
Abstract
Multi-color fluorescence emission from leaf tissues is presented as a powerful reporter on plant biochemistry and physiology that can be applied both at macro- and micro-scales. The blue–green fluorescence emission is typically excited by ultraviolet (UV) excitation. However, this approach cannot be applied in investigating intact leaf interior because the UV photons are largely absorbed in the epidermis of the leaf surface. This methodological barrier is eliminated by replacing the UV photon excitation by excitation with two infra-red photons of the same total energy. We demonstrate this approach by using two-photon excitation for microscopy of Arabidopsis thaliana leaves infected by pathogenic bacterium Pseudomonas syringae. The leaf structures are visualized by red chlorophyll fluorescence emission reconstructed in 3-D images while the bacteria are detected by the green emission of engineered fluorescence protein.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Photoautotrophic organisms are unique by their capacity to use light for driving their metabolism by photosynthesis. Light is also a powerful reporter signal probing the plant structure and function by reflection, scattering, and absorption that is followed by fluorescence emission. Here, we focus on the fluorescence emission of higher plants that is excited by photons of the solar spectrum. The UV-A and short-wavelength blue photons are strongly absorbed by aromatic compounds covalently bound to cell walls of epidermis and found in vacuoles (Buschmann et al. 2000). Another class of short-wavelength absorbing compounds comprises pyridine and flavine nucleotides in the mesophyll tissue (Cerovic et al. 1999). Leading to the photosynthetic energy conversion, the blue photons are strongly absorbed by chlorophylls a and b and by carotenes and xanthophylls of the thylakoid membranes (Frank et al. 1997). Green photons are absorbed by chlorophyll less strongly than blue or red light and penetrate deep into the plant tissue (Vogelmann and Evans 2002). Red photons are absorbed by chlorophyll a and b but not by carotenoids and, thus, penetrate the tissue deeper than the blue photons. A more exhaustive overview of plant fluorophores can be found in Ross (1995).
Either of the plant fluorophores is brought by the photon absorption into an excited singlet state. The excitonic energy can be dissipated into heat or emitted as light in form of fluorescence photons. The energy absorbed by photosynthetic pigments of thylakoid membranes (Lichtenthaler 1987) is primarily used to drive photochemistry in the reaction centers (reviewed in Blankenship (2002)) while only a small fraction of the absorbed light, is de-excited through fluorescence emission or lost as heat (Kitajima and Butler 1975, reviewed recently in Maxwell and Johnson 2000).
The fluorescence photons are always of lower energy than the ones absorbed (Condon 1926; Franck and Dymond 1926). The blue and green fluorescence (400–630 nm) is excited by UV-A and blue photons (Cerovic et al. 1999), while the red and infrared fluorescence (630–800 nm) (Strasser and Butler 1977) is emitted by chlorophyll a (ChlF) of reaction center that can be excited directly or receives the energy absorbed in blue and red absorption bands of the light harvesting complexes.
Recently, spectrum of the fluorescence reporter signals was extended by a variety of fluorescent proteins (FPs) that can be fused to proteins of interest to report on their cellular localization or biosynthesis (Rizzo and Piston 2004; Shaner et al. 2005). The FP variants can be traced back to the original, green fluorescence protein (GFP) that was isolated from the jellyfish Aequorea victoria and that is specific by its typical green fluorescence emission occurring upon UV excitation (Chalfie et al. 1994; Tsien 1998). Mutagenic interventions yielded a number of FP variants with different absorbance spectra extending to blue and cyan in the short-wavelength range and to yellow and red emission in the long-wavelength range (Lippincott-Schwartz and Patterson 2003).
Both the autofluorescence and the emission of FP are typically heterogeneous in space. Fluorescence emission of interveinal leaf segments, of veins, of fruits, and other organs can differ significantly. Further heterogeneity can occur due to ontogeny of the organism or due to abiotic and biotic stresses (Berger et al. 2007; Endo and Omasa 2007; Pineda et al. 2008). The heterogeneity of the fluorescence emission of plants can be correctly captured by using fluorescence imaging techniques (Nedbal and Whitmarsh 2004). Fluorescence imaging is currently possible on a wide range of scales, from trees (Omasa et al. 2007) to resolving emission of individual chloroplasts (Vácha et al. 2007).
A de-convolution of the fluorescence signals emitted from different cellular structures has been traditionally achieved by cutting the leaf into sections and by conventional fluorescence microscopy (Ross 1995). Non-invasive optical sectioning is possible by the confocal microscopy that is based on an iris diaphragm (pinhole) that eliminates the out-of-focus signals (Pawley 1995). However, the use of confocal microscopy to image plant leaves is limited by high tissue opacity (Feijó and Moreno 2004). Moreover, acquiring a high contrast confocal image requires a strong UV-A or blue laser excitation leading to bleaching and destruction of the inherently photosensitive samples. In addition, single-photon fluorescence excitation by UV wavelengths, that is typically required in confocal microscopy, is restricted to surface leaf layers because the leaf interior is shielded by a protective UV-blocking chromophore barrier (Cerovic et al. 2002).
Some of these obstacles can be avoided by using two-photon microscopy (TPM) (Bhawalkar et al. 1996; Diaspro and Robello 2000; König 2000). Figure 1 compares the principles of microscopy based on the single-photon and two-photon excitation. The blue arrow in Fig. 1a shows a single-photon excitation from the ground state S to a singlet excited state S* while the two red arrows represent two infra-red photons that lead to the same process when absorbed simultaneously. The single-photon excitation occurs in the entire double cone illuminated by the microscope objective (blue in Fig. 1b) and, thus, photodamage and photobleaching occur with confocal, single-photon microscopy in a large tissue volume (green in Fig. 1b). In contrast, the infrared photons used in two-photon microscopy pass the leaf tissue without being absorbed (red double cone in Fig. 1b). Only in the objective focus, the photon density is high enough to elicit simultaneous two-photon absorption (Diaspro and Robello 2000). Thereby, only fluorophores in the focus, corresponding to sub-femtoliter focal volume, are excited (the green spot in Fig. 1b) and photodamage is limited. Also, by the same argument, fluorescence emission can be attributed to a small tissue volume. This optical cell layer sectioning and reduced photo-bleaching are the major strengths of TPM. Another advantage is due to lower scattering of the infra-red photons compared to blue or UV photons that are used in the single-photon microscopy. Less scattered, the infra-red photons penetrate deeper into the specimen. Imaging of brain up to 500 μm was reported by Oheim et al. (2001). Such an imaging depth is hard to achieve in leaves that are containing densely packed chromophores making passage of photons difficult even though these are in the infra-red range of low absorption. In spite of these complications, two-photon images of chloroplasts 50 μm deep in a young leaf tissue were published (Feijó and Moreno 2004). Last but not the least, the TPM makes possible to image fluorescence emission from fluorophores that are deep in the leaf tissue where UV or blue photons cannot reach because of the screening epidermis barrier.

a The Jablonski diagram showing single (blue arrow) and two-photon excitation (red arrows), both leading to the same fluorescence emission (green arrow). b Irradiance profiles near the microscope focus for the single-photon excitation (left panel) and for the two-photon excitation (right panel). The single-photon absorption is directly proportional to the photon flux density of incident light resulting in double-cone profile formed by the objective lens (the green double cone left). Two-photon absorption requires coincidence of two photons and, thus, depends on the square of the incident light intensity. Thus, only molecules that reside in sub-femtoliter focal volume generated by high-numerical-aperture objective are excited (the green ellipse right)
Here, we present macroscopic as well as microscopic fluorescence images of plants that were captured in green as well as red emission bands. The reported fluorescence emission is excited both by single and two-photon absorption. We also demonstrate the power of combining measurements of autofluorescence emission from the intrinsic fluorophores with fluorescent proteins in plant invading pathogen Pseudomonas syringae.
Materials and methods
Macroscopic fluorescence imaging
Macroscopic fluorescence images of whole leaves were captured using FluorCam 800-C (P.S.I., Brno Czech Republic, www.psi.cz) adopted for multicolor detection. Both green fluorescence and ChlF in Fig. 2 were elicited with blue excitation generated by panels of light emitting diodes (LED, λmax ≈ 455 nm, LXHL-PR09, Lumileds Lighting, LLC, San Jose, California, USA). The excitation spectrum was restricted by a short-pass filter (<480 nm) placed in front of the LEDs. The images of green fluorescence and ChlF were acquired with high resolution CCD of 1,024 × 768 pixels. A filter wheel mounted in front of the CCD allowed alternative detection of both emission signals using different sets of filters. The green fluorescence was detected using the combination of the color glass GG495 (Schott, Mainz, Germany), the short-pass filter (<660 nm) and the band-pass 505–560 nm. Chlorophyll fluorescence was selected by the red-edge filter RG695 (Schott, Mainz, Germany) combined with the short-pass (<765 nm). The band-pass and all the short-pass filters were custom made.

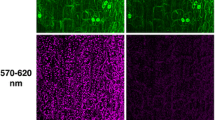
a Scheme of a leaf cross section. Different light colors penetrate into different depth of the leaf tissue. UV light is effectively absorbed in the top layers, including cuticle and epidermis, giving rise to the blue-green fluorescence shown in panel b. Blue and red lights are absorbed by photosynthetic pigments, utilized for photosynthesis and, in small extent, re-emitted as chlorophyll a fluorescence (ChlF, panel c). Red photons reach the deeper mesophyll layers than blue ones which are more readily absorbed by chlorophylls closer to the surface. Chlorophylls absorb only weakly green light, which penetrates deep into the leaf tissue. b An image of green fluorescence excited by blue light in N. tabacum leaf, two senescent A. thaliana leaves and in A. alba shoot. c An image of the steady-state chlorophyll fluorescence emission elicited by the blue light in the same samples as in (b)
Two-photon microscopic imaging
The green leaf fluorescence emission was imaged using i-MIC 2000 digital platform (Till-Photonics, Gräfelfing, Germany) with excitation by infrared light of 745 nm by Ti–Sapphire laser (Tsunami, Spectra Physics, Mountain View, USA). Laser beam was directed to the sample by dichroic mirror FF665-Di02 (Semrock, Rochester, USA, www.semrock.com) and fluorescence was selected by the band-pass emission filter FF01-542/50 (Semrock, Rochester, USA, www.semrock.com). Images were obtained with Olympus PlanApoChromat 60×, water immersion objective (Olympus, Hamburg, Germany).
The 3-dimensional distribution of pathogenic bacteria in leaf tissue was visualized by the two-photon microscope Leica DM IRE2 HC Fluo TCS 1-B (Leica Microsystems, Wetzlar, Germany). The infrared laser Chameleon Ultra (Coherent, Santa Clara, USA) was tuned to 900 nm to excite both GFP and ChlF. Emission bands for GFP (500–540 nm) and ChlF (680–700 nm) were selected by acousto-optical beam splitter. Samples were observed using a 63× water immersion objective HCX PL APO, NA 1.2 (Zeiss, Göttingen, Germany).
Detached Arabidopsis leaves were mounted in a custom made microscopic chamber equipped with corrected cover slips no. 1.5 (Assistent, Glaswarenfabrik Karl Hecht Gmbh+Co, Sondheim, Germany), dipped in 10 mM MgCl2 solution under a block of agarose.
Plant material and bacteria
Plants of Arabidopsis thaliana, accession Col gl1 and tobacco plants (Nicotiana tabacum) were grown in a controlled chamber Sanyo Gallenkamp (Leicester, UK). Plants were grown with 9/15 h photoperiod at 150 μmol (photons) m−2 s−1 of white light. Cultivation conditions were 25°C and 50% relative humidity during a day and 20°C and 65% at night. Approximately 5–6 week old plants (from sowing) were used for experiments.
Pathogenic bacteria P. syringae pv. tomato DC3000 was kindly provided by Thomas Roitsch, University of Würzburg, Germany. The cells were electroporated with EGFP carrying plasmid (pTB93F), which was a gift from Prof. Sharon R. Long, Stanford University, California. Bacteria were cultured in Kings B medium supplemented with antibiotics rifampicin (50 mg/l), chloramphenicol (50 mg/l) and spectinomycin (50 mg/l). An overnight culture was diluted 1:50 with fresh King’s B medium prior to the experiment and cultivated for another 2 h at 28°C. Bacterial cells were centrifuged (4°C, 1,000g, 10 min), washed once and dissolved in 10 mM MgCl2. The inoculum corresponding to 0.5 × 107 colony forming units per ml (OD 600 = 0.02) was gently infiltrated from abaxial leaf side with a needleless plastic syringe.
Results and discussion
Macroscopic fluorescence imaging
The relative intensity of individual emission bands depends on intrinsic leaf properties such as concentration of fluorophores, their distribution in tissues and leaf architecture (Fig. 2a). These determine penetration of excitation photons into different layers of leaf tissue as well as the fate of fluorescence photons emanating outward. UV photons are rapidly absorbed in the top cuticle and epidermis layers, whereas, the blue and red photons penetrate deeper and are largely absorbed by photosynthetic pigments in the leaf mesophyll. The deepest penetration occurs with green photons that are only weakly absorbed by chlorophylls.
Different origin and non-uniform lateral distribution of plant fluorophores in the tissues affect distribution of various fluorescence signals over the leaf area. Images of a young tobacco leaf (Nicotiana tabacum), two senescent A. thaliana leaves, and a silver fir shoot (Abies alba) captured in the green fluorescence band are shown in Fig. 2b. The same samples imaged in the red chlorophyll fluorescence (ChlF) emission band are shown in Fig. 2c. Fluorescence emission was excited by blue light (455 nm) for both the green and the red chlorophyll emission. The green fluorescence was measured between 505 and 560 nm and the chlorophyll fluorescence in the near infrared 690–750 nm spectral band. In the healthy young tobacco leaf, the image of green (Fig. 2b) and red chlorophyll fluorescence (Fig. 2c) shows largely uniform distribution of the emission over the leaf area. In the fir shoot, the red fluorescence was detected only in the chlorophyll containing needles while the green fluorescence was strongly emitted also from the woody shoot tissue. It was shown earlier by confocal microscopy (Hutzler et al. 1998), that conifers emit the red fluorescence from needle chloroplasts while the UV-induced blue and green fluorescence is attributed to cuticle, endodermis, cell walls of subepidermal cells, phloem, and xylem of the needle (for an early reference on the blue–green fluorescence see Chappelle et al. 1984).
The two A. thaliana senescent leaves emitted steady-state chlorophyll fluorescence comparable to that of the tobacco leaf in the healthy green leaf segments. The low chlorophyll fluorescence emission of the apical part of the left leaf and of the two lateral lesions of the right leaf (Fig. 2c) corresponds to yellowish senescent leaf tissue (not shown). In contrast, the green fluorescence emission of these senescent segments is markedly increased when compared to the surrounding healthy green tissue (Fig. 2b) (Meyer et al. 2003).
Two-photon fluorescence imaging of A. thaliana leaves
The capacity of the two-photon microscopy to visualize UV-induced green fluorescence from different tissue depths is demonstrated in Fig. 3. The fluorescence was excited by two 745 nm photons leading to a singlet excited state that can alternatively be reached by the single-photon absorption approximately at 370 nm. The fluorescence emission was detected in the green range 517–567 nm selected for low re-absorption by photosynthetic pigments.

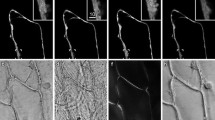
a Two-photon micrograph of UV induced green fluorescence excited by 745 nm infra-red laser light. Adaxial (top) and abaxial (bottom) leaf sides of A. thaliana are compared for green fluorescence signal emanating from different depth of the leaf tissue: the leaf surface and 10, 20, and 30 μm beneath epidermis. Stomata are shown by the white arrows in the surface images. Scale bar represents 20 μm. b Depth cross section images corresponding to the vertical white lines in the panels a. Scale bar represents 20 μm. c The depth intensity profile of the green fluorescence. The fluorescence intensity was averaged over 5 × 5 μm area centered over an arbitrarily chosen mesophyll chloroplast. Black squares correspond to adaxial side and gray triangles to abaxial side
The two top row images in panel a of Fig. 3 show the green fluorescence emission from the surface layer of adaxial and abaxial leaf sides with clearly visible stomata. The second row images show fluorescence signal emitted from epidermal cells 10 μm deep into the leaf. In the 20 μm images, there are mesophyll cells with clearly visible chloroplasts of palisade parenchyma of the adaxial leaf side and of the spongy mesophyll of the abaxial side. This panel documents that the mesophyll tissue emits not only red chlorophyll fluorescence but also significant green fluorescence. TPM images leading to the same conclusion were shown by Cheng et al. (2001). Although the blue–green fluorescence of chloroplasts is a known phenomenon (Cerovic et al. 1994), it has not been visualized from an intact leaf. Confocal microscopy attributed most of the blue–green fluorescence to surface leaf layers (Hideg et al. 2002). Only faint green fluorescence could be detected from leaf interior. This was primarily caused by the absorption of the UV excitation in the epidermis, and partly by re-absorption of the blue–green fluorescence by photosynthetic pigments (Cerovic et al. 1998). Several chloroplast fluorophores can be suspected for origin of this emission (Cerovic et al. 1999). Among them pyridine nucleotide NAD(P)H and flavins can be excited by two near-infrared photons (Xu et al. 1996; Zipfel et al. 2003). NAD(P)H exhibits a wide emission spectrum in the 400–600 nm range (Zipfel et al. 2003) and flavins (FAD and FMN) are also known to be green emitters (Kao et al. 2002).
The bottom images of Fig. 3a show that the TPM can capture fluorescence emission from cells located 30 μm deep in the leaf. The dashed vertical lines in Fig. 3a indicate position in the leaf, in which fluorescence from different leaf depths was combined to reconstruct the depth cross section that is shown in Fig. 3b. The green fluorescence emission is relatively weak in epidermis and is strongest in mesophyll chloroplasts close to the leaf surface. This observation is confirmed in Fig. 3c that integrates fluorescence in different leaf depths and maximum green emission at ca. 20 μm deep in the tissue. Fluorescence emission from cells deeper than 30 μm is hardly detectable, probably because light scattering limits the two-photon absorption and re-absorption of fluorescence photons prevents their detection.
Microscopic imaging of P. syringae infection in A. thaliana leaf tissue
Two-photon microscopy introduced in Figs. 1 and 3 was used to examine three-dimensional distribution of pathogenic bacteria in the leaf tissue. Figure 4 presents two-photon fluorescence emission from A. thaliana leaf infected with EGFP-tagged bacteria P. syringae. Green pseudo-color is assigned to EGFP-expressing bacteria and red visualizes chlorophyll fluorescence emission emanating from chloroplasts of palisade mesophyll cell layer. The single excitation wavelength of 900 nm was used to excite both chlorophyll and EGFP (Zipfel et al. 2003). The advantage of using a single excitation wavelength to elicit multiple color fluorescence minimizes differential effects of the objective chromatic aberration. One can be sure that the signal obtained from spectrally separated detecting channels can be assigned to the same focal plane.

Three-dimensional distribution of pathogenic bacteria (P. syringae) relative to steady-state chlorophyll auto-fluorescence of mesophyll cells visualized with two-photon excitation microscope. The images were captured 24 h after the pathogen infiltration. Images are presented in false colors: EGFP-expressing bacteria are shown green and chlorophyll fluorescence red. a A single optical section acquired 25 μm beneath the adaxial epidermis at the center of the infection lesion. Individual bacteria (white arrow) are dispersed in air spaces of mesophyll tissue. b The three-dimensional scene was reconstructed from multiple successive optical sections. The depth separation between individual slices was 1 μm. Reconstruction was performed using direct volume rendering in ImageJ software (Abramoff et al. 2004). Scale bar represents 20 μm
Figure 4a shows the EGFP-tagged Pseudomonas cells detected 25 μm deep in the leaf tissue. The green emission of bacteria was detected in the 500–540 nm range and the ChlF in the 680–700 nm emission window. Although, some level of auto-fluorescence contaminated also the green spectral channel, the bacteria can be easily distinguished as rod-shaped objects (arrow in Fig. 4a) of several micrometers length. The chlorophyll autofluorescence originated mostly from the stomata and mesophyll chloroplasts, whereas, the EGFP emission from the pathogen cells was found dominantly in large intercellular spaces.
A stack of the optical sections (one shown in Fig. 4a) from different focal planes from leaf surface to the depth of about 30 μm was combined to generate reconstruction shown in Fig. 4b. The imaged volume corresponds to the space between the epidermis characterized by the green auto-fluorescence of stomata and the upper cell layer of palisade parenchyma. The reconstructed image shows that the bacteria were mainly in the space between epidermis and the first layer of mesophyll cells. The bacterial colonies were preferentially formed in the air spaces under the stomata. The stomata are not well visible in the reconstructed image and their position is indicated in Fig. 4b by the letters ‘S’. P. syringae is an aerobic organism and the observed preferential growth of the pathogen under stomata might imply that the gas exchange is important factor for a faster pathogen growth (Wilson and Lindow 1994).
The two-photon microscopy was shown to image cellular structures and pathogen cells that are, in the traditional single-photon techniques, shielded by the epidermis barrier that effectively prevents UV excitation of deep leaf tissue. We anticipate that this capacity will also become instrumental in monitoring NAD(P)H in situ—a reporter that will be reflecting redox dynamics in mesophyll cells.
Abbreviations
- ChlF:
-
Chlorophyll fluorescence
- EGFP:
-
Enhanced green fluorescent protein variant
- FP:
-
Fluorescent protein
- GFP:
-
Green fluorescent protein
- TPM:
-
Two-photon microscopy
- UV-A:
-
Ultraviolet A radiation
References
Abramoff MD, Magelhaes PJ, Ram SJ (2004) Image processing with ImageJ. Biophotonics Int 11:36–42
Berger S, Benediktyova Z, Matous K, Bonfig K, Müller MJ, Nedbal L, Roitsch T (2007) Visualization of dynamics of plant-pathogen interaction by novel combination of chlorophyll fluorescence imaging and statistical analysis: differential effects of virulent and avirulent strains of P. syringae and of oxylipins on A. thaliana. J Exp Bot 58:797–806
Bhawalkar JD, Shih A, Pan SJ, Liou WS, Swiatkiewicz J, Reinhardt BA, Prasad PN, Cheng PC (1996) Two-photon laser scanning fluorescence microscopy—from a fluorophore and specimen perspective. Bioimaging 4:168–178
Blankenship RE (2002) Molecular mechanisms of photosynthesis. Blackwell Science, London, UK
Buschmann C, Langsdorf G, Lichtenthaler HK (2000) Imaging of the blue, green, and red fluorescence emission of plants: an overview. Photosynthetica 38:483–491
Cerovic ZG, Morales F, Moya I (1994) Time-resolved spectral studies of blue-green fluorescence of leaves, mesophyll and chloroplasts of sugar beet (Beta vulgaris L). Biochim Biophys Acta 1188:58–68
Cerovic ZG, Langrand E, Latouche G, Morales F, Moya I (1998) Spectral characterization of NAD(P)H fluorescence in intact isolated chloroplasts and leaves: Effect of chlorophyll concentration on reabsorption of blue-green fluorescence. Photosynth Res 56:291–301
Cerovic ZG, Samson G, Morales F, Tremblay N, Moya I (1999) Ultraviolet-induced fluorescence for plant monitoring: present state and prospects. Agronomie 19:543–578
Cerovic ZG, Ounis A, Cartelat A, Latouche G, Goulas Y, Meyer S, Moya I (2002) The use of chlorophyll fluorescence excitation spectra for the non-destructive in situ assessment of UV-absorbing compounds in leaves. Plant Cell Environ 25:1663–1676
Chalfie M, Tu Y, Euskirchen G, Ward WW, Prasher DC (1994) Green fluorescent protein as a marker for gene-expression. Science 263:802–805
Chappelle EW, Wood FM, McMurtrey YE, Newcomb WW (1984) Laser induced fluorescence of green plants. 1: a technique for remote detection of plant stress and species differentiation. Appl Opt 23:134–138
Cheng PC, Lin BL, Kao FJ, Gu M, Xu MG, Gan XS, Huang MK, Wang YS (2001) Multi-photon fluorescence microscopy—the response of plant cells to high intensity illumination. Micron 32:661–669
Condon E (1926) A theory of intensity distribution in band systems. Phys Rev 28:1182–1201
Diaspro A, Robello M (2000) Two-photon excitation of fluoresnce for three-dimensional optical imaging of biological structures. J Photochem Photobiol B 55:1–8
Endo R, Omasa K (2007) 3-D cell-level chlorophyll fluorescence imaging of ozone-injured sunflower leaves using a new passive light microscope system. J Exp Bot 58:765–772
Feijó JA, Moreno N (2004) Imaging plant cell by two-photon excitation. Protoplasma 223:1–32
Franck J, Dymond EG (1926) Elementary processes of photochemical reactions. Trans Faraday Soc 21:536–542
Frank H, Chynwat V, Desamero R, Farhoosh R, Erickson J, Bautista J (1997) On the photosynthetics and photochemical properties of carotenoids and their role as light-harvesting pigments in photosynthesis. Pure Appl Chem 69:2117–2124
Hideg E, Juhasz M, Bornman J, Asada K (2002) The distribution and possible origin of blue–green fluorescence in control and stressed barley leaves. Photochem Photobiool Sci 1:934–941
Hutzler P, Fischbach R, Heller W, Jungblut TP, Reuber S, Schmitz R, Veit M, Weissenbock G, Schnitzler JP (1998) Tissue localization of phenolic compounds in plants by confocal laser scanning microscopy. J Exp Bot 49:953–965
Kao FJ, Wang YM, Chen JC, Cheng PC, Chen RW, Lin BL (2002) Micro-spectroscopy of chloroplasts in protoplasts from Arabidopsis thaliana under single- and multi-photon excitations. J Lumin 98:107–114
Kitajima M, Butler WL (1975) Excitation spectra for photosystem I and photosystem II in chloroplasts and spectral characteristics of the distribution of quanta between the two photosystems. Biochim Biophys Acta 408:297–305
König K (2000) Multiphoton microscopy in life sciences. J Microsc 200:83–104
Lichtenthaler HK (1987) Chlorophylls and carotenoids—pigments of photosynthetic biomembranes. Method Enzymol 148:350–382
Lippincott-Schwartz J, Patterson GH (2003) Development and use of fluorescent protein markers in living cells. Science 300:87–91
Maxwell K, Johnson GN (2000) Chlorophyll fluorescence—a practical guide. J Exp Bot 51:659–668
Meyer S, Cartelat A, Moya I, Cerovic Z (2003) UV-induced blue-green and far-red fluorescence along wheat leaves: a potential signature of leaf ageing. J Exp Bot 54:757–769
Nedbal L, Whitmarsh J (2004) Chlorophyll fluorescence imaging of leaves and fruits. In: Papageorgiu GC, Govindjee (eds) Chlorophyll a fluorescence: a signature of photosynthesis. Springer, Netherlands
Oheim M, Beaurepaire E, Chaigneau E, Metrz J, Charpak S (2001) Two-photon microscopy in brain tissue: parameters influencing the imaging depth. J Neurosci Methods 111:29–37
Omasa K, Hosoi F, Konishi A (2007) 3D lidar imaging for detecting and understanding plant responses and canopy structure. J Exp Bot 58:881–898
Pawley JB (1995) Handbook of biological confocal microscopy. Plenum, New York
Pineda M, Gáspár L, Morales F, Szigeti Z, Barón M (2008) Multicolor fluorescence imaging of leaves—a useful tool for visualizing systemic viral infection in plants. Photochem Photobiol 84:1048–1060
Rizzo MA, Piston DW (2004) Fluorescent protein tracking and detection. In: Goldman RD, Spector DL (eds) Live cell imaging: a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Ross FWD (1995) Fluorescence microscopy, vol 2. Cambridge University Press, Cambridge
Shaner NC, Steinbach PA, Tsien RY (2005) A guide to choosing fluorescent proteins. Nat Methods 2:905–909
Strasser RJ, Butler WL (1977) Florescence emission spectra of photosystem I, photosystem II and the light-harvesting chlorophyll a/b complex of higher plants. Biochim Biophys Acta 462:307–313
Tsien RY (1998) The green fluorescent protein. Annu Rev Biochem 67:509–544
Vácha F, Sarafis V, Benediktyová Z, Bumba L, Valenta J, Vácha M, Sheue CR, Nedbal L (2007) Identification of photosystem I and photosystem II enriched regions of thylakoid membrane by optical microimaging of cryo-fluorescence emission spectra and of variable fluorescence. Micron 38:170–175
Vogelmann TC, Evans JR (2002) Profiles of light absorption and chlorophyll within spinach leaves from chlorophyll fluorescence. Plant Cell Environ 25:1313–1323
Wilson M, Lindow SE (1994) Inoculum density dependent mortality and colonization of the phylosphere by Pseudomonas syringae. Appl Environ Microbiol 60:2232–2237
Xu C, Williams RM, Zipfel W, Webb WW (1996) Multiphoton excitation cross-sections of molecular fluorophores. Bioimaging 4:198–207
Zipfel WR, Williams RM, Christie R, Nikitin AY, Hyman BT, Webb WW (2003) Live tissue intrinsic emission microscopy using multiphoton-excited native fluorescence and second harmonic generation. Proc Natl Acad Sci USA 100:7075–7080
Acknowledgments
This study was supported by “AUTOSCREEN for cell-based high-throughput and high-content gene function analysis and drug discovery screens” project of the Framework 6 program of the European Community (STREP 037897) and by the grant AV0Z60870520 of the Academy of Sciences of the Czech Republic.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Benediktyová, Z., Nedbal, L. Imaging of multi-color fluorescence emission from leaf tissues. Photosynth Res 102, 169–175 (2009). https://doi.org/10.1007/s11120-009-9498-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-009-9498-z