
Abstract
The cyanobacterial small CAB-like proteins (SCPs) are one-helix proteins with compelling similarity to the first and third transmembrane helix of proteins belonging to the CAB family of light-harvesting complex proteins in plants. The SCP proteins are transiently expressed at high light intensity and other stress conditions but their exact function remains largely unknown. Recently we showed association of ScpD with light-stressed, monomeric Photosystem II in Synechocystis sp. PCC 6803 (Yao et al. J Biol Chem 282:267–276, 2007). Here we show that ScpB associates with Photosystem II at normal growth conditions. Moreover, upon introduction of a construct into Synechocystis so that ScpB is expressed continuously under normal growth conditions, ScpE was detected under non-stressed conditions as well, and was copurified with tagged ScpB and Photosystem II. We also report on a one-helix protein, Slr1544, that is somewhat similar to the SCPs and whose gene is cotranscribed with that of ScpD; Slr1544 is another member of the extended light-harvesting-like (Lil) protein family, and we propose to name it LilA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The first step in photosynthesis is the absorption of sunlight by chlorophylls and other pigments. To keep them in their proper location and orientation, the pigments are bound to carotenoid-containing proteins to ensure efficient and rapid energy transfer, and effective quenching of toxic triplet states. In plants the majority of pigments, including chlorophyll a, chlorophyll b, and various carotenoids are bound to a family of membrane-integral proteins, called the light-harvesting complex (LHC). Most abundant is LHCII, the main light-harvesting complex of Photosystem II (PSII), which is known to consist of three transmembrane helices, each of which is comprised of 20–34 amino acids (Kühlbrandt et al. 1994; Liu et al. 2004; Standfuss et al. 2005). The sequences of helices I and III of LHCII are very similar to each other and comprise a motif of approximately 25 amino acid residues that includes the chlorophyll-binding domain and is known as the CAB (chlorophyll a/b binding) motif (Jansson 1999). Proteins sharing the CAB motif display a high degree of sequence similarity and are believed to have a common evolutionary origin (Durnford et al. 1999; Heddad and Adamska 2002). The members of this family also include the antenna proteins located around Photosystem I (PSI), the stress-induced early light-induced proteins (ELIPs) (see Adamska 2001), and the PsbS protein (reviewed by Funk 2001) that has an important function in non-photochemical quenching (Li et al. 2000). Moreover, related genes coding for polypeptides with one or two transmembrane α-helices have been detected in the genomes of Arabidopsis thaliana (Heddad and Adamska 2000; Jansson et al. 2000), rice, poplar (Klimmek et al. 2006), the green alga Chlamydomonas reinhardtii (Teramoto et al. 2004), and the red alga Cyanidioschyzon merolae (Ohta et al. 2003).
In contrast to plants, cyanobacteria lack multi-helical CAB proteins. Instead, many cyanobacteria use the extrinsic phycobilisome as a major peripheral light-harvesting complex. However, the genomes of both marine and freshwater cyanobacteria encode for a family of small CAB-like proteins with masses of less than 8 kDa (reviewed in Bhaya et al. 2002). These proteins are predicted to have a single membrane-spanning α-helix that displays significant sequence similarity to the first and third membrane-spanning region of the CAB proteins of green plants. This common characteristic has given them the name small CAB-like proteins (SCPs) (Funk and Vermaas 1999). They were also designated high-light inducible proteins (HLIPs) since the level of the corresponding transcripts was found to increase after transfer of cells to high light intensity and other stress conditions (He et al. 2001; Mikami et al. 2002).
In the cyanobacterium Synechocystis sp. PCC 6803 (hereafter Synechocystis 6803), five SCPs were identified (Funk and Vermaas 1999); four of them (ScpB to ScpE) encode proteins of around 6 kDa, while the fifth one (ScpA, Slr0839) is the C-terminal extension of ferrochelatase. The genes coding for ScpB–ScpE are induced under various different stress conditions, including very high light intensity (>500 μmol m−2 s−1), low temperature, and N- and S-starvation (He et al. 2001; Mikami et al. 2002). A mutant with these four genes inactivated was sensitive to high-intensity illumination and showed alteration in pigmentation and in the ability to perform non-photochemical dissipation of absorbed light energy (Havaux et al. 2003). SCPs may prevent the formation of reactive oxygen species (ROS) by serving as transient carriers of chlorophyll (Xu et al. 2004; Xu et al. 2002). Furthermore, the SCPs appear to participate in tetrapyrrole biosynthesis and to regulate pigment availability (Xu et al. 2004; Xu et al. 2002). Interestingly, ScpC (Ssl2542, HliA) and ScpD (Ssr2595, HliB) seem to be functionally complementary (Xu et al. 2004). These two genes are most similar (87.1% identity, He et al. 2001), indicating a rather recent gene duplication event (Bhaya et al. 2002) or a strict requirement for conserved primary structure. Using 2D PAGE, N-terminally His-tagged ScpD was immunologically detected to comigrate with PSII (Promnares et al. 2006). Consistently, His-ScpD copurified with PSII upon affinity chromatography and its closest neighbor was CP47 (Yao et al. 2007). The very similar ScpC protein also comigrated with PSII (Yao et al. 2007) but no compelling experimental data on the subcellular location of ScpB and ScpE are available yet.
In this study, we overexpressed Strep-tagged ScpB (Ssl1633, HliC) in Synechocystis 6803. We observed association of both Strep-tagged ScpB and ScpE with Photosystem II in cells grown under normal growth conditions, while neither ScpC nor ScpD were detectable. In contrast, high-light stressed cells expressed slr1544, the product of which copurified with PSII and resembles the SCPs.
Materials and methods
Growth conditions. Synechocystis 6803 was cultivated at 30°C at a light intensity of 40 μmol photons m−2 s−1 in BG-11 medium (Rippka et al. 1979), in which sodium nitrate was partially substituted by ammonium nitrate so that the final concentration was 13.2 mM and 4.4 mM respectively. The growth medium was supplemented with 10 mM TES/NaOH buffer, pH 8.2, and 5 mM glucose. Antibiotics were added where appropriate. Liquid cultures were grown while being shaken at 120 rpm. Solid medium was supplemented with 1.5% (w/v) agar, 0.3% (w/v) sodium thiosulfate, and 10 mM TES/NaOH buffer, pH 8.2. The optical density of the cell cultures was determined at 730 nm using T90+ UV/VIS Spectrometer by PG Instruments. For all experiments cells were harvested from liquid cultures at an optical density of 0.5–0.7, which corresponds to mid-log phase.
DNA isolation and genetic manipulations. Chromosomal DNA was isolated from the wild-type strain by a standard method of N-lauryl sarcosine lysis and phenol-chloroform extraction (Joset 1988; Maniatis et al. 1982). PCR amplification of the locus containing scpB (ssl1633) was conducted using the primers BF and BR (Supplementary Table 1). The BF primer sequence contained the Strep-tag II and linker sequences, which allowed introduction of this tag at the N-terminus of ScpB. The PCR product contained restriction sites for NcoI and PstI, which were used for cloning the tag-scpB fusion in lieu of lhcb in the pA3lhcgA3 plasmid (He et al. 1999). In the resulting construct scpB was under the psbA3 promoter that allows continuous expression of this gene under non-stressed growth conditions. This construct (Supplementary Fig. 1) was used to transform a wild-type strain of Synechocystis sp. PCC 6803, and the resulting mutant continuously expressed a Strep-tagged ScpB under normal growth conditions whereas in its original locus, scpB is only expressed under stress conditions (Funk and Vermaas 1999; He et al. 2001). The gentamicin resistance cassette downstream of scpB provided a convenient means of mutant selection and segregation.
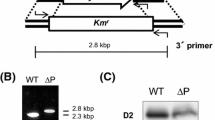
To attach the Strep-tagII (IBA GmbH) to the N-terminus of Slr1544 the following strategy was used: A 2,160 bp PCR fragment was generated using the primers D1 and D2 (see Supplementary Table 1); this fragment included the coding region of scpD and slr1544 as well as flanking regions. This fragment was ligated into pUC18 using the HindIII and SacI restriction sites resulting in plasmid pDF that is shown in Supplementary Fig. 1B. Both genes (scpD and slr1544) were then replaced by a kanamycin resistance cassette to generate the plasmid pDKF. After transforming wild type Synechocystis cells with pDKF a scpD − /slr1544 − kanamycin-resistant mutant was obtained. The PCR product encoding for the Strep-tagII using primers ST1 and ST2 (Supplementary Table 1), was inserted into pUC18; the new plasmid was called pStag (Supplementary Fig. 1B). The reverse primer (ST2) contained a SpeI site in addition to the BamHI site, allowing cloning of slr1544 in frame with the Strep-tag, using the PCR product obtained with primers F1 (SpeI) and 18R (PstI), creating the plasmid pStagF. Using pDF (primers 18F and DR) and pStagF (primers F2 and 18R) as templates a new plasmid called pDStagF was generated, in which the N-terminal strep-tagged slr1544 is in frame with its natural promoter (the scpD sequence was excluded in the PCR). A spectinomycin resistance cartridge (Eaton-Rye and Vermaas 1991) (2 kb, restricted by BamHI in pDStagF using the BclI restriction site) was inserted and the resulting plasmid was used for transformation of the double-knockout Synechocystis mutant scpD − /slr1544 −. Cells were allowed to segregate in the presence of antibiotics. Complete segregation of the mutants was confirmed by PCR using the primers D1 and D2 (Supplementary Fig. 1C).
Total RNA purification
Cell cultures grown to mid-logarithmic phase (O.D. 0.5–0.8) were transferred to 50 ml sterile Sarstedt tubes, chilled on ice to 0°C, and immediately centrifuged at 4,500g for 5 min. The cell pellet was resuspended in RLT buffer (RNeasy Mini Kit, Qiagen) and mixed with an equal volume of 0.1-mm-diameter glass beads. Cells were broken in the Bead-Beater using four breakage cycles (each cycle consisted in 1 min breakage followed by 1 min cooling on ice). RNA was purified from lysed cells according to the RNeasy Mini Kit (Qiagen) protocol.
Northern Blot analysis
Total RNA (5 μg) was resolved by gel electrophoresis (2% agarose/2% formaldehyde) and transferred to a positively charged nylon membrane (Roche Diagnostics). DNA probes were synthesized using a digoxigenin (DIG) DNA Labeling and Detection kit (Roche). To synthesize the scpD and slr1544 probes the following primers were used for amplification: scpD forward 5′-ATGACTAGCCGCGGATTTC-3′, scpD reverse 5′- TTAGAGAGAGAGCAACC-3′, slr1544 forward 5′-ATGAACTACCAAAGGACTG-3′, slr1544 reverse 5′-TTAGAGAAAGTCAGCCTGAAG-3′. Hybridization and detection were performed according to the DIG hybridization (Roche) standard protocol.
Isolation of protein complexes containing StrepII-tagged ScpB
Thylakoids were isolated from cultures of the Synechocystis ScpB-Strep mutant according to Funk (2000). Thylakoids were solubilized with dodecyl β-d-maltoside (β-dM) at a final detergent/chlorophyll ratio of 8:1 (w/w) in a medium consisting of 100 mM Tris/HCl, pH 8.0, 150 mM NaCl, and 1 mM EDTA. Purification of ScpB-Strep-II-containing protein complexes was conducted using a strep-tactin sepharose gravity flow column according to the manufacturer’s recommendations (www.iba-go.com).
Isolation of a PSII-enriched fraction
Thylakoid membranes were extracted according to Rögner et al. (1990) with modifications: Cells in logarithmic growth phase (OD730 0.5–0.8) were pelleted by centrifugation at 6,000g for 7 min at 4°C, and washed twice in washing buffer (20 mM MES/NaOH pH 6.5, 10 mM CaCl2, 10 mM MgCl2). The cells were resuspended in thylakoid buffer (20 mM MES/NaOH pH 6.5, 10 mM CaCl2, 10 mM MgCl2, 1 M sucrose) containing proteases inhibitors, keeping the chlorophyll concentration at about 1 μg/ml. One ml of cell suspension was transferred to 2 ml screw-capped microcentrifuge tubes half-filled with pre-wetted glass beads. Cells were broken by BeadBeater at 4°C using 0.1 mm glass beads, by 4 cycles of 30 s each, with 5 min cooling intervals between cycles. Unbroken cells and glass beads were precipitated at 550g for 6 min. The supernatant containing the thylakoid membranes was centrifuged at 18,000g for 30 min; the soluble fraction containing phycobilins was discarded, and the membranes were resuspended in Dissolving Buffer (20 mM MES/NaOH pH 6.5, 10 mM CaCl2, 10 mM MgCl2, 0.5 M mannitol, and 20% glycerol) at a chlorophyll concentration of 1 mg/ml. Thylakoid membranes at a concentration of 1 mg Chl/ml were stirred with an equal volume of Dissolving Buffer containing 4% (w/v) β-DM for about 40 min at 4°C and 10 min at room temperature. Extracted complexes were separated from the remaining membranes by centrifugation (18,000g, for 20 min); the supernatant was diluted at least 2 times with Binding Buffer (20 mM MES/NaOH pH 6.5, 10 mM CaCl2, 10 mM MgCl2, 0.5 M mannitol, 0.04% (w/v) β-DM).
PSI and PSII were separated on a FPLC (ÄKTA machine) anion-exchange column Hitrap Q FF (GE Healthcare), equilibrated with Binding Buffer (5 mM MgSO4 and 0.03% (w/v) β-DM added). The thylakoid suspension was eluted by a non-linear gradient of 5–200 mM MgSO4 at a flow rate of 0.5 ml/min. The different fractions were analyzed by 77K fluorescence (Fluoromax2) to monitor the enrichment in PSII.
Non-denaturing PAGE
Analysis of the pigment-protein composition of the complex co-isolating with Strep-ScpB was conducted according to Anderson et al. (1978) with modifications (Kufryk 1997).
SDS-PAGE
Denaturing PAGE was performed according to Schägger and von Jagow (1987). The gels contained 6 M urea. The samples were loaded on a chlorophyll basis (5 μg chlorophyll per lane).
Pigment analysis
Chlorophyll content was determined in 80% acetone and was calculated as described by Porra et al. (1989).
Immunoblotting
After electrophoresis, the proteins were electrotransferred to PVDF membranes and immunostained according to Towbin et al. (1979). The Strep-tag II was detected using StrepMAB-Classic HRP conjugate (IBA) according to the manufacturer’s protocol. Chemiluminescence was detected using a LAS3000 Luminescent image analyzer (Fuji), and the images were analyzed by Image Reader LAS3000 software.
Protein analysis by mass spectrometry
Peptides for mass spectrometry analysis were prepared by in-gel digestion using trypsin according to the principal steps of standard methods (Shevchenko et al. 1996, Pandey et al. 2000). Bands from a green gel were excised, cut into small cubes and destained by extracting the gel pieces four times for about 30 min using 0.2 ml of 20 mM ammonium bicarbonate in 35% (v/v) acetonitrile. The gel pieces were dried by two wash steps using 0.1 ml of pure acetonitrile, and the dry white gel pieces were reconstituted on ice with a solution containing 20 mM ammonium bicarbonate, 10% (v/v) acetonitrile, and 2 ng/μl of sequencing grade, modified trypsin (Promega). The amount of trypsin solution depended on the volume of the gel pieces and was just enough to cover the gel pieces. After about 1 h the in-gel digest samples were transferred from ice to an incubation room that was set to 37°C; the amount of trypsin solution was checked after 10 min and, if needed, adjusted to ensure that the gel pieces did not get dry during the incubation. The trypsin treatment was continued overnight at 37°C.
To prepare samples for analysis by reverse-phase LC-MS/MS the liquid phase of the in-gel digest samples was diluted with Milli-Q water to final concentration of 5% (v/v) acetonitrile; the solutions were acidified by adding 10% (v/v) formic acid to a final concentration of 1% (v/v) and centrifuged for 15 min at 20,000g at 4°C. Analysis by LC-MS/MS was performed using a nano Acquity UltraPerformance LC (Waters) coupled to a Q-TOF Ultima mass spectrometer (Waters). Peptides were separated by nanoflow reverse-phase chromatography using a BEH C18 1.7 μm column (100 μm × 100 mm) that was equilibrated at a flow rate of 350 nl min−1 with 95% of solvent A (5% (v/v) acetonitrile in 0.1% (v/v) formic acid) and 5% of solvent B (95% (v/v) of acetonitrile in 0.1% (v/v) formic acid). Peptide separation was achieved by applying a linear gradient from 15% solvent B at 1 min to 45% solvent B at 20 min. The total time for a single LC run was 35 min. Spectra acquisition was performed using typical settings for data-directed analysis of the MassLynx 4.1 software. Peptides eluted on-line into the ion source were monitored using a MS-TOF survey scan (m/z 400 to m/z 1,200, 0.8 s scan time, and 0.2 s inter delay), and the three most abundant signals were selected by charge state for MS/MS analysis (50–2,000 m/z range, 1.0 s scan time, 0.1 s inter delay). To obtain a better coverage of the peptides present in the samples additional runs under identical conditions were performed that employed smaller mass windows for the MS-TOF survey scan (m/z 400–620, m/z 600–820, and m/z 800–1,020).
The raw data files were processed using the ProteinLynx Global Server 2.2.5 (Waters) and settings for fast deisotoping combined with 50% baseline subtraction. The resulting PKL files were used for searches in the Synechocystis sp. PCC 6803 database of the EBI (v 41) on an in-house Mascot server (version 2.1.04) licensed to Umeå University by Matrix Science (www.matrixscience.com). Searches were carried out by Mascot Daemon using the merged PKL files for one sample. The search parameters allowed mass errors up to 100 ppm for MS data, and up to 0.06 Da for MS/MS data. Additional settings permitted two missed cleavage sites for trypsin and included the variable modifications deamidation (N, Q), oxidation (M), phosphorylation (S, T, Y) and propionamide derivation (C). The threshold for individual peptide identifications was set to 99.5% confidence and redundant identifications were excluded using the bold red function.
Results
The introduction of affinity tags has proven to be a successful strategy to study the subcellular localization of the ScpC and ScpD proteins of Synechocystis and to discover their PSII association in light-stressed cells (Promnares et al. 2006, Yao et al. 2007). In this study we used a related approach to determine the location of other members of the SCP family. Of particular interest was ScpB as earlier observations indicated that this protein might also be PSII-associated (Yao et al. 2007). To investigate the subcellular location of ScpB, we constructed a mutant, in which the scpB gene was under the control of the psbA3 promoter that is continuously active but is strongest at high light intensity. Using this design we were able to study ScpB not only under light stress but also under normal growth conditions. To facilitate immunodetection of ScpB and copurification with other complex forming proteins, ScpB was fused at its N-terminus to a Strep-tag-II. Mutant cells that expressed Strep-tagged ScpB in addition to the ScpB wild type (the native gene was not disturbed) displayed a phenotype similar to wild type and showed no significant differences in their chlorophyll content and growth rate under photoautotrophic and photomixotrophic conditions (data not shown).
To identify the proteins that bind to ScpB we employed streptavidin affinity chromatography that specifically binds Strep-ScpB and allows a straightforward purification of proteins that form complexes with ScpB. As the result of a single-step affinity purification may contain considerable cross-contamination, we employed a tandem approach in which the affinity chromatography was followed by a separation using a non-denaturing electrophoresis gel. Figure 1 shows the efficiency of this purification. Lane T displays the separation of complexes of a total membrane fraction of wild type cells, while lane S shows the components of the fraction of proteins co-purifying with StrepII-tagged ScpB. Band 1 (lane T) representing PSI trimer core complexes, and band 4 containing PsaA/PsaB complexes (Ivanov et al. 2006) were missing in the ScpB-StrepII fraction (lane S). However, the broad band visible in lane S was comigrating rather with band 3, representing Photosystem II (PS II) in the thylakoid fraction (lane T). The localization of StrepII-tagged ScpB was determined by immunoblotting that specifically labeled the StrepII-tag in the lower region of band 3 (Fig. 1, lane I). This observation indicates that StrepII-ScpB expressed under the psbA3 promoter accumulates in mutant cells under normal growth conditions, and StrepII-ScpB might co-isolate with PSII complexes.

Non-denaturing “green” PAGE of thylakoid membranes (T) from Synechocystis sp. PCC 6803 and ScpB-strep (S) co-isolate from mutant cells, both grown under normal conditions. Bands 1 and 2 are designated to the PSI complex; band 3 is designated to the PS II reaction center complex; band 4 contains the PsaA/PsaB core complex of PSI; band 5 represents free pigments (Ivanov et al. 2006). I, Immunodetection of strep-tagged ScpB using the StrepMAB-Classic HRP conjugate
To obtain more insight into the proteins that copurified with StrepII-tagged ScpB the protein components from the corresponding part of band 3 in lane S were analyzed using tandem mass spectrometry. As shown in Table 1, the proteins copurifying with StrepII-tagged ScpB include subunits of PSII, phycobilisome components, and other proteins related to photosynthesis. The inner antenna protein CP47 was one of the most abundant proteins that associated with ScpB under our experimental conditions. In addition, we found the PSII reaction center proteins (D1 and D2), a cytochrome b 559 subunit, CP43 and PsbH. Also, the orange carotenoid-binding protein (OCP, Slr1963), regulating the efficiency of energy transfer between the phycobilisome and PSII (Wilson et al. 2006), was detected. Two ATP-dependent proteases were identified in the StrepII-ScpB/PSII complexes: one of these belongs to the Clp family of serine proteases, the other is a FtsH metalloprotease. Interestingly, some proteins of unknown function co-isolated with this fraction and possibly might associate with PS II (see Table 1). An important finding of our mass spectrometry analysis was the identification of ScpE among the proteins that comigrated with StrepII-tagged ScpB.
While ScpD and ScpC associate with PSII upon exposure to high light intensity, at normal light intensity these two proteins could not be detected, even when a constitutive promoter, psbA2, was used (Promnares et al. 2006; Yao et al. 2007 and this work). However, accumulation of ScpB (under the control of the psbA3 promoter) led to detectable accumulation of ScpE under its natural promoter, whereas the scpE promoter has been found to be inactive unless stress is applied (Funk and Vermaas 1999, He et al. 2001). This suggests that accumulation of ScpB in the cell induces expression of wild-type scpE.
A similar coregulation has been detected for scpD (ssr2595), which by microarray analysis was found in a gene cluster together with the adjacent slr1544 (Suzuki et al. 2001). Both transcripts were found to be responsive to cellular redox state (Hihara et al. 2003) and were enhanced at low temperature (Suzuki et al. 2001), and upon osmotic (Mikami et al. 2002) and salt stress (Marin et al. 2003). Genome clustering of scpD and slr1544 was found not only in Synechocystis, but also in the cyanobacteria Anabaena variabilis, Nostoc punctiforme, and Nodularia spumigena (Fig. 2A). In all four strains ScpD as well as Slr1544 have high sequence similarity (Fig. 2B). Northern blot analysis showed that the transcript detected with a slr1544 probe was around 600 bases long and suggests cotranscription of scpD (210 bases) and slr1544 (309 bases) (Fig. 2C). Slr1544, similar to CAB-family proteins, contains the conserved amino acid pair Gly-Arg of the CAB motif in its predicted membrane-spanning helix (Fig. 2D).

(A) Ortholog neighborhood regions of Synechocystis, Anabaena variabilis, Nostoc punctiforme and Nodularia spumigena, obtained using the Integrated Microbial Genomes (IMG) genomic data analysis system (DOE Joint Genome Institute). The genes scpD and slr1544 and their orthologs in other cyanobacteria are highlighted in the rectangle. (B) Sequence alignments of the proteins encoded by the scpD and slr1544 genes of Synechocystis sp. PCC 6803, Anabaena variabilis, Nostoc punctiforme, and Nodularia spumigena, (C) Northern blot probed with slr1544. (D) Alignment of the third membrane-spanning helix of Lhcb3 and ELIP1 of Arabidopsis thaliana (Heddad and Adamska 2002) with the predicted transmembrane regions of the ScpB-E and Slr1544
The conserved genomic adjacency of the scpD and slr1544 genes in cyanobacteria indicates that the corresponding gene products may be functionally related, and share similarity to other members of the light-harvesting like (Lil) family (Jansson 2005). To study the Slr1544 protein in Synechocystis a mutant was constructed, in which ScpD and Slr1544 were deleted, and a StrepII-tagged Slr1544 was reintroduced under its natural promoter; cells of the resulting strain were subjected to light stress according to Yao et al. (2007). Two fractions of this mutant, one containing total membranes and the other one enriched in PSII, were separated by SDS-PAGE and either stained with Coomassie Brilliant Blue or immunostained using Strep-specific antibodies. Lane A of Fig. 3 shows immunodetection of the Strep-tagII in the total membrane fraction. The immunoblot displayed in Lane C shows that the Strep-tagII signal also was detected in the fraction enriched in PSII. Lanes B and D show the corresponding protein fractions using Coomassie Brilliant Blue staining. The apparent mass of the Strep-tagII signals in the total membrane fraction and in the PSII enriched membrane fraction was very close to the theoretical mass of 11.9 kDa that was expected for StrepII-tagged Slr1544 indicating that StrepII-tagged Slr1544 was expressed in light-stressed cells even in the absence of ScpD. The results also indicate that Slr1544 copurifies with PSII suggesting that it might fulfill a function related to that of the SCPs.

Thylakoid membranes (lanes 1 and 2) and PSII-enriched fractions (lanes 3 and 4) were loaded on to a Tricine-SDS-PAGE gel containing 6M Urea (5 μg Chl per lane). After SDS PAGE the gels were stained with Coomassie Brilliant Blue (lanes 1 and 3) or immunostained using the monoclonal StrepMAB-Classic HRP conjugate (IBA) (lanes 2 and 4)
Discussion
Cyanobacterial Small CAB-like proteins are members of the extended family of light-harvesting like (Lil) proteins (Jansson 1999, Klimmek et al. 2006). In contrast to the chlorophyll a/b binding antenna proteins, the Lil proteins do not seem to function in the absorption of light as they are regulated opposite to LHC proteins and are induced by stress. In addition, they play roles in the tetrapyrrole biosynthesis and chlorophyll stability (Xu et al. 2002, 2004; Tzvetkova-Chevolleau et al. 2007) indicating that they might rather be involved in photoprotection (reviewed in Jansson 2005) and function as pigment-carrier proteins (Adamska 1997; Funk and Vermaas 1999; Xu et al. 2002, 2004).
Synechocystis 6803 has five clear small CAB-like proteins (Funk and Vermaas 1999). Little was known about their location until ScpC and ScpD were found to associate with PSII (Promnares et al. 2006; Yao et al. 2007). This study provides evidence that ScpB and ScpE also associate with PSII. However, the expression of ScpC/ScpD and ScpB/ScpE may be distinctly different. When the ScpD-His protein was expressed under the light-inducible psbA2 promoter (Promnares et al. 2006; Yao et al. 2007) that, unlike the native scpD promoter, leads to high expression levels even at low light intensity (Mohamed et al. 1993), the ScpD-His protein accumulated only in cells grown at high light intensity, consistent with the expression of native ScpD (Funk and Vermaas 1999). An explanation for this phenomenon may be provided in He et al. (2001), who found that native ScpC and ScpD were degraded immediately after transfer of cells from high light intensity to normal growth conditions. Therefore, the lack of accumulation of ScpD under conditions of high constitutive expression may be due to regulation at the post-translational level. In this study, we observed StrepII-tagged ScpB and ScpE in PSII of cells grown at normal conditions using the psbA3 promoter that allows high expression under normal light. StrepII-tagged ScpB appeared to be sufficiently stable for detection when cells were grown at moderate light intensity, whereas ScpC and ScpD were not detected under these conditions.
Surprisingly, the accumulation of StrepII-ScpB under normal growth conditions coincided with the expression of ScpE. Since the promoter of ScpE was found to be inactive unless stress is applied (Funk and Vermaas 1999; He et al. 2001) ScpB either may be a positive regulator of scpE transcription under its natural promoter, or may stabilize ScpE so that it can accumulate even when transcript levels are low. In support of the former explanation, He et al. (2001) showed that ScpE expression upon high light intensity treatment followed ScpB expression with a time lag of 3 h: while ScpB accumulated after 1 h of exposure to high light intensity, reaching maximum abundance at 3 h, ScpE was immunodetected only after 3 h of high-light exposure and reached its highest level at 6 to 9 h after the onset of the high intensity light.
Based on the discussion above, ScpC/D and ScpB/E might function in pairs, but have different roles. Whereas we do not have experimental data whether ScpC and ScpD or ScpB and ScpE form a complex, a heterodimeric complex—if any—is not expected to be a prerequisite for function as deletion of single scp genes generally does not have a major phenotype (Xu et al. 2002) whereas the effects of the deletion of pairs are much more pervasive (Xu et al. 2004). Therefore, members of a pair may be functionally complementary. Moreover, if complex formation would be required, similar amounts of the two members of the pair would be expected. Based on immunological studies the amount of ScpC and ScpD seem to be similar during high-light-intensity stress (Yao et al. 2007), whereas the ratio of ScpB/ScpE may be less constant as discussed above. In addition, expression of C-terminally His-tagged versions of ScpB and ScpE has been found to differ greatly: ScpE accumulated to a much lower extent under all conditions tested (He et al. 2001). Nonetheless, SCP dimers are conceptually attractive as a pair of two membrane-spanning helices could form a structure similar to helix 1 and 3 of the light-harvesting complex, which would enable transient pigment binding.
The expression patterns of ScpC and ScpD and their high sequence similarity (87% identity) suggest that they play a similar role in the cell, demanding a tight coregulation. Moreover, cotranscription of the scpD and slr1544 genes (Suzuki et al. 2001, Fig. 2C) suggests coregulation. Under the control of the scpD promoter slr1544 is transcribed upon various stress conditions. The two genes are separated by 49 nucleotides in Synechocystis (59 nt in Anabaena, 65 nt in Nostoc, and 60 nt in Nodularia, see Fig. 2) thus excluding the formation of a two-helix protein similar to the SEPs (Heddad and Adamska 2002) (see also Yao et al. 2007). The slr1544 gene codes for a protein with molecular weight of 12 kDa, predicted to have a single transmembrane helix. Similar to the CAB-protein family it contains the amino acids Gly-Arg of the CAB-motif inside this membrane spanning region. Whereas Slr1544 is insufficiently similar to the SCP family to be considered a bona-fide SCP, Slr1544 appears to share an evolutionary ancestry with the SCPs and fits in with the broader family of light-harvesting-like proteins (Lils) (Jansson 1999). While in higher plants Lil proteins are numbered (Klimmek et al. 2006), we will follow the established prokaryotic nomenclature and suggest Slr1544 LilA. The SCPs and also LilA appear to be associated specifically with PSII, possibly particularly under conditions of complex turnover. Our mass spectrometry analysis of proteins comigrating with StrepII-tagged ScpB not only showed the presence of PSII proteins but also of the ATP-dependent FtsH protease (Sll1463). Of the four FtsH proteases in Synechocystis, two are crucial for cell viability (Slr1390 and Slr1604) and two are dispensable (Slr0228 and Sll1463) (Mann et al. 2000). However, Slr0228 is required for normal rates of D1 degradation during light stress (reviewed in Nixon et al. 2005); no phenotype was detected for a Δsll1463 mutant. Sll1463 is associated with PSII during light stress (Yao et al. 2007) and here we could detect it in the PSII fraction in cells grown under normal conditions.
It is noteworthy that PsbH was found in the fraction of StrepII-ScpB. Previous studies detected ScpD-His in a complex of CP47 and PsbH in cells grown at high light intensity (Promnares et al. 2006).
The results presented in this study further emphasize the association of SCPs with PSII, and show that such associations persist even under unstressed conditions if ScpB is expressed under a constitutive promoter.
Abbreviations
- CAB:
-
Chlorophyll a/b binding
- ELIP:
-
Early-light induced protein
- HLIP:
-
High-light induced protein
- LHC:
-
Light-harvesting complex
- Lil:
-
Light-harvesting like
- PAGE:
-
Polyacrylamide gel electrophoresis
- PS:
-
Photosystem
- SCP:
-
Small CAB-like protein
- LC-MS/MS:
-
Liquid chromatography combined with tandem mass spectrometry
- β-dM:
-
β-d-maltoside
References
Adamska I (1997) ELIPs: light induced stress proteins. Physiol Plant 100:798–805
Adamska I (2001) The Elip family of stress proteins in the thylakoid membrane of pro- and eukaryota. In: Aro E-M, Andersson B (eds) Advances in photosynthesis and respiration: regulation of photosynthesis, vol 11. Kluwer Academic Publishers, Dordrecht, pp 487–505
Anderson JM, Waldron JC, Thorne SW (1978) Chlorophyll-protein complexes of spinach and barley thylakoids: spectral characterization of six complexes resolved by an improved electrophoretic procedure. FEBS Lett 92:227–233
Bhaya D, Dufresne A, Vaulot D, Grossman A (2002) Analysis of the hli gene family in marine and freshwater cyanobacteria. FEMS Microbiol Lett 215:209–219
Durnford DG, Deane JA, Tan S, McFadden GI, Gantt E, Green BR (1999) A phylogenetic assessment of the eurkaryotic light-harvesting antenna proteins with implications for plastid evolution. J Mol Evol 48:59–68
Eaton-Rye JJ, Vermaas WFJ (1991) Oligonucleotide-directed mutagenesis of psbB, the gene encoding CP47, employing a deletion mutant strain of the cyanobacterium Synechocystis sp. PCC 6803. Plant Mol Biol 17:1165–1177
Funk C (2000) Functional analysis of the PsbX protein by deletion of the corresponding gene in Synechocystis sp. PCC 6803. Plant Mol Biol 44:815–827
Funk C (2001) The PsbS protein: a Cab-protein with a function of its own. In: Aro E-M, Andersson B (eds) Advances in photosynthesis and respiration: regulation of photosynthesis, vol 11. Kluwer Academic Publishers, Dordrecht, pp 453–467
Funk C, Vermaas W (1999) A cyanobacterial gene family coding for single-helix proteins resembling part of the light-harvesting proteins from higher plants. Biochemistry 38:9397–9404
Havaux M, Guedeney G, He Q, Grossman AR (2003) Elimination of high-light-inducible polypeptides related to eukaryotic chlorophyll a/b-binding proteins results in aberrant photoacclimation in Synechocystis PCC6803. Biochim Biophys Acta 1557:21–33
He Q, Schlich T, Paulsen H, Vermaas W (1999) Expression of a higher plant light-harvesting chlorophyll a/b-binding protein in Synechocystis sp. PCC 6803. Eur J Biochem 263:561–570
He Q, Dolganov N, Bjorkman O, Grossman AR (2001) The high light-inducible polypeptides in Synechocystis PCC6803. Expression and function in high light. J Biol Chem 276:306–314
Heddad M, Adamska I (2000) Light stress-regulated two-helix proteins in Arabidopsis thaliana related to the chlorophyll a/b-binding gene family. Proc Natl Acad Sci USA 97:3741–3746
Heddad M, Adamska I (2002) The evolution of light stress proteins in photosynthetic organisms. Comp Funct Genom 3:504–510
Hihara Y, Sonoike K, Kanehisa M, Ikeuchi M (2003) DNA microarray analysis of redox-responsive genes in the genome of the cyanobacterium Synechocystis sp. strain PCC 6803. J Bacteriol 185:1719–1725
Ivanov AG, Krol M, Sveshnikov D, Selstam E, Sandstrom S, Koochek M, Park YI, Vasil’ev S, Bruce D, Oquist G, Huner NP (2006) Iron deficiency in cyanobacteria causes monomerization of photosystem I trimers and reduces the capacity for state transitions and the effective absorption cross section of photosystem I in vivo. Plant Physiol 141:1436–1445
Jansson S (1999) A guide to the Lhc genes and their relatives in Arabidopsis. Trends Plant Sci 4:236–240
Jansson S (2005) A protein family saga: from photoprotection to light-harvesting (and back?) In: Demming-Adams B (ed) Photoprotection, photoinhibition, gene regulation and environment. Springer, The Netherlands, pp 145–153
Jansson S, Andersson J, Kim SJ, Jackowski G (2000) An Arabidopsis thaliana protein homologous to cyanobacterial high-light-inducible proteins. Plant Mol Biol 42:345–351
Joset F (1988) Transformation in Synechocystis PCC 6714 and PCC 6803:preparation of chromosomal DNA. Methods Enzymol 167:712–714
Klimmek F, Sjödin A, Noutsos C, Leister D, Jansson S (2006) Abundantly and rarely expressed Lhc protein genes exhibit distinct regulation patterns in plants. Plant Physiol 140:793–804
Kufryk G (1997) Changes in pigment-protein composition of maize subchloroplast fragments under phosphorylation. Ukr Biochem J 69:66–71
Kuhlbrandt W, Wang DN, Fujiyoshi Y (1994) Atomic model of plant light-harvesting complex by electron crystallography. Nature 367:614–621
Li XP, Bjorkman O, Shih C, Grossman AR, Rosenquist M, Jansson S, Niyogi KK (2000) A pigment-binding protein essential for regulation of photosynthetic light harvesting. Nature 403:391–395
Liu Z, Yan H, Wang K, Kuang T, Zhang J, Gui L, An X, Chang W (2004) Crystal structure of spinach major light-harvesting complex at 2.72 A resolution. Nature 428:287–292
Maniatis T, Frisch EF, Sambrook J (1982) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
Mann NH, Novac N, Mullineaux CW, Newman J, Bailey S, Robinson C (2000) Involvement of an FtsH homologue in the assembly of functional photosystem I in the cyanobacterium Synechocystis sp. PCC 6803. FEBS Lett 479:72–77
Marin K, Suzuki I, Yamaguchi K, Ribbeck K, Yamamoto H, Kanesaki Y, Hagemann M, Murata N (2003) Identification of histidine kinases that act as sensors in the perception of salt stress in Synechocystis sp. PCC 6803. Proc Natl Acad Sci USA 100:9061–9066
Mikami K, Kanesaki Y, Suzuki I, Murata N (2002) The histidine kinase Hik33 perceives osmotic stress and cold stress in Synechocystis sp PCC 6803. Mol Microbiol 46:905–915
Mohamed A, Eriksson J, Osiewacz HD, Jansson C (1993) Differential expression of the psbA genes in the cyanobacterium Synechocystis 6803. Mol Gen Genet 238:161–168
Nixon PJ, Barker M, Boehm M, de Vries R, Komenda J (2005) FtsH-mediated repair of the photosystem II complex in response to light stress. J Exp Bot 56:357–363
Ohta N, Matsuzaki M, Misumi O, Miyagishima SY, Nozaki H, Tanaka K, Shin IT, Kohara Y, Kuroiwa T (2003) Complete sequence and analysis of the plastid genome of the unicellular red alga Cyanidioschyzon merolae. DNA Res 10:67–77
Pandey A, Andersen JS, Mann M (2000) Use of mass spectrometry to study signaling pathways. Sci STKE 2000(37):pl1
Porra RJ, Thompson WA, Kriedemann PE (1989) Determination of accurate extinction coefficients and simultaneous equations for assaying chlorophylls a and b extracted with four different solvents: verification of the concentration of chlorophyll standards by atomic absorption spectroscopy. Biochim Biophys Acta 975:385–394
Promnares K, Komenda J, Bumba L, Nebesarova J, Vacha F, Tichy M (2006) Cyanobacterial small chlorophyll binding protein ScpD (HliB) is located on the periphery of photosystem II in the vicinity of PsbH and CP47 subunits. J Biol Chem 281:32705–32713
Rippka R, Deruelles J, Waterbury JB, Herdman M, Stanier RT (1979) Generic assignments, strain histories and properties of pure cultures of cyanobacteria. J Gen Microbiol 111:1–61
Rögner M, Nixon PJ, Diner BA (1990) Purification and characterization of Photosystem I and Photosystem II core complexes from wild-type and phycocyanin-deficient strains of the cyanobacterium Synechocystis PCC 6803. J Biol Chem 265:6189–6196
Schagger H, von Jagow G (1987) Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal Biochem 166:368–379
Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins from silver-stained polyacrylamide gels. Anal Chem 68:850–858
Standfuss J, Terwisscha van Scheltinga AC, Lamborghini M, Kuhlbrandt W (2005) Mechanisms of photoprotection and nonphotochemical quenching in pea light-harvesting complex at 2.5 A resolution. EMBO J 24:919–928
Suzuki I, Kanesaki Y, Mikami K, Kanehisa M, Murata N (2001) Cold-regulated genes under control of the cold sensor Hik33 in Synechocystis. Mol Microbiol 40:235–244
Teramoto H, Itoh T, Ono TA (2004) High-intensity-light-dependent and transient expression of new genes encoding distant relatives of light-harvesting chlorophyll a/b proteins in Chlamydomonas reinhardtii. Plant Cell Physiol 45:1221–1232
Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76:4350–4354
Tzvetkova-Chevolleau T, Franck F, Alawady AE, Dall’Osto L, Carriere F, Bassi R, Grimm B, Nussaume L, Havaux M (2007) The light stress-induced protein ELIP2 is a regulator of chlorophyll synthesis in Arabidopsis thaliana. Plant J 50:795–809
Wilson A, Ajlani G, Verbavatz JM, Vass I, Kerfeld CA, Kirilovsky D (2006) A soluble carotenoid protein involved in phycobilisome-related energy dissipation in cyanobacteria. Plant Cell 18:992–1007
Xu H, Vavilin D, Funk C, Vermaas W (2002) Small Cab-like proteins regulating tetrapyrrole biosynthesis in the cyanobacterium Synechocystis sp. PCC 6803. Plant Mol Biol 49:149–160
Xu H, Vavilin D, Funk C, Vermaas W (2004) Multiple deletions of small Cab-like proteins in the cyanobacterium Synechocystis sp. PCC 6803:consequences for pigment biosynthesis and accumulation. J Biol Chem 279:27971–27979
Yao D, Kieselbach T, Komenda J, Promnares K, Prieto MA, Tichy M, Vermaas W, Funk C (2007) Localization of the small CAB-like proteins in photosystem II. J Biol Chem 282:267–276
Acknowledgments
The authors would like to thank the Swedish Foundation for International Cooperation in Research and Higher Education (STINT) for supporting the collaboration between the two research groups. C.F. is very grateful to the Royal Swedish Academy of Sciences (KVA) for granting her research position and to the Swedish Research Council (VR) for financial support. W.V. acknowledges research grant support from the US Department of Energy (DE-FG02-04ER15543).
Author information
Authors and Affiliations
Corresponding author
Additional information
Galyna Kufryk and Miguel Hernandez-Prieto have contributed equally to this work.
Electronic supplementary material
Below are the link to the electronic supplementary materials.
Rights and permissions
About this article
Cite this article
Kufryk, G., Hernandez-Prieto, M.A., Kieselbach, T. et al. Association of small CAB-like proteins (SCPs) of Synechocystis sp. PCC 6803 with Photosystem II. Photosynth Res 95, 135–145 (2008). https://doi.org/10.1007/s11120-007-9244-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11120-007-9244-3