bull Math Biol 54:999–1021
Baake E, Strasser RJ (1990) A differential equation model for the description of the fast fluorescence rise (O-I-D-P-transient) in leaves. In: Current research in photosynthesis vol.I. Kluwer, The Netherlands
Belyaeva NE, Pashchenko VZ, Renger G, Riznichenko GY, Rubin AB (2006) Application of a photosystem II model for analysis of fluorescence induction curves in the 100 ns to 10 s time domain after excitation with a saturating light pulse. Biophysics 51:860–872
Belyaeva NE, Bulychev AA, Riznichenko GY, Rubin AB (2011) A model of photosystem II for the analysis of fast fluorescence rise in plant leaves. Biophysics Pleiades Publ 56:464–477
Belyaeva NE, Schmitt F-J, Paschenko VZ, Riznichenko GY, Rubin AB, Renger G (2014) Model based analysis of transient fluorescence yield induced by actinic laser flashes in spinach leaves and cells of green alga Chlorella pyrenoidosa Chick. Plant Physiol Biochem 77:49–59
Belyaeva NE, Schmitt F-J, Paschenko VZ, Riznichenko GY, Rubin AB (2015) Modeling of the redox state dynamics in photosystem II of Chlorella pyrenoidosa Chick cells and leaves of spinach and Arabidopsis thaliana from single flash-induced fluorescence quantum yield changes on the 100 ns–10 s time scale. Photosynth Res 125:123–140
Bowes JM, Crofts AR (1980) Binary oscillations in the rate of oxidation of the primary acceptor of Photosystem II. Biochim Biophys Acta 590:373–384
Christen G, Reifarth F, Renger G (1998) On the origin of the ‘35-µs kinetics’ of P680+· reduction in photosystem II with an intact water oxidizing complex. FEBS Lett 429:49–52
Crofts AR, Wraight CA (1983) The electrochemical domain of photosynthesis. Biochim Biophys Acta 726(2):149–185
de Wijn R, van Gorkom HJ (2001) Kinetics of electron transfer from QA to QB in photosystem II. BioChemistry 40:11912–11922
Delosme R (1967) Étude de l’induction de fluorescence des algues vertes et des chloroplastes an début d’une illumination intense. Biochim Biophys Acta 143:108–128
Duysens LNM, Sweers HE (1963) Mechanisms of two photochemical reactions in algae as studied by means of fluorescence. In: Physiologists JSoP (ed) Studies on microalgae and photosynthetic bacteria, special issue of plant and cell physiology. University of Tokyo Press, Tokyo, pp 353–372
Haferkamp S, Haase W, Pascal AA, van Amerongen H, Kirchhoff H (2010) Efficient light harvesting by photosystem II requires an optimized protein packing density in grana thylakoids. J Biol Chem 285:17020–17028
Jablonsky J, Lazar D (2008) Evidence for intermediate S-states as initial phase in the process of oxygen-evolving complex oxidation. Biophysical J 94:2725–2736
Joliot A, Joliot P (1964) Étude cinétique de la réaction photochimique libérant l’oxygène au cours de la photosynthése. C R Acad Sc Paris 258:4622–4625
Joliot P, Joliot A (1977) Evidence for a double hit process in photosystem II based on fluorescence studies. Biochim Biophys Acta 462:559–574
Joliot P, Joliot A (1981) Double photoreactions induced by laser flash as measured by oxygen emission. Biochim Biophys Acta 638:132–140
Joliot P, Lavergne J, Béal D (1992) Plastoquinone compartmentation in chloroplasts. I. Evidence for domains with different rates of photo-reduction. Biochim Biophys Acta 1101:1–12
Kirchhoff H, Horstmann S, Weis E (2000) Control of the photosynthetic electron transport by PQ diffusion microdomains in thylakoids of higher plants. Biochim Biophys Acta 1459:148–168
Koblížek M, Kaftan D, Nedbal L (2001) On the relationship between the non-photochemical quenching of the chlorophyll fluorescence and the photosystem II light harvesting efficiency. A repetitive flash fluorescence induction study. Photosynth Res 68:141–152
Kolber Z, Prasil O, Falkowski PG (1998) Measurements of variable chlorophyll fluorescence using fast repetition rate technique. I. Defining methodology and experimental protocols. Biochim Biophys Acta 1367:88–106
Laisk A, Oja V (2013) Thermal phase and excitonic connectivity in fluorescence induction. Photosynth Res 117:431–448
Laisk A, Oja V, Eichelmann H (1994) Dependence between the quantum yield of photosystem II and chlorophyll fluorescence at constant nonphotochemical quenching. In: BBSRC Second Robert Hill symposium on photosynthesis. Imperial College of Science, Technology and Medicine, London, pp 59–60
Laisk A, Rasulov BH, Loreto F (1998) Thermoinhibition of photosynthesis as analyzed by gas exchange and chlorophyll fluorescence. Russian J Plant Physiol 45(4):412–421
Laisk A, Nedbal L, Govindjee (eds) (2009) Photosynthesis in silico. Understanding complexity from molecules to ecosystems. Advances in photosynthesis and respiration, vol 29. Springer, Dordrecht
Laisk A, Eichelmann H, Oja V (2012) Oxygen evolution and chlorophyll fluorescence from multiple turnover light pulses: charge recombination in photosystem II in sunflower leaves. Photosynth Res 113:145–155
Laisk A, Eichelmann H, Oja V (2015) Oxidation of plastohydroquinone by photosystem II and by dioxygen in leaves. Biochim Biophys Acta 1847:565–575
Laisk A, Oja V, Eichelmann H (2016) Kinetics of plastoquinol oxidation by the Q-cycle in leaves. Biochim Biophys Acta 1857:819–830
Lavergne J, Joliot P (1991) Restricted diffusion in photosynthetic membranes. TIBS 16:129–134
Lazár D (2003) Chlorophyll a fluorescence rise induced by high light illumination of dark-adapted plant tissue studied by means of a model of photosystem II and considering photosystem II heterogeneity. J Theor Biol 220:469–503
Lazár D (2009) Modelling of light-induced chlorophyll a fluorescence rise (O-J-I-P transient) and changes in 820 nm-transmittance signal of photosynthesis. Photosynthetica 47:483–498
Lazár D (2013) Simulations show that a small part of variable chlorophyll a fluorescence originates in photosystem I and contributes to overall fluorescence rise. J Theor Biol 335:249–264
Lazár D, Jablonsky J (2009) On the approaches applied in formulation of a kinetic model of photosystem II: different approaches lead to different simulations of the chlorophyll a fluorescence transients. J Theor Biol 257:260–269
Lazár D, Schansker G (2009) Models of chlorophyll a fluorescence transients. In: Laisk A, Nedbal L, Govindjee (eds) Photosynthesis in silico. Understanding complexity from molecules to ecosystems. Springer Science + Business Media B.V., Dordrecht, pp 85–123
Lebedeva GV, Belyaeva NE, Demin OV, Riznichenko GY, Rubin AB (2002) Kinetic model of primary photosynthetic processes in chloroplasts. Description of the fast phase of chlorophyll fluorescence induction under different light intensities. Biophysics 47(966):968–980 Translated from Russian Biofizika, Vol. 947, No. 966, 2002, pp. 1044–1058
Moser CC, Page CC, Dutton PL (2005) Tunneling in PSII. Photochem Photobiol Sci 4:933–939
Neubauer C, Schreiber U (1987) The polyphasic rise of chlorophyll fluorescence upon onset of strong continuous illumination: I. Saturation characteristics and partial control by the photosystem II acceptor side. Z Naturforschung 42c:123–131
Novoderezhkin VI, Romero E, van Grondelle R (2015) How exciton-vibrational coherences control charge separation in the photosystem II reaction center. Phys Chem Chem Phys 17:30828–30841
Oja V, Eichelmann H, Anijalg A, Rämma H, Laisk A (2010) Equilibrium or disequilibrium? A dual-wavelength investigation of photosystem I donors. Photosynth Res 103:153–166
Oja V, Eichelmann H, Laisk A (2011) Oxygen evolution from single- and multiple-turnover light pulses: temporal kinetics of electron transport through PSII in sunflower leaves. Photosynth Res 110:99–109
Osmond B, Chow WS, Wyber R, Zavafer A, Keller B, Pogson BJ, Robinson SA (2017) Relative functional and optical absorption cross-sections of PSII and other photosynthetic parameters monitored in situ, at a distance with a time resolution of a few seconds, using a prototype light induced fluorescence transient (LIFT) device. Functional Plant Biol. doi:10.1071/FP17024
Paillotin G, Geactinov NE, Breton J (1983) A master equation theory of fluorescence induction, photochemical yield, and singlet-triplet exciton quenching in photosynthetic systems. Biophys J 44:65–77
Peterson RB, Oja V, Eichelmann H, Bichele I, Dall’Osto L, Laisk A (2014) Fluorescence F0 of photosystems II and I in developing C3 and C4 leaves, and implications on regulation of excitation balance. Photosynth Res 122:41–56
Rappaport F, Blanchard-Desce M, Lavergne J (1994) Kinetics of electron transfer and electrochromic change during the redox transitions of the photosynthetic oxygen-evolving complex. Biochom Biophys Acta 1184:178–192
Renger G (2010) The light reactions of photosynthesis. Current Sci 98:1305–1319
Robinson H, Crofts AR (1983) Kinetics of the oxidation-reduction reactions of the photosystem II quinone acceptor complex, and the pathway for deactivation. FEBS Lett 153:221–226
Samson G, Bruce D (1996) Origin of the low yield of chlorophyll fluorescence induced by single turnover flash in spinach thylakoids. Biochim Biophys Acta 1276:147–153
Schansker G, To’th S, Kova’cs L, Holzwarth AR, Garab G (2011) Evidence for a fluorescence yield change driven by a light-induced conformational change within photosystem II during the fast chlorophyll a fluorescence rise. Biochim Biophys Acta 1807:1032–1043
Schansker G, Tóth SC, Holzwarth AR, Garab G (2014) Chlorophyll a fluorescence: beyond the limits of the QA model. Photosynth Res 120:43–58
Schreiber U (2002) Assessment of maximal fluorescence yield. Donor-side dependent quenching and QB-quenching. In: Van Kooten O, Snel J (ed) Plant spectrophotometry: applications and basic research. Rozenberg, Amsterdam, pp 23–47
Schreiber U, Neubauer C (1987) The polyphasic rise of chlorophyll fluorescence upon onset of strong continuous illumination: II. Partial control by the photosystem II donor side and possible ways of interpretation. Z Naturforsch 42c:132–141
Schreiber U, Klughammer C, Kolbowski J (2012) Assessment of wavelength-dependent parameters of photosynthetic electron transport with a new type of multi-color PAM chlorophyll fluorometer. Photosynth Res 113:127–144
Shinkarev VP (2004) Photosystem II: oxygen evolution and chlorophyll a fluorescence induced by multiple flashes. In: Papageorgiou GC, Govindjee (eds) Chlorophyll a fluorescence: a signature of photosynthesis. Springer, Dordrecht, pp 197–229
Steffen R, Eckert H-J, Kelly AA, Dörmann PG, Renger G (2005) Investigations on the reaction pattern of photosystem II in leaves from Arabidopsis thaliana by time-resolved fluorometric analysis. BioChemistry 44:3123–3132
Stirbet A, Govindjee (2012) Chlorophyll a fluorescence induction: a personal perspective of the thermal phase, the J-I-P rise. Photosynth Res 113:15–61
Strasser BJ (1997) Donor side capacity of photosystem II probed by chlorophyll a fluorescence transients. Photosynth Res 52:147–155
Strasser RJ, Govindjee (1991) The F 0 and the O-J–I–P fluorescence rise in higher plants and algae. In: A-A JH (ed) Regulation of chloroplast biogenesis. Plenum Press, New York, pp 423–426
Strasser R, Tsimili-Michael M, Srivastava A (2004) Analysis of the chlorophyll a fluorescence transient. In: Papageorgiou GC, Govindjee (eds) Chlorophyll a fluorescence. A signature of photosynthesis. Springer, Dordrecht, pp 321–362
Sušila P, Lazár D, Ilik P, Tomek P, Nauš P (2004) The gradient of exciting radiation within a sample affects the relative height of steps in the fast chlorophyll a fluorescence rise. Photosynthetica 42:161–172
Tóth SZ, Schansker G, Strasser RJ (2005a) In intact leaves, the maximum fluorescence level (F M) is independent of the redox state of the plastoquinone pool: a DCMU-inhibition study. Biochim Biophys Acta 1708:275–282
Tóth SZ, Schansker G, Strasser RJ (2005b) In intact leaves, the maximum fluorescence level (FM) is independent of the redox state of the plastoquinone pool: a DCMU-inhibition study. Biochim Biophys Acta 1708:275–282
Tóth SZ, Schansker G, Garab G, Strasser RJ (2007a) Photosynthetic electron transport activity in heat-treated barley leaves: the role of internal alternative electron donors to photosystem II. Biochim Biophys Acta 1767:295–305
Tóth SZ, Schansker G, Strasser RJ (2007b) A non-invasive assay of the plastoquinone pool redox state based on the OJIP-transient. Photosynth Res 93:193–203
Umena Y, Kawakami K, Shen J-R, Kamiya N (2011) Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 473:55–61
Vredenberg VJ (2008a) Algorithm for analysis of OJDIP fluorescence induction curves in terms of photo- and electrochemical events in photosystems of plant cells. Derivation and application. J Photochem Photobiol B 91:58–65
Vredenberg WJ (2008b) Analysis of initial chlorophyll fluorescence induction kinetics in chloroplasts in terms of rate constants of donor side quenching release and electron trapping in photosystem II. Photosynth Res 96:83–97
Vredenberg W (2015) A simple routine for quantitative analysis of light and dark kinetics of photochemical and non-photochemical quenching of chlorophyll fluorescence in intact leaves. Photosynth Res 124:87–106
Vredenberg W, Durchan M, Prášil O (2007) On the chlorophyll a fluorescence yield in chloroplasts upon excitation with twin turnover flashes (TTF) and high frequency flash trains. Photosynth Res 93:183–192
Vredenberg W, Durchan M, Prášil O (2009) Photochemical and photoelectrochemical quenching of chlorophyll fluorescence in photosystem II. Biochim Biophys Acta 1787:1468–1478
Xu Q, Baciou L, Sebban P, Gunner MR (2002) Exploring the energy landscape for QA – to QB electron transfer in bacterial photosynthetic reaction centers: effect of substrate position and tail length on the conformational gating step. BioChemistry 41:10021–10025
Zhu X-G, Govindjee, Baker NR, deSturler E, Ort DR, Long SP (2005) Chlorophyll a fluorescence induction kinetics in leaves predicted from a model describing each discrete step of excitation energy and electron transfer associated with photosystem II. Planta 223:114–133
Acknowledgements
This work was supported by Personal Grant No. 393P from Estonian Ministry of Education and Science and by personal support to A.L. from Estonian Academy of Science. Comments and help by R. Peterson (Scientist Emeritus, The Connecticut Agricultural Experiment Station) are highly appreciated.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Laisk, A., Oja, V. Kinetics of photosystem II electron transport: a mathematical analysis based on chlorophyll fluorescence induction. Photosynth Res 136, 63–82 (2018). https://doi.org/10.1007/s11120-017-0439-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-017-0439-y