
Abstract
Field pea (Pisum sativum L.) is a cool-season legume that is cultivated worldwide for both human consumption and stockfeed. Genetic improvement is essential for enhanced crop production and management of field pea, especially to deliver superior varieties adapted to various biotic and abiotic stresses. A detailed understanding of the genetic basis of such stress tolerances is hence desirable. Genetic linkage maps based on single nucleotide polymorphisms (SNP) and simple sequence repeat (SSR) markers have been developed from two recombinant inbred line (RIL) populations generated by crossing phenotypically divergent parental genotypes. The Kaspa × Yarrum map contained 428 loci across 1910 cM, while the Kaspa × ps1771 map contained 451 loci across 1545 cM. Data from these maps were combined through bridging markers with those from previously published studies to generate a consensus structure including 2028 loci distributed across seven linkage groups (LGs), with a cumulative length of 2387 cM at an average density of one marker per 1.2 cM. Trait dissection of powdery mildew resistance was performed for both RIL populations, identifying a single genomic region of large magnitude in the same genomic region on Ps VI, which were inferred to correspond to the er1 gene. Equivalent studies of the Kaspa × ps1771 RIL population identified a major quantitative trait locus (QTL) for boron tolerance that coincided with the disease resistance-controlling locus, permitting strategies of co-selection for these desirable traits. Resequencing of the PsMLO1 candidate gene from resistant and susceptible genotypes allowed design and validation of a putative diagnostic marker for powdery mildew resistance. The availability of a highly saturated consensus map, linked markers for key biotic and abiotic stress tolerances and a diagnostic marker for the agronomically important er1 gene provide important resources for field pea molecular breeding programs.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Field pea (Pisum sativum L.) is an important grain legume, which is consumed both as human food and animal feed. Field pea is the fourth most extensively cultivated pulse crop on a global basis and is widely grown in Europe (Rubiales et al. 2009; Pavan et al. 2011). Of various abiotic and biotic stresses, toxicity effects due to soil boron (B) and damaging infection by powdery mildew are among the most serious threats to pea cultivation. B toxicity is a major problem in several major production zones, including southern Australia, India, Pakistan, Iraq, Peru and the USA (Yau and Ryan 2008). High B concentrations occur at depths between 40 and 100 cm in the soil profile, inhibiting both crop growth and grain yield (Cartwright et al. 1984; Nuttall et al. 2003). In Australia, typical patterns of low rainfall exacerbate B toxicity. For instance, about 15 % of agricultural soils in Western Australia are at moderate to high risk of toxicity effects (Lacey and Davies 2009), and high B concentrations are also frequently associated with elevated salinity, levels of each stress varying seasonally according to precipitation. However, in low-rainfall environments and on clay soils, B is leached more slowly than salt (Bennett 2012). Due to these factors, B toxicity is difficult to manage by manipulation of agricultural systems, creating an incentive for solutions based on genetic improvement.
To date, limited progress has been made in understanding the genetics of B tolerance in field pea. Bagheri et al. (1996) detected two major genes for tolerance to high B concentrations, which interact with each other with incomplete dominance at each locus. However, recent studies in closely related species, such as the model legume Medicago truncatula Gaertn. (Bogacki et al. 2013) and cultivated lentil (Kaur et al. 2014), have indicated the effects of a single dominant locus. Several resistant cultivars have been identified within field pea breeding programs through phenotypic screening (Bennett 2012; A. Leonforte, personal communication), but no information on genomic regions associated with B tolerance in field pea is currently available.
Powdery mildew of pea, due to infection by the fungal pathogen Erysiphe pisi DC, causes yield losses of 25–50 % (Munjal et al. 1963; Warkentin et al. 1996; Janila and Sharma 2004; Katoch et al. 2010). The alternate pathogens Erysiphe baeumleri (Magnus) U, Braun & S. Takam. 2000 and Erysiphe trifolii Grev. have also been reported to cause powdery mildew of pea (Ondřej et al. 2005; Attanayake et al. 2010). Protection by treatment with fungicides provides a temporary control option. However, identification of resistance sources and incorporation into contemporary cultivars remain the most effective method for disease control (Janila and Sharma 2004; Katoch et al. 2010). The genetics of resistance to powdery mildew in pea is relatively well understood, three major loci (er1, er2 and Er3) having been described (Smýkal et al. 2012). The majority of resistant cultivars rely on the presence of the recessive er1 gene (Harland 1948; Timmerman et al. 1994; Vaid and Tyagi 1997; Janila and Sharma 2004). However, some studies have also detected a second recessive gene, er2 (Heringa et al. 1969; Sokhi et al. 1979; Kumar and Singh 1981). Slightly differing mechanisms have been reported for the two genes, such that er1 confers systemic resistance under field and controlled conditions, by prevention of epidermal cell penetration resulting in formation of very few haustoria or colonies. In contrast, the effects of er2 are largely confined to leaves, in which expression is influenced by both temperature and leaf age, such that complete resistance is only observed at 25 °C or in mature leaves. The er2-derived resistance is primarily based on reduced penetration rate, along with post-penetration cell death in mature leaves (Fondevilla et al. 2006; Marx 1986; Tiwari et al. 1997). Only er1 has been extensively used in breeding programs, and it is considered as stable and effective. In contrast, Er3 is a dominant resistance locus recently characterised from a related species, tawny pea (Pisum fulvum Sibth. & Sm.) and is known to segregate independently of er1 and er2 (Fondevilla et al. 2011).
DNA-based genetic markers provide powerful tools for identification and location of genes for agronomically important characters, and subsequent selection for trait introgression in plant breeding programs. As previously noted, no such studies have yet been performed for B tolerance in field peas, but in the case of powdery mildew resistance, markers belonging to various classes such as restriction fragment length polymorphism (RFLP), randomly amplified polymorphic DNA (RAPD), sequence characterised region (SCAR), sequence-tagged site (STS), SSR and SNP have been linked to er1 on LG VI (Dirlewanger et al. 1994; Ek et al. 2005; Janila and Sharma 2004; Timmerman et al. 1994; Tiwari et al. 1998; Katoch et al. 2010; Tonguç and Weeden 2010; Pavan et al. 2013). The er2 gene was localised to pea LG III, in linkage with various amplified fragment length polymorphism (AFLP) and SCAR markers (Tiwari et al. 1999; Katoch et al. 2010). In addition, SCAR markers in linkage with the Er3 gene have been reported (Fondevilla et al. 2008). SNP markers are highly prevalent, usually biallelic and co-dominant in nature, and amenable to development of cost-effective and high-throughput marker systems that can provide sufficiently dense genome coverage for the dissection of key traits. In addition, discovery of SNPs from genic sequences, such as expressed sequence tags (ESTs), permits marker development from functionally associated sequences. Large-scale resources for field pea have been obtained from transcriptome sampling (Kaur et al. 2012). Consequently, SNPs provide the marker class of choice for determination of the genetic basis of agronomically important traits.
Genetic linkage maps are an essential prerequisite for the identification and localisation of genes for agronomically important characters. Several linkage maps have been constructed for pea based on different kinds of markers, including morphological markers, isoenzymes, RFLPs, RAPDs, SSRs, and SNPs (Weeden et al. 1996; Weeden and Boone 1999; Timmerman-Vaughan et al. 2000; Ellis and Poyser 2002; Loridon et al. 2005; Aubert et al. 2006; Deulvot et al. 2010). Recently, SNP-based linkage maps have been developed for field pea, which have been used for dissection of the genetic basis for salinity stress tolerance (Leonforte et al. 2013). However, individual linkage maps may suffer limitations in terms of practical application due to low marker density. Consensus linkage maps, which combine information from multiple mapping populations, have been developed for many crop species, including pea (Cloutier et al. 2012; Millan et al. 2010; Weeden et al. 1998; Duarte et al. 2014). Consensus maps offer the following advantages: higher marker density in a single map, and hence more complete genome coverage; opportunities to determine the relative position of common markers across mapping populations; determination of conserved marker locus location; and identification of chromosomal rearrangements and degree of gene duplication (Milczarski et al. 2011; Blenda et al. 2012). Consensus maps have been constructed for several crops using software programs such as JoinMap (Shirasawa et al. 2013) and MergeMap (Gautami et al. 2012). JoinMap accounts for both sizes and structures of populations in order to estimate marker order and genetic distance using common or bridging markers (Stam 1993). In MergeMap, individual maps are first converted to directed acyclic graphs (DAGs) internally with nodes representing the mapped markers and edges defining the order of adjacent markers (Wang et al. 2011). DAGs are then merged into a consensus graph on the basis of shared vertices. Conflicts between the individual maps are apparent as cycles in the consensus graph, and MergeMap attempts to resolve such conflicts by deletion of minimum set of marker occurrences (Wu et al. 2011).
Complementary trait-specific mapping families will permit analysis of characters such as B toxicity tolerance and powdery mildew resistance, along with their mutual interactions, when these key stress tolerance traits are present in the same germplasm. SNP-based genetic maps also permit comparative analysis with related species for identification of candidate genes. In the case of cool-season legumes, species with full genome-sequence species such as the models M. truncatula and Lotus japonicus L., and the cultivated species soybean (Glycine max L. [Merr.]) and chickpea (Cicer arietinum L.) provide appropriate comparators. Candidate gene information may be further used to develop potential diagnostic markers directly applicable to selection and gene pyramiding in breeding programs. Recently, through the study of a novel chemically induced allele of the er1 gene, co-segregation with a loss-of-function mutation at the PsMLO1 (mildew resistance locus O) gene was reported. Analysis of the gene sequence from several known powdery mildew-resistant cultivars further supported the hypothesis that a loss-of-function in PsMLO1 is responsible for the trait, and indicated that the molecular basis is shared with well-known powdery mildew immunity determinants in barley (mlo) and tomato (ol-2) (Pavan et al. 2010; Humphry et al. 2011).
The present study describes the generation of high-density genetic linkage maps, based exclusively on SSRs and SNPs, from two biparental field pea RIL mapping populations (Kaspa × Yarrum and Kaspa × ps1771) that exhibit variation for both B tolerance and powdery mildew resistance; development of a consensus map of field pea combining the maps from these trait-specific populations, a previously described RIL family (Leonforte et al. 2013) and an existing integrated pea map; identification of genomic regions controlling the target traits, based on phenotypic data collected from glasshouse-based nursery screens in Victoria, Australia; and development of a potential diagnostic marker for powdery mildew resistance based on resequencing analysis of the PsMLO1 gene.
Materials and Methods
Plant Materials
Crosses were made between single genotypes selected from cultivar Kaspa (sensitive to B toxicity, susceptible to powdery mildew) with each of Yarrum (resistant to powdery mildew) and ps1771 (tolerant to B toxicity and resistant to powdery mildew) at DEPI-Horsham, Victoria, Australia, and F2 generation progeny were produced. Single seed descent was undertaken from F2 progeny-derived genotypes for four generations in the glasshouse, and a total of 106 F6 RILs were subsequently generated from each of the Kaspa × Yarrum and Kaspa × ps1771 populations.
Plants were grown under glasshouse conditions at 20 ± 2 °C under a 16/8-h (light/dark) photoperiod. Genomic DNA was extracted from young leaves using the DNeasy® 96 Plant Kit (QIAGEN, Hilden, Germany) according to manufacturer’s instructions. Approximately one to two leaves per sample were used for each extraction and were ground using a Mixer Mill 300 (Retsch®, Haan, Germany). DNA was resuspended in Milli-Q water to a concentration of 50 ng/μl and stored at −20 °C until further use.
SSR and SNP Genotyping
A total of 242 genomic DNA- and EST-derived SSRs (Loridon et al. 2005; Kaur et al. 2012) were screened on the mapping parents for polymorphism detection. Primer synthesis and PCR amplifications were performed as described previously (Schuelke 2000; Kaur et al. 2012). PCR products were combined with the ABI GeneScan LIZ500 size standard and analysed using an ABI3730xl (Life Technologies Australia Pty Ltd, Victoria, Australia) capillary electrophoresis platform according to the manufacturer’s instructions. Allele sizes were scored using GeneMapper® 3.7 software package (Life Technologies Australia Pty Ltd).
For SNP genotyping, a previously described set of 768 SNPs (Leonforte et al. 2013) was selected for genotyping using the GoldenGate™ oligonucleotide pooled assay (OPA). A total of 250 ng of genomic DNA from each genotype was used for amplification, after which PCR products were hybridised to bead chips via the address sequence for detection on an Illumina iSCAN Reader. On the basis of obtained fluorescence, allele call data were viewed graphically as a scatter plot for each marker assayed using GenomeStudio software v2011.1 (Illumina) with a GeneCall threshold of 0.20.
Genetic Linkage Mapping
Data obtained from SNP and SSR genotyping were tested for goodness-of-fit to the expected Mendelian ratio of 1:1 using χ 2 analysis (P < 0.05). The genetic linkage map was generated using Map Manager Software version QTXb19 (Manly et al. 2001). Map distances were calculated using the Kosambi mapping function (Kosambi 1944) at a threshold LOD score of 3. LGs were assigned on the basis of marker locus commonality with publicly available linkage maps of pea (Loridon et al. 2005), and by comparison with chromosomes of M. truncatula (Choi et al. 2004; Kalo et al. 2004). LGs were drawn using Mapchart software v 2.2 (Voorrips 2002).
Consensus Map Construction
The Kaspa × Yarrum- and Kaspa × ps1771-derived maps from the current study were combined with the Kaspa × Parafield map of Leonforte et al. (2013), which shared a high proportion of common markers, in order to generate a preliminary composite map. MergeMap (Wu et al. 2011) converted the individual maps into DAGs that were merged in a consensus graph on the basis of shared vertices (Yap et al. 2003). A comparative analysis of this preliminary composite map with the integrated pea map of Duarte et al. (2014) was performed using BLAST-based sequence analysis. Similarity searches were performed with DNA sequences underlying SNP markers assigned to the preliminary composite map against transcriptome sequencing data (DDBJ/EMBL/GenBank under the accession GAMJ00000000) with a threshold E value of 10−10. After the identification of common sequences, the composite and integrated maps were melded into a single consensus structure using MergeMap (Wu et al. 2011). The consensus map for each LG was visualised by MapChart (Voorrips 2002).
Phenotypic Screening
Boron Toxicity
All individuals from the Kaspa × ps1771 RIL mapping populations were screened for response to B-induced stress applied at the seedling stage. Screening was undertaken by sowing three replicates of two plants each from each RIL-derived line at equidistant spacing in PVC cores using sandy clay soil. Two B concentration regimes were applied in soluble form as boric acid: 0 ppm (control) and 10 ppm. B toxicity was measured as visual assessment of leaf and stem necrotic symptoms on a 0–10 scale (Additional file 1a) performed over three different time intervals. Phenotyping data was analysed to estimate means after adjustments for any spatial effects within the trial. This was performed by calculating the averages for plant symptom score from individual plant assessments, which were then used to estimate genotype-specific average values for symptom score using residual maximum likelihood (REML) spatial row-column analysis. An index was used to quantify genotypic boron tolerance values and to describe tolerance levels according to sensitivity based on weighted symptom scores and final biomass. Models were fitted using REML as implemented in GenStat (GenStat Committee 2002 and previous releases). Best linear unbiased predictions (BLUP) analysis was used to calculate narrow-sense heritability. Means of symptom rating from each data set were used to construct distribution histograms in order to deduce Mendelian inheritance models for the trait.
Powdery Mildew Resistance
The parents and RIL progeny of the Kaspa × Yarrum and Kaspa × ps1771 populations were sown in pots under controlled environment conditions at DEPI-Horsham, Victoria, with three replicates. Plants were left in the glasshouse during the spring season to allow natural incidence of powdery mildew due to infection by E. pisi. The identity of the pathogen, and absence of other Erysiphe species (such as E. trifolii, which is capable of overcoming er1 and Er3, but not er2: Fondevilla et al. (2013)), was confirmed by the Senior Pathologist, Dr. Grant Holloway (pers. comm.). For the Kaspa × Yarrum mapping population, the disease reaction was recorded on a 0–5 scale (Additional file 1b) based on the percentage of the infected foliage area. For the Kaspa × ps1771 population, whole plant symptom status was recorded as either resistant or susceptible reaction. Phenotypic assessment data were analysed to estimate means after adjustment for any spatial patterning within the trial. Models were fitted using REML as implemented in GenStat (GenStat Committee 2002 and previous releases). Means of symptom ratings from each individual of the mapping populations were used to construct distribution histograms in order to determine the mode of inheritance for the trait.
QTL Analysis
QTL detection was performed using marker regression, simple interval mapping (SIM) and composite interval mapping (CIM) in QTL Cartographer v 2.5 (Wang et al. 2012). For SIM, an arbitrary LOD threshold of 2.5 was used to determine significance, while for CIM, significance levels for LOD thresholds were determined using 1000 permutations.
Development of a Diagnostic Marker for Powdery Mildew Resistance
Primers for amplification of the PsMLO1 genomic sequence (5′-ATGGCTGAAGAGGGAGTT-3′ and 5′- GGTAGCAGCTTGATTTGTGGATA -3′) were designed using Sequencher 4.7 (Gene Codes Corporation, USA) and OligoCalc: Oligonucleotide Properties Calculator (http://www.basic.northwestern.edu/biotools/oligocalc.html); on the basis of the published sequence (Santo et al. 2013: www.ncbi.nlm.nih.gov/nuccore/KC466597.1). PCR amplification was performed in a 20 μl reaction containing 20 ng DNA, 1× PCR buffer (Bioline), 0.4 μM of each primer, 0.2 mM of each dNTP and 0.5 U IMMOLASE (Bioline). PCR conditions included a hot start at 95 °C for 10 min, followed by 35 cycles of 94 °C for 30 s, 46–50 °C for 30 s and 72 °C for 30 s, and a final elongation step at 72 °C for 10 min.
PCR products were purified in a 15-μl reaction containing 2.5 U of shrimp alkaline phosphatase (USB-VWR International, Pennsylvania, USA) and 2.5 U of exonuclease I (at 20 U/μl) (New England Biolabs) and 5 μl of PCR product. Purified PCR products were analysed using a sequencing primer and BigDye Terminator v3.1 sequencing chemistry following the manufacturer’s instructions. Final PCR products were purified using ethanol precipitation and resuspended in 12 μl Hi–Di formamide for sequence determination using an ABI3730xl (Applied Biosystems) capillary electrophoresis platform according to the manufacturer’s instructions. Sequence analysis and assembly of the resulting electropherograms were performed in Sequencher 4.7 (Gene Codes Corporation, USA), allowing alignment and visual identification of sequence variants. Full-length sequence was generated using the Sanger sequencing-primer walking approach.
A specific PCR test, using the amplification primer pairs—5′-TGGTTCAATCGTCCTCACCT -3′ and 5′-TGCAAGTTGAAAGGCATTCT -3′—was designed to detect the presence of amplicon length variation due to insertion–deletion (indel) events, and the corresponding SCAR allele variation was determined in the mapping populations using PCR conditions as described for SSR analysis by Kaur et al. (2012).
Results
Polymorphic Markers for Map Construction
A total of 242 publicly available SSR markers (96 EST-SSRs and 146 genomic DNA-derived SSRs) were screened for polymorphism detection. A total of 41 (28 %) and 45 (31 %) of the genomic DNA-derived SSRs detected polymorphisms in the Kaspa × Yarrum and Kaspa × ps1771 mapping populations, respectively. Lower numbers of the EST-SSR markers detected polymorphism in the Kaspa × Yarrum (27 %) and Kaspa × ps1771 (26 %) populations (Table 1). All marker data were tested for conformity to the expected Mendelian ratio, and markers with χ 2 score >10 (P < 0.05) were removed from further analysis. Residual sets of 41 (Kaspa × Yarrum) and 51 (Kaspa × ps1771) segregating marker alleles were used for linkage mapping.
Sub-sets of 424 and 422 SNPs from the total of 768 detected polymorphism in the Kaspa × Yarrum and Kaspa × ps1771 RILs, respectively (Table 1). A total of 314 polymorphic loci were found to be common between the two mapping populations. For each SNP, three main clusters were identified, corresponding to AA homozygotes, AB heterozygotes and BB homozygotes. The majority of the SNP markers produced two major clusters representing the homozygous genotypes, with a minor extra cluster corresponding to the heterozygous class. As both mapping populations were descended to the F6 level, the frequency of heterozygous combinations was expected to be low, as was observed in practice (about 5 % in both populations). The χ 2 test (P < 0.05) identified 5 % (Kaspa × Yarrum) and 4.2 % (Kaspa × ps1771) of the SNP markers that did not segregate in accordance with the expected Mendelian inheritance ratio. All markers exhibiting such significant segregation distortion were excluded from the final analysis.
Genetic Linkage Mapping
For the Kaspa × Yarrum mapping population, χ 2 analysis and missing data led to the exclusion of 48 markers (8.27 %) from further analysis. Of the remaining 443 loci (41 SSRs and 402 SNPs) (Table 1), 428 (35 SSRs and 393 SNPs) were assigned to 13 LGs (Additional file 2a). Five RILs were excluded from analysis due to marker heterozygosity levels between 25 and 35 %. For Kaspa × ps1771, 37 (6.6 %) markers were excluded from further analysis due to χ 2 values (P < 0.05) and missing data. Of the remaining 455 loci (51 SSRs and 404 SNPs) (Table 1), 451 (50 SSRs and 401 SNPs) were assigned to 9 LGs (Additional file 2b). Comparisons between the two genetic maps revealed substantial commonality of marker order, although specific map distances were not always in similar proportion (Additional file 3a–3g).
The proportion of loci assigned to LGs was 97 and 99 % for the Kaspa × Yarrum and Kaspa × ps1771 maps, respectively, while the remaining markers were unlinked. In total, 11 LGs and 2 satellite were generated for Kaspa × Yarrum, and 7 LGs and 2 satellites were obtained for Kaspa × ps1771 (Additional file 4a and 4b). The cumulative length of the Kaspa × Yarrum map was 1910 cM, with an average distance of 4.4 cM between loci, while the Kaspa × ps1771 map spanned a total length of 1545 cM, with an average marker density of 1 locus per 3.4 cM (Table 2). As high levels of co-linearity were observed with previously published maps, most of the satellite LGs, when identity was predictable on the basis of common loci, could be reasonably expected to coalesce with the corresponding intact LG through the use of a larger number of markers (data not shown). In total, 308 markers were common between the Kaspa × Yarrum and Kaspa × ps1771 maps. Instances of markers assigned to different LGs between the two maps were rare, with the exception of Kaspa × Yarrum, for which 10 SNP markers (SNP_100000353, SNP_100000801, SNP_100000802, SNP_100000347, SNP_100000674, SNP_100000220, SNP_100000293, SNP_100000150, SNP_100000224, SNP_100000183) were located in a segment of Ps IV, while for Kaspa × ps1771, the corresponding region was on Ps III.
Consensus Linkage Map Construction
Data from three F6 RIL populations was used to construct the preliminary composite map. The number of individual marker loci ranged from 429 (Kaspa × Yarrum) to 452 (Kaspa × ps1771) and 458 (Kaspa × Parafield). A total of 764 marker loci (680 SNPs and 84 SSRs) were assembled into 7 LGs (Additional file 5), of which 286 markers (37.4 %) were unique to single populations (Kaspa × Parafield, 157; Kaspa × Yarrum, 81; Kaspa × ps1771. 48), the remaining 478 (62.6 %) providing bridging loci between two or more maps. In total, 160 markers were common across all three maps. The highest number of common markers (308) was between the Kaspa × Yarrum and Kaspa × ps1771 maps, followed by the Kaspa × Parafield–Kaspa × ps1771 comparison with 261 markers. The total length of the preliminary composite linkage map was 2555 cM, lengths of major LGs ranging from 249 cM (Ps V) to 421 cM (Ps III), with an average density of one marker per 3.4 cM. The largest numbers of markers (135) were assigned to Ps VII, while the lowest numbers (83) were on Ps V. The marker order was largely co-linear with the three individual maps, although a few local inversions and marker rearrangements over short intervals were observed. A total of 33 markers (30 SNPs and 3 SSRs) were assigned to single loci on different LGs across mapping populations. Such loci were not considered as identical and were consequently reckoned as unique in the preliminary composite genetic map.
Based on BLASTN analysis of corresponding DNA sequences to the 768 SNPs, 767 detected significant sequence similarity matches to recently available pea transcriptome sequences (Additional file 6). This analysis supported establishment of 135 bridging loci between the field pea preliminary composite map and the available integrated map of Duarte et al. (2014). Comparison of these maps revealed the highest number of matches (29) between Ps VII and LG 7 and the lowest (7) between Ps V and LG 5. Minor discrepancies occurred in some markers (especially terminal locations), but marker order was generally co-linear. However, the global orientation was reversed for four of the seven LGs between the composite and integrated maps.
Merger of the two datasets obtained a new consensus map containing 2028 markers on 7 LGs spanning 2387 cM (Fig. 1; Table 3). A total of 535 SNP markers from the preliminary composite map were derived from transcript sequences common with the SNP markers from Duarte et al. (2014): Ps III and Ps VII containing the highest number (102) and Ps V the lowest (56) (Additional file 7). The marker order of the final consensus map was largely co-linear between the participating structures, although several inversions and local rearrangements were observed.

Consensus map of field pea, with marker loci shown on the right-hand side of LGs, and distances between markers indicated in cM on the left. For presentation purposes, not all genetic markers are shown on the map. Specific details of marker locus identity and location are provided in Additional File 7
Phenotypic Analysis and QTL Detection
For B toxicity, in the Kaspa × ps1771 RIL population, a high degree of correlation was observed for symptom rating obtained at different time intervals (r 2 = 0.86). Averages for plant symptom score (calculated from individual plant assessments), and boron index were used to generate frequency distribution histograms. Although the distribution pattern was not indicative of continuous variation, it did not conform to a bimodal structure arising from a single gene effect (Additional file 8a). Narrow-sense heritability values for each measurement ranged from 0.83 to 0.85. Marker regression analysis identified two markers (AB71 and PBA_PS_0398) that were significantly associated with variation for the trait. SIM identified one genomic region on Ps VI, accounting for 58 % of the phenotypic variance (V p) (Additional file 2b).
In the case of powdery mildew resistance, significant differences between RILs for each population were observed for both symptom score and percentage of leaf cover. Frequency distribution patterns obtained from both populations, due to bimodal structures, indicated the presence of a single gene responsible for powdery mildew resistance (Additional file 8b). The locations and magnitudes of effect for each genomic region were estimated, and for each mapping population, both marker regression analysis and SIM detected a single genomic region on Ps VI (Table 4). CIM identified the same region, accounting for 93 and 81 % of V p for the Kaspa × Yarrum and Kaspa × ps1771 populations, respectively (Additional file 2a and 2b).
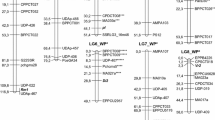
The same genomic region on the Kaspa × ps1771 map contained major QTLs for both powdery mildew resistance and boron tolerance region, indicating that the respective causal polymorphisms are closely linked in coupling phase (Fig. 2).

Co-linearity of common markers between individual genetic linkage maps (Kaspa × ps1771 and Kaspa × Yarrum) and consensus map on Ps VI at the QTL-containing regions of the field pea. LGs or chromosomes are shaded in colours for presentation purposes. Marker loci are shown on the right of the linkage groups, and map distances between markers are indicated in cM on the left. Coloured lines represent the corresponding positions of common markers
Development of a Diagnostic Marker for the er1 Powdery Mildew Resistance Gene
The full-length PsMLO1 coding sequence was used a template for primer design to support amplicon generation from the Kaspa, Yarrum and ps1771 genotypes. The DNA sequence of the PsMLO1-specific amplicon was determined (Additional file 9), and sequence analysis revealed that the Yarrum and ps1771-derived PsMLO1 allele containing a 2-bp insertion event in intron 11 of the gene as compared to the equivalent from Kaspa, the powdery mildew susceptible parental genotype (Fig. 3). A SCAR marker (Fig. 3) was designed to identify the allelic difference and was screened across both RIL populations. The PsMLO1 SCAR locus was confirmed as coinciding with the major powdery mildew resistance QTLs that were identified in both populations.

Schematic diagram of intron-exon structure in the PsMLO1 gene in the region surrounding the 2-bp indel polymorphism. The sequence of exon 11 is shown in red text, sequence of intron 11 shown in black text and exon 12 in blue, and the 2-bp insertion characteristic of Yarrum and Ps1771 is highlighted. The SCAR primer sequences and location are underlined
Discussion
Attributes of Genetic Linkage Maps and Consensus Map Construction
During a lengthy history of genetic mapping studies in field pea, different types of genetic marker systems have been successively used (Weeden et al. 1996; Weeden and Boone 1999; Laucou et al. 1998; Timmerman-Vaughan et al. 2000; Ellis and Poyser 2002; Loridon et al. 2005; Aubert et al. 2006; Deulvot et al. 2010). However, to date, only one high-density genetic linkage map has been constructed predominantly through the use of gene-associated SSR and SNP markers (Leonforte et al. 2013). In the present study, the same marker sets have been used to generate another two maps, both of which displayed a relatively uniform marker distribution, with average marker densities of one per 4.4 cM (Kaspa × Yarrum) and 3.4 cM (Kaspa × ps1771). The former map was significantly longer (1910 cM) than the latter (1545 cM). The reason for excess map length could be due to recombination events or missing data in the Kaspa × Yarrum RIL population (Knox and Ellis 2002). Based on comparison through common markers, most LGs were consistent between the two individual maps, with a number of minor exceptions.
A preliminary composite map was constructed in this study from the three trait-specific RIL populations, which was then combined with a previously generated integrated map to obtain a final consensus. The MergeMap software that was used for this purpose has previously been used for construction of consensus maps in crops such as Phaseolus vulgaris (Galeano et al. 2012), Arachis hypogaea L. (Gautami et al. 2012) and Vicia faba L. (Satovic et al. 2013) based on 3, 11 and 3 populations, respectively. The preliminary composite map provided the opportunity to assign a larger number of loci than in individual maps, providing increased reliability of marker location prediction. Comparison to individual component maps revealed only minor inconsistencies of marker order, generally within the same LGs. Such discrepancies may be due to mapping errors due to missing data, limited linkage in individual maps, chromosome rearrangements or the influence of paralogous sequences. In general, composite maps provide one of many possible non-conflicting linear representations of the consensus DAGs (Close et al. 2009). Consequently, the order of markers in an integrated structure map may not match the order of corresponding nucleotides in a genome sequence, but marker order over longer distances should generally be preserved.
Developments in high-throughput genotyping based on SNPs facilitated comparison between the preliminary composite and integrated maps of pea. In general, small rearrangements were observed when comparing the two maps, especially near the distal ends of LGs. The observed differences in map length and marker order could be attributable to the use of different software to generate each map (Muñoz-Amatriaín et al. 2011). Comparison between the transcriptome datasets underlying each sequence-based marker led to the identification of common mRNA-encoding sequences. The newly generated consensus map for pea features integration of a large number of coding regions, providing an effective framework for downstream analyses, including comparisons between the locations of major genes for important traits or QTL positions between populations from different crosses. A high-density genetic map based on gene-based markers also provides an important foundation for QTL mapping and for anchoring of sequence scaffolds. Linkage maps have previously been used for anchoring and orientation of scaffolds in whole genome sequencing projects for many crop species, including soybean (Hyten et al. 2010), watermelon (Ren et al. 2012), grape (Jaillon et al. 2007) and cucumber (Huang et al. 2009). The linkage maps described here would hence be highly useful for the future genome assembly expected from the field pea genome sequencing consortium (http://www.coolseasonfoodlegume.org/pea_genome).
Phenotypic Analysis and Identification of QTLs for B Tolerance and Powdery Mildew Resistance
Physiological mechanisms for B tolerance are relatively well understood for some plant species such as Arabidopsis thaliana, and for cereal crops like wheat and barley. One major mechanism is the elimination of excess B from the root system based on transporter activity. Several different types of B transporter have been characterised in A. thaliana (Takano et al. 2002), and a corresponding gene has been cloned and characterised from barley (Hayes and Reid 2004; Schnurbusch et al. 2010). However, no equivalent studies have so far been conducted to determine the genetic basis of B tolerance in field pea. The frequency distributions for boron index and mean symptom score were not consistent with the contributions of a large number of genetic loci (as they did not conform to normal distributions), but equally, were not clearly attributable to single gene effects. Nonetheless, the present study identified a single genomic region of major effect on Ps VI. This finding is consistent with the outcomes of similar studies of other legume species such as lentil (Kaur et al. 2014) and M. truncatula (Bogacki et al. 2013), which also reported single gene models. It is possible that one or more minor gene also contributes to B tolerance in the Kaspa × ps1771 mapping population, but was below the threshold level for detection in the present study.
Resistance to powdery mildew in field pea is also well understood, the recessive er1 gene being the most common cause (Timmerman et al. 1994; Vaid and Tyagi 1997; Janila and Sharma 2004. The present study obtained evidence for single gene resistance in both populations, based on both the bimodal nature of the frequency distribution data, and QTL analysis. The genomic location on Ps VI, in the vicinity of er1, was consistent with the outcomes of prior studies (Ek et al. 2005; Janila and Sharma 2004; Timmerman et al. 1994; Tiwari et al. 1998; Katoch et al. 2010). In a previous study, two SSR loci (AB71 and AD59) were found to be located on either side of er1, 4.6 and 4.3 cM distant, respectively (Loridon et al. 2005). In the present study, these loci were also identified in the QTL-containing interval. The results of the present study were not consistent with presence of er2 in the resistant parental genotypes, as no QTL effects were identified on pea LG III, to which this locus has been previously attributed (Katoch et al. 2010).
A major implication arising from parallel assessment of B toxicity tolerance and powdery mildew resistance is that both traits in Kaspa × ps1771 population are controlled by single QTLs of large magnitude within the same interval. Although there is no reason to suspect a causal association, the respective genes are presumably in close linkage, consistent with observations from local field pea breeding trials (A. Leonforte, unpublished data). The ps1771 genotype can hence function as a common donor for the two traits. Fortunately, both the linked traits are favourable in nature, as otherwise recombination events within the QTL-containing region would be required to separate the determinants.
Development of a Diagnostic Marker for Powdery Mildew Resistance
Recent studies based on sequence analysis in four field pea accessions have reported that PsMLO1 provides the functional basis for allelic variation of the field pea er1 gene (Humphry et al. 2011). Loss-of-function of PsMLO1 is further known to provide powdery mildew resistance. Following mutagenesis of the resistant genotypes Solara and Frilene with ethylnitrosourea (ENU), susceptible derivatives were shown to contain point mutations in coding sequences leading to drastic truncation of the PsMLO1 gene product (Santo et al. 2013). In the present study, a small indel was identified that differentiates the PsMLO1 alleles from resistant and susceptible genotypes. Conversion of the indel polymorphism into a SCAR marker demonstrated coincidence with the powdery mildew resistance QTLs reported in this study. Based on previous research, it may be reasonably concluded that pathogen resistance in Yarrum and ps1771 is due to the er1 gene and that the SCAR provides a diagnostic molecular marker for trait variation in this germplasm. However, the origin of functional variation in PsMLO1 is not so clear, as, in contrast, to the outcomes of mutagenesis studies, the susceptibility-associated allele in Kaspa is not obviously impaired in terms of translation product structure, given that the 2-bp indel is located within an intron. No obvious exonic changes were identified across the full-length gene (Additional file 9). Functional variation for powdery mildew resistance must hence be due either to effects of the intron-located change on transcript splicing, or possibly the presence of a regulatory mutation outside the coding sequence, presumably in the 5′-proximal region of the gene, in linkage disequilibrum with the observed polymorphism. Further sequencing studies are required to test these hypotheses.
Implications for Field Pea Breeding Programs
Molecular markers linked to important agronomic traits have been demonstrated to be highly applicable to selection for desirable gene variants in different breeding programs. Marker-assisted selection (MAS) is cost and time efficient, non-destructive in nature and less error-prone than phenotypic selection. The major benefit of MAS for B toxicity tolerance and powdery mildew resistance in field pea would be to co-select genes for tolerance in multiple different genetic backgrounds. The Ps VI-located QTLs account for large percentages of V p, providing the capacity for introgression into elite parental background by donor-recipient backcrossing with minimal linkage drag. In addition, the development of a diagnostic marker for powdery mildew resistance will further facilitate selection processes in field pea breeding programs by direct identification of donor genotypes in germplasm collections and hence reduce duration of the breeding cycle.
References
Attanayake RN, Glawe DA, McPhee KE, Dugan FM, Chen W (2010) Erysiphe trifolii—a newly recognized powdery mildew pathogen of pea. Plant Pathol 59:712–720
Aubert G, Morin J, Jacquin F, Loridon K, Quillet MC, Petit A, Rameau C, Lejeune-Hénaut I, Huguet T, Burstin J (2006) Functional mapping in pea, as an aid to the candidate gene selection and for investigating synteny with the model legume Medicago truncatula. Theor Appl Genet 112:1024–1041
Bagheri A, Paull JG, Rathjen AJ (1996) Genetics of tolerance to high concentrations of soil boron in peas (Pisum sativum L.). Euphytica 87:69–75
Bennett SJ (2012) Early growth of field peas under saline and boron toxic soils. In Proceedings of 16th Agronomy Conference 2012, University of New England, Armidale, NSW, 14–18th October 2012
Blenda A, Fang DD, Rami JF, Garsmeur O, Luo F, Lacape JM (2012) A high density consensus genetic map of tetraploid cotton that integrates multiple component maps through molecular marker redundancy check. PLoS ONE 7:e45739
Bogacki P, Peck DM, Nair RM, Howie J, Oldach KH (2013) Genetic analysis of tolerance to boron toxicity in the legume Medicago truncatula. BMC Plant Biol 13:54
Cartwright B, Zarcinas BA, Mayfield AH (1984) Toxic concentrations of boron in a red-brown earth at Gladstone, South Australia. Soil Res 22:261–272
Choi HK, Mun JH, Kim DJ, Zhu H, Baek JM, Mudge J, Roe B, Ellis N, Doyle J, Kiss GB, Young ND, Cook DR (2004) Estimating genome conservation between crop and model legume species. Proc Natl Acad Sci U S A 101:15289–15294
Close TJ, Bhat PR, Lonardi S, Wu Y, Rostoks N, Ramsay L, Druka A, Stein N, Svensson JT, Wanamaker S, Bozdag S, Roose ML, Moscou MJ, Chao S, Varshney RK, Szűcs P, Sato K, Hayes PM, Matthews DE, Kleinhofs A, Muehlbauer GJ, Young JD, Marshal DF, Madishetty K, Fenton RD, Condamine P, Graner A, Waugh R (2009) Development and implementation of high-throughput SNP genotyping in barley. BMC Genomics 10:582
Cloutier S, Ragupathy R, Miranda E, Radovanovic N, Reimer E, Walichnowski A, Ward K, Rowland G, Duguid S, Banik M (2012) Integrated consensus genetic and physical maps of flax (Linum usitatissimum L.). Theor Appl Genet 125:1783–1795
Deulvot C, Charrel H, Marty A, Jacquin F, Donnadieu C, Lejeune-Henaut I, Burstin J, Aubert G (2010) Highly-multiplexed SNP genotyping for genetic mapping and germplasm diversity studies in pea. BMC Genomics 11:48
Dirlewanger E, Isaac PG, Ranade S, Belajouza M, Cousin R, de Vienne D (1994) Restriction fragment length polymorphism analysis of loci associated with disease resistance genes and developmental traits in Pisum sativum L. Theor Appl Genet 88:17–27
Duarte J, Rivière N, Baranger A, Aubert G, Burstin J, Cornet L, Lavaud C, Lejeune-Hénaut I, Martinant J-P, Pichon J-P, Pilet-Nayel M-L, Boutet G (2014) Transcriptome sequencing for high throughput SNP development and genetic mapping in pea. BMC Genomics 15:126
Ek M, Eklund M, von Post R, Dayteg C, Henriksson T, Weibull P, Ceptilis A, Issac P, Tuvesson S (2005) Microsatellite markers for powdery mildew resistance in pea (Pisum sativum L.). Hereditas 142:86–91
Ellis THN, Poyser SJ (2002) An integrated and comparative view of pea genetic and cytogenetic maps. New Phytol 153:17–25
Fondevilla S, Carver TLW, Moreno MT, Rubiales D (2006) Macroscopic and histological characterisation of genes er1 and er2 for powdery mildew resistance in pea. Eur J Plant Pathol 115:309–321
Fondevilla S, Rubiales D, Moreno MT, Torres AM (2008) Identification and validation of RAPD and SCAR markers linked to the gene Er3 conferring resistance to Erysiphe pisi DC in pea. Mol Breed 22:193–200
Fondevilla S, Cubero JI, Rubiales D (2011) Confirmation that the Er3 gene, conferring resistance to Erysiphe pisi in pea, is a different gene from er1 and er2 genes. Plant Breed 130:281–282
Fondevilla S, Chattopadhyay C, Kahre N, Rubiales D (2013) Erysiphe trifolii is able to overcome er1 and Er3, but not er2 resistance genes in pea. Eur J Plant Pathol 136:557–563
Galeano CH, Cortés AJ, Fernandez AC, Soler A, Franco-Herrera N et al (2012) Gene-based single nucleotide polymorphism markers for genetic and association mapping in common bean. BMC Genet 13:48
Gautami B, Foncéka D, Pandey MK, Moretzsohn MC, Sujay V et al (2012) An international reference consensus genetic map with 897 marker loci based on 11 mapping populations for tetraploid groundnut (Arachis hypogaea L.). PLoS ONE 7:e41213
GenStat Committee (2002) The Guide to GenStat Release 6.1—Part 2: Statistics. VSN International, Oxford
Harland SC (1948) Inheritance of immunity to mildew in Peruvian forms of Pisum sativum. Heredity 2:263–269
Hayes JE, Reid RJ (2004) Boron tolerance in barley is mediated by efflux of boron from the roots. Plant Physiol 136:3376–3382
Heringa RJ, Van Nore A, Tazelaar MF (1969) Resistance to powdery mildew (Erysiphe polygoni D.C.) in peas (Pisum sativum L.). Euphytica 18:163–169
Huang S, Li R, Zhang Z, Li L, Gu X, Fan W, Lucas WJ, Wang X, Xie B, Ni P, Ren Y, Zhu H, Li J, Lin K, Jin W, Fei Z, Li G, Staub J, Kilian A, van der Vossen EAG, Wu Y, Guo J, He J, Jia Z, Ren Y, Tian G, Lu Y, Ruan J, Qian W, Wang M, Huang Q, Li B, Xuan Z, Cao J, Asan WZ, Zhang J, Cai Q, Bai Y, Zhao B, Han Y, Li Y, Li X, Wang S, Shi Q, Liu S, Cho WK, Kim J-Y, Xu Y, Heller-Uszynska K, Miao H, Cheng Z, Zhang S, Wu J, Yang Y, Kang H, Li M, Liang H, Ren X, Shi Z, Wen M, Jian M, Yang H, Zhang G, Yang Z, Chen R, Liu S, Li J, Ma L, Liu H, Zhou Y, Zhao J, Fang X, Li G, Fang L, Li Y, Liu D, Zheng H, Zhang Y, Qin N, Li Z, Yang G, Yang S, Bolund L, Kristiansen K, Zheng H, Li S, Zhang X, Yang H, Wang J, Sun R, Zhang B, Jiang S, Wang J, Du Y, Li S (2009) The genome of the cucumber, Cucumis sativus L. Nat Genet 41:1275–1281
Humphry M, Reinstädler A, Ivanov S, Bisseling T, Panstruga R (2011) Durable broad‐spectrum powdery mildew resistance in pea er1 plants is conferred by natural loss‐of‐function mutations in PsMLO1. Mol Plant Pathol 12:866–878
Hyten DL, Cannon SB, Song Q, Weeks N, Fickus EW, Shoemaker RC, Specht JE, Farmer AD, May GD, Cregan PB (2010) High-throughput SNP discovery through deep resequencing of a reduced representation library to anchor and orient scaffolds in the soybean whole genome sequence. BMC Genomics 11:38
Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, Choisne N, Aubourg S, Vitulo N, Jubin C, Vezzi A, Legeai F, Hugueney P, Dasilva C, Horner D, Mica E, Jublot D, Poulain J, Bruyere C, Billault A, Segurens B, Gouyvenoux M, Ugarte E, Cattonaro F, Anthouard V, Vico V, Del Fabbro C, Alaux M, Di Gaspero G, Dumas V, Felice N, Paillard S, Juman I, Moroldo M, Scalabrin S, Canaguier A, Le Clainche I, Malacrida G, Durand E, Pesole G, Laucou V, Chatelet P, Merdinoglu D, Delledonne M, Pezzotti M, Lecharny A, Scarpelli C, Artiguenave F, Pe ME, Valle G, Morgante M, Caboche M, Adam-Blondon AF, Weissenbach J, Quetier F, Wincker P, French-Italian P (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449:463–467
Janila P, Sharma B (2004) RAPD and SCAR markers for powdery mildew resistance gene er in pea. Plant Breed 123:271–274
Kalo P, Seres A, Taylor SA, Jakab J, Kevei Z, Kereszt A, Endre G, Ellis THN, Kiss GB (2004) Comparative mapping between Medicago sativa and Pisum sativum. Mol Gen Genomics 272:235–246
Katoch V, Sharma S, Pathania S, Banayal DK, Sharma SK, Rathour R (2010) Molecular mapping of pea powdery mildew resistance gene er2 to pea linkage group III. Mol Breed 25:229–237
Kaur S, Pembleton LW, Cogan NOI, Savin KW, Leonforte T, Paul J, Materne M, Forster JW (2012) Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genomics 13:104
Kaur S, Cogan NOI, Stephens A, Noy D, Butsch M, Forster JW, Materne M (2014) EST-SNP discovery and dense genetic mapping in lentil (Lens culinaris Medik.) enable candidate gene selection for boron tolerance. Theor Appl Genet 127:703–713
Knox MR, Ellis THN (2002) Map expansion in pea. Genetics 162:861–873
Kosambi DD (1944) The estimation of map distance from recombination values. Ann Eugen 12:172–175
Kumar H, Singh RB (1981) Genetic analysis of adult plant resistance to powdery mildew in pea (Pisum sativum L.). Euphytica 30:147–154
Lacey A, Davies S (2009) Boron toxicity in WA soils. Farm note 388, Department of Agriculture and food, Western Australia
Laucou V, Haurogne K, Ellis N, Rameau C (1998) Genetic mapping in pea: I. RAPD-based linkage map of Pisum sativum. Theor Appl Genet 97:905–915
Leonforte A, Sudheesh S, Cogan NOI, Salisbury PA, Nicolas ME, Materne M, Forster JW, Kaur S (2013) SNP marker discovery, linkage map construction and identification of QTLs for enhanced salinity tolerance in field pea (Pisum sativum L.). BMC Plant Biol 13:161
Loridon K, Mcphee K, Morin J (2005) Microsatellite marker polymorphism and mapping in pea (Pisum sativum L). Theor Appl Genet 111:1022–1031
Manly KF, Cudmore RH Jr, Meer JM (2001) Map Manager QTX, cross-platform software for genetic mapping. Mamm Genome 12:930–932
Marx GA (1986) Location of er proving elusive. Pisum Newsl 18:39–41
Milczarski P, Bolibok-Bragoszewska H, Myśków B, Stojałowski S, Heller-Uszyńska K, Góralska M, Brągoszewski P, Uszyński G, Kilian A, Rakoczy-Trojanowska M (2011) A high density consensus map of rye (Secale cereale L.) based on DArT markers. PLoS ONE 7:e28495
Millan T, Winter P, Jüngling R, Gil J, Rubio J, Cho S, Cobos MJ, Iruela M, Rajesh PN, Tekeoglu M, Kahl G, Muehlbauer FJ (2010) A consensus genetic map of chickpea (Cicer arietinum L.) based on 10 mapping populations. Euphytica 175:175–189
Munjal RL, Chenulu VV, Hora TS (1963) Assessment of losses due to powdery mildew (Erysiphe polygoni) on pea. Indian Phytopathol 19:260–267
Muñoz-Amatriaín M, Moscou MJ, Bhat PR, Svensson JT, Bartoš J, Suchánková P, Šimková H, Endo TR, Fenton RD, Lonardi S, Castillo AM, Chao S, Cistué L, Cuesta-Marcos A, Forrest KL, Hayden MJ, Hayes PM, Horsley RD, Makoto K, Moody D, Sato K, Vallés MP, Wulff BBH, Muehlbauer GJ, Doležel J, Close TJ (2011) An improved consensus linkage map of barley based on flow-sorted chromosomes and single nucleotide polymorphism markers. Plant Genome 4:238–249
Nuttall JG, Armstrong RD, Connor DJ, Matassa VJ (2003) Interrelationships between edaphic factors potentially limiting cereal growth on alkaline soils in north-western Victoria. Soil Res 41:277–292
Ondřej M, Dostálová R, Odstrčilová L (2005) Response of Pisum sativum germplasm resistant to Erysiphe pisi to inoculation with Erysiphe baeumleri, a new pathogen of peas. Plant Prot Sci 41:95–103
Pavan S, Jacobsen E, Visser RGF, Bai Y (2010) Loss of susceptibility as a novel breeding strategy for durable and broad-spectrum resistance. Mol Breed 25:1–12
Pavan S, Schiavull A, Appiano M, Marcotrigiano AR, Cillo F, Visser RGF, Bai Y, Lotti C, Ricciardi L (2011) Pea powdery mildew er1 resistance is associated to loss-of-function mutations at a MLO homologous locus. Theor Appl Genet 123:1425–1431
Pavan S, Schiavulli A, Appiano M, Miacola C, Visser RG, Bai Y, Lotti C, Ricciardi L (2013) Identification of a complete set of functional markers for the selection of er1 powdery mildew resistance in Pisum sativum L. Mol Breed 31:247–253
Ren Y, Zhao H, Kou Q, Jiang J, Guo S, Zhang H, Hou W, Zou X, Sun H, Gong G, Levi A, Xu Y (2012) A high resolution genetic map anchoring scaffolds of the sequenced watermelon genome. PLoS ONE 7:e29453
Rubiales D, Fernandez-Aparicio M, Moral A, Barilli E, Sillero JC, Fondevilla S (2009) Disease resistance in pea (Pisum sativum L.) types for autumn sowings in Mediterranean environments. Czech J Genet Plant Breed 45:135–142
Santo T, Rashkova M, Alabaça C, Leitão J (2013) The ENU-induced powdery mildew resistant mutant pea (Pisum sativum L.) lines S (er1mut1) and F (er1mut2) harbour early stop codons in the PsMLO1 gene. Mol Breed 32:723–727
Satovic Z, Avila CM, Cruz-Izquierdo S, Díaz-Ruíz R, García-Ruíz GM, Palomino C, Gutiérrez N, Vitale S, Ocaña-Moral S, Gutiérrez MV, Cubero JI, Torres AM (2013) A reference consensus genetic map for molecular markers and economically important traits in faba bean (Vicia faba L). BMC Genomics 14:932
Schnurbusch T, Hayes J, Hrmova M, Baumann U, Ramesh SA, Tyerman SD, Langridge P, Sutton T (2010) Boron toxicity tolerance in barley through reduced expression of the multifunctional aquaporin HvNIP2; 1. Plant Physiol 153:1706–1715
Schuelke M (2000) An economic method for the fluorescent labelling of PCR fragments. Nat Biotechnol 18:233–234
Shirasawa K, Bertioli DJ, Moretzsohn MC, Leal-Bertioli SCM, Varshney RK et al (2013) An integrated consensus map of cultivated peanut and wild relatives reveals structures of the A and B genomes of Arachis and divergences with other legume genomes. DNA Res 20:173–184
Smýkal P, Aubert G, Burstin J, Coyne CJ, Ellis NTH, Flavell AJ, Ford R, Hýbl M, Macas J, Neumann P, McPhee KE, Redden RJ, Rubiales D, Weller JL, Warkentin TD (2012) Pea (Pisum sativum L.) in the genomic era. Agronomy 2:74–115
Sokhi SS, Jhooty JS, Bains SS (1979) Resistance in pea against powdery mildew. Indian Phytopathol 32:571–574
Stam P (1993) Construction of integrated genetic linkage maps by means of a new computer package: Join Map. Plant J 3:739–744
Takano J, Noguchi K, Yasumori M, Kobayashi M, Gajdos Z, Miwa K, Hayashi H, Yoneyama T, Fujiwara T (2002) Arabidopsis boron transporter for xylem loading. Nature 420:337–340
Timmerman GM, Frew TJ, Weeden NF, Miller AL, Goulden DS (1994) Linkage analysis of er-1, a recessive Pisum sativum gene for resistance to powdery mildew fungus (Erysiphe pisi D.C.). Theor Appl Genet 88:1050–1055
Timmerman-Vaughan G, Frew TJ, Weeden NF (2000) Characterization and linkage mapping of R-gene analogous DNA sequences in pea (Pisum sativum L.). Theor Appl Genet 101:241–247
Tiwari KR, Penner GA, Warkentin TD (1997) Inheritance of powdery mildew resistance in pea. Can J Plant Sci 77:307–310
Tiwari KR, Penner GA, Warkentin TD (1998) Identification of coupling and repulsion phase RAPD markers for powdery mildew resistance gene er1 in pea. Genome 41:440–444
Tiwari KR, Penner GA, Warkentin TD (1999) Identification of AFLP markers for powdery mildew resistance gene er2 in pea. Pisum Genet 31:27–29
Tonguç M, Weeden NF (2010) Identification and mapping of molecular markers linked to er1 gene in pea. J Plant Mol Biol Biotechnol 1:1–5
Vaid A, Tyagi P (1997) Genetics of powdery mildew resistance in pea. Euphytica 96:203–206
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:7–78
Wang J, Lydiate DJ, Parkin IA, Falentin C, Delourme R et al (2011) Integration of linkage maps for the Amphidiploid Brassica napus and comparative mapping with Arabidopsis and Brassica rapa. BMC Genomics 12:101
Wang S, Basten CJ, Zeng ZB (2012) Windows QTL Cartographer 2.5. Department of Statistics, North Carolina State University, Raleigh, NC. (http://statgen.ncsu.edu/qtlcart/WQTLCart.htm)
Warkentin TD, Rashid KY, Xue AG (1996) Fungicidal control of powdery mildew in field pea. Can J Plant Sci 76:933–935
Weeden NF, Boone WE (1999) Mapping the Rb locus on linkage group III using long PCR followed by endonuclease digestion. Pisum Genet 31:36
Weeden NF, Swiecicki WK, Timmerman-Vaughan GM, Ellis THN, Ambrose M (1996) The current pea linkage map. Pisum Genet 28:1–4
Weeden NF, Ellis THN, Timmerman-Vaughan GM, Swiecicki WK, Rozov SM, Berdnikov VA (1998) A consensus linkage map for Pisum sativum. Pisum Genet 30:1–3
Wu Y, Close TJ, Lonardi S (2011) Accurate construction of consensus genetic maps via integer linear programming. IEEE/ACM Trans Comput Biol Bioinforma 8:381–394. doi:10.1109/TCBB.2010.35
Yap IV, Schneider D, Kleinberg J, Matthews D, Cartinhour S, McCouch SR (2003) A graph-theoretic approach to comparing and integrating genetic, physical and sequence-based maps. Genetics 165:2235–2247
Yau SK, Ryan J (2008) Boron toxicity tolerance in crops: a viable alternative to soil amelioration. Crop Sci 48:854–865
Acknowledgements
This work was supported by funding from the Victorian Department of Environment and Primary Industries and the Grains Research and Development Council, Australia.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Additional File 1
a Assessment scale for powdery mildew infection in field pea: This file contains details of the assessment scale for severity of powdery mildew infection in field pea. (DOCX 13 kb)
11105_2014_837_MOESM2_ESM.docx
b Assessment scale for boron tolerance in field pea: This file contains details of the assessment scale for levels of boron tolerance in field pea (DOCX 12 kb)
Additional File 2
a Kaspa × Yarrum linkage map of field pea: This file contains the visual representation of linkage maps of field pea based on RILs of Kaspa × Yarrum showing the location of the QTL-containing region for powdery mildew resistance. The name is provided at the top of each LG. Distances of the loci (cM) are shown to the left, and the names of loci are shown to the right side of the linkage groups. For presentation purposes, only one of a set of co-located genetic markers are shown on the map. (PPT 570 kb)
11105_2014_837_MOESM4_ESM.ppt
b Kaspa × ps1771 linkage map of field pea: This file contains the visual representation of linkage maps of field pea based on RILs of Kaspa × ps1771 showing the location of QTL containing region for powdery mildew resistance and boron tolerance. The name is provided at the top of each LG. Distances of the loci (cM) are shown to the left, and the names of loci are shown to the right side of the linkage groups. For presentation purposes, only one of a set of co-located genetic markers are shown on the map. (PPT 502 kb)
Additional File 3
a Comparison between linkage group (Ps I) of Kaspa × Yarrum and Kaspa × ps1771: This file shows the visual representation of Ps I from both the Kaspa × Yarrum and Kaspa × ps1771 maps, and the common marker loci between them. Red coloured lines represent the corresponding positions of common markers. (PPTX 93 kb)
11105_2014_837_MOESM6_ESM.pptx
b Comparison between linkage group (Ps II) of Kaspa × Yarrum and Kaspa × ps1771: This file shows the visual representation of Ps II from both Kaspa × Yarrum and Kaspa × ps1771 maps, and the common marker loci between them. Red coloured lines represent the corresponding positions of common markers. (PPTX 58 kb)
11105_2014_837_MOESM7_ESM.pptx
c Comparison between linkage group (Ps III) of Kaspa × Yarrum and Kaspa × ps1771: This file shows the visual representation of Ps III from both Kaspa × Yarrum and Kaspa × ps1771 maps, and the common marker loci between them. Red coloured lines represent the corresponding positions of common markers. (PPTX 78 kb)
11105_2014_837_MOESM8_ESM.pptx
d Comparison between linkage group (Ps IV) of Kaspa × Yarrum and Kaspa × ps1771: This file shows the visual representation of Ps IV from both Kaspa × Yarrum and Kaspa × ps1771 maps, and the common marker loci between them. Red coloured lines represent the corresponding positions of common markers. (PPTX 77 kb)
11105_2014_837_MOESM9_ESM.pptx
e Comparison between linkage group (PS V) of Kaspa × Yarrum and Kaspa × ps1771: This file shows the visual representation of Ps V from both Kaspa × Yarrum and Kaspa × ps1771 maps, and the common marker loci between them. Red coloured lines represent the corresponding positions of common markers. (PPTX 64 kb)
11105_2014_837_MOESM10_ESM.pptx
f Comparison between linkage group (Ps VI) of Kaspa × Yarrum and Kaspa × ps1771: This file shows the visual representation of Ps VI from both the Kaspa × Yarrum and Kaspa × ps1771 maps, and the common marker loci between them. Red coloured lines represent the corresponding positions of common markers. (PPTX 62 kb)
11105_2014_837_MOESM11_ESM.pptx
g Comparison between linkage group (Ps VII) of Kaspa × Yarrum and Kaspa × ps1771: This file shows the visual representation of Ps VII from both the Kaspa × Yarrum and Kaspa × ps1771 maps, and the common marker loci between them. Red coloured lines represent the corresponding positions of common markers. (PPTX 76 kb)
Additional File 4
a Linkage map statistics from Kaspa × Yarrum RIL mapping population: This file contains details of different markers (SSRs and SNPs) and their corresponding positions on different LGs. (XLSX 17 kb)
11105_2014_837_MOESM13_ESM.xlsx
b Linkage map statistics from Kaspa × ps1771 RIL mapping population: This file contains details of different markers (SSRs and SNPs) and their corresponding positions on different LGs. (XLSX 18 kb)
Additional File 5
Preliminary composite map statistics: This file contains details of different markers (SSRs and SNPs) and their corresponding positions on different LGs. (XLSX 23 kb)
Additional File 6
BLAST results of the integrated map: The data represent the BLAST-based sequence analysis of DNA sequences underlying SNP markers assigned to the preliminary composite map against transcriptome sequencing data. (XLSX 95 kb)
Additional File 7
Consensus genetic map statistics: This file contains details of different markers (SSRs and SNPs) and their corresponding positions on different LGs. (XLSX 48 kb)
Additional File 8
a Frequency distribution histogram: This file contains frequency histograms generated for boron index and boron plant symptom score in the Kaspa × ps1771 mapping population. (PPTX 105 kb)
11105_2014_837_MOESM18_ESM.pptx
b Frequency distribution histogram: This file contains frequency histograms generated for powdery mildew symptom scores and percentage leaf necrosis in the Kaspa × Yarrum mapping population. (PPTX 66 kb)
Additional File 9
PsMLO1 gene sequence: This file contains sequence of PsMLO1 gene obtained from resequencing analysis. (DOCX 14 kb)
Rights and permissions
About this article

Cite this article
Sudheesh, S., Lombardi, M., Leonforte, A. et al. Consensus Genetic Map Construction for Field Pea (Pisum sativum L.), Trait Dissection of Biotic and Abiotic Stress Tolerance and Development of a Diagnostic Marker for the er1 Powdery Mildew Resistance Gene. Plant Mol Biol Rep 33, 1391–1403 (2015). https://doi.org/10.1007/s11105-014-0837-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-014-0837-7