
Abstract
Fruit firmness and weight are among the most important fruit quality traits in fruit species. Understanding the control of fruit firmness and weight is essential for the development of domestication research approaches and for the implementation of new breeding strategies. A forward genetic study for these traits was performed using two F1 sweet cherry (Prunus avium) progenies derived from modern cultivars. Quantitative trait locus (QTL) analysis allowed the identification of genomic regions accounting for most of the phenotypic variation in both families. In addition, screening the Prunus persica genome v1.0 permitted the identification of putative candidate genes underlying the QTL with the major effect for fruit weight (LG5) and the one for firmness (LG6). A colocalization of QTLs and candidate genes was found in peach, apple, and tomato. These results give new insights of the interaction between fruit firmness and fruit weight and provide new cues for the identification of genes implicated in the control of these traits. The colocalization of genomic regions between progenies issued from modern cultivars and from modern cultivars × wild individuals suggests the absence of allele fixation within genes controlling fruit firmness and size, two traits potentially involved in domestication/diversification in sweet cherry.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Plant domestication is an outstanding example of plant-animal coevolution and is a far richer model for studying evolution than is generally appreciated (Purugganan and Fuller 2009). Plant domestication is the genetic modification of a wild species to create a new form of plant altered to meet human needs (Doebley et al. 2006), whereas diversification has been referred as the subsequent evolution of new varieties, including greater improvement in yield, adaptation, or quality in crop species (Meyer and Purugganan 2013). Domesticated perennials are an important element of agricultural economies around the globe (Schreckenberg et al. 2006). However, large gaps exist in understanding the genetic basis of perennial fruit crop domestication (Miller and Gross 2011) and diversification. It is well known in the case of annuals that traits such as shattering, apical dominance, and grain size have been impacted during domestication (Doebley et al. 2006). Identification of wild ancestors using a comparison to domesticated annuals is difficult due to the differentiation during domestication (i.e., teosinte vs. maize). On the contrary, in domesticated fruit perennials as sweet cherry, differences between wild mazzards and domesticated sweet cherry are less evident (Frankel et al. 1995). Quantitative trait loci (QTLs) have served as a major avenue for understanding the genetic basis of domestication and diversification in plants (Miller and Gross 2011). In perennial fruit crops, QTL studies have traditionally lagged behind those in annual crops due to limitations associated to the development and maintenance of mapping populations (i.e., long juvenile period, long-lived organisms, etc.). In annual crops, molecular genetics of domestication is fairly more advanced than that in perennials; early QTL studies indicated that only a few genes of large effect controlled many domestication traits (Koinange et al. 1996). Further studies showed that these genes tend to be clustered in the genome and to be conserved across different taxa (Paterson 2002). Recently, it has been suggested that this pattern might not be universal, and many multiple genomic regions may be involved for some domestication traits (Gross and Olsen 2010). In sweet cherry, little is known about the molecular genetics of domestication. Previous studies have shown a marked genetic bottleneck between wild and cultivated cherries (Mariette et al. 2010; Panda et al. 2003), a population structure with three clusters: wild cherry, landraces, and modern sweet cherry cultivars (Mariette et al. 2010) and linkage disequilibrium (LD) similar to the one detected in peach and grapevine (Arunyawat et al. 2012). In addition, along the evolutionary history of the species, several domestication events may have happened in sweet cherry, and/or intense gene flow from local wild cherry was probably maintained (Mariette et al. 2010; Tavaud 2000).
The majority of domesticated perennials are long-lived, woody species cultivated for their edible fruits (Miller and Gross 2011; Van Tassel et al. 2010). Commonly observed traits in fruit crops accompanying domestication and diversification have included larger fruit size and softer fruits (Meyer and Purugganan 2013). The same traits can also undergo parallel selection in multiple crop species and may be a recurring target of selection (Meyer and Purugganan 2013). One example is the fruit-weight locus FW2.2 (Doganlar et al. 2002) in tomato, chili pepper, and eggplant (Meyer and Purugganan 2013). The effect of fw2.2 is due to a single gene that controls carpel cell number (Cong et al. 2002; Frary et al. 2000). Additionally, differences in fruit weight, either between cultivars (Olmstead et al. 2007) or between thinning treatments (Goffinet et al. 1995), are related to cell number in different Rosaceae species. Previous work showed clustering of QTLs of mesocarp cell number and fruit size in sweet cherry on LG2, suggesting that these two traits were linked (Zhang et al. 2010). In peach, a total of 23 fw2.2/cell number regulator (CNR) family members were identified spanning the eight Prunus chromosomes (De Franceschi et al. 2013). A common regulation of cell number and organ size driven by fw2.2/CNR, located on LG2, has been suggested for both sweet and sour cherry (De Franceschi et al. 2013). Other genomic regions than LG2 have been also shown to carry QTLs for fruit weight within Prunus: LG1 on sweet cherry (Rosyara et al. 2013) and on peach (Eduardo et al. 2011); LG3 on sweet cherry (Rosyara et al. 2013) and peach (Yamamoto et al. 2001); LG4 on Prunus davidiana × Prunus persica (Quilot et al. 2004), sour cherry (Wang et al. 2000), and peach (Cantin et al. 2010a; Eduardo et al. 2011); LG6 on sweet cherry (Cao et al. 2012a; Rosyara et al. 2013; Zhang et al. 2010) and peach (Cao et al. 2012a; Dirlewanger et al. 1999; Eduardo et al. 2011; Yamamoto et al. 2001); and LG8 on peach (Cao et al. 2012a).
Fruit firmness and weight appear to be inversely correlated in different crops. For example, a positive correlation between fruit size and the proportion of intercellular spaces in apple has been described (Ruess and Stosser 1993). However, to date, no data on the genetic determinism in sweet cherry is available for both fruit firmness and size. In peach, QTLs for flesh firmness were identified on LG1, LG2, LG5, LG6, and LG7 (Cao et al. 2012a), and candidate genes (CG) for texture were mapped on all eight LGs (Illa et al. 2011; Ogundiwin et al. 2009). The locus responsible for the melting vs. non-melting (M/m) flesh character has a major effect on fruit texture and firmness in peach and has been mapped on LG4 (Cantin et al. 2010a; Dirlewanger et al. 2004; Martinez-Garcia et al. 2013; Peace et al. 2005). This region is syntenic with LG10 of apple, where a cluster of QTLs for fruit quality traits, including firmness and fruit weight, has been described (Kenis et al. 2008). Also, in apple, a polygalacturonase gene (Md-PG1), known to be involved in cell wall metabolism processes, is mapped on this interval (Longhi et al. 2012).
In addition, firmness and weight are two main objectives in modern breeding strategies. Fruit size, a proxy of fruit weight, is the main attribute for sale grading for the fresh market in sweet cherry (Whiting et al. 2006). Fruit firmness is one of the most important fruit attributes that consumers use in judging acceptability of sweet cherries (Guyer et al. 1993; Romano et al. 2006) and directly relates to fruit susceptibility to mechanical damage during handling and packaging. Despite the importance of fruit weight and firmness in sweet cherry breeding, genetic control is still not completely understood.
Advances in molecular technology, such as the development of SNP chip arrays for Rosaceae species (Chagne et al. 2012; Peace et al. 2012; Verde et al. 2012), have permitted a high throughput genotyping, easing the detection of QTLs for several fruit crops. Additionally, thanks to the high synteny between Prunus (Dirlewanger et al. 2004), the release of the peach genome (Verde et al. 2013) provides an excellent framework for CG research within the Prunus genus and other related species. On the contrary, the solid and multiyear phenotyping needed for QTL studies has recently arisen as the bottleneck of this approach. This bottleneck is even harder to overcome for fruit traits in long juvenility species, such as sweet cherry. In this species, the first fruit harvest occurs after 4 to 6 years of growing seasons.
For the first time, firmness and fruit weight, the two main breeding traits, potentially involved in sweet cherry domestication/diversification, were evaluated over 4 and 5 years for two mapping progenies. The objectives of the present study in sweet cherry were (1) to shed light on the genetic basis of two correlated traits, fruit firmness, and weight; and (2) to look for CGs, potentially involved in domestication or diversification events, located in the region of the identified QTLs using the available information of the genome in several domesticated fruit crops: tomato (Sato et al. 2012), apple (Velasco et al. 2010), and peach (Verde et al. 2013).
Material and Methods
Plant Material
Two segregating F1 adult families (Prunus avium L.) were used in this study: (1) The first consisted of 122 individuals derived from the cross between “Regina” and “Lapins” (R × L); (2) the second consisted of 117 individuals from the cross between “Regina” and “Garnet” (R × G). Both families were planted on 2001, raised and evaluated at Toulenne, located 30 km south-west from Bordeaux, France. Parents of the progeny were situated in the same orchard as controls. Trees were cultivated on their own roots; hence, only one replicate per genotype was available. The plot used was highly homogeneous in terms of soil composition, and horticultural practices such as pruning, irrigation, fertilization, and control of insects and diseases were consistently performed. Fruit maturity was determined every year by the same technicians based on observations of color, firmness (by “hand” appreciation), and flavor carried out in the orchard.
Trait Measurement
Fruit firmness was measured using a Durofel® (Setop Giraud technologie, Cavaillon, France) texture analyzer on the day of harvest. A 3-mm probe was applied at two points on the fruit equator, the movement of the probe was recorded, and the average of the two measures on ten fruits was used. Fruit weight was determined and averaged for 100 fruits. Measurements were performed during 7 years (2006–2012) and 4 years (2009–2012) for R × L and R × G, respectively. On 2007, no R × L firmness data were available.
Statistical Analysis for Firmness and Fruit Weight
Mean, range, standard deviation, and the skewness of the population distributions were calculated for the different years of evaluation. Analysis of variance was performed using the data in a single year as one replication. Broad-sense heritability (H BS) was estimated with the following equation: \( {H}^2=\frac{\sigma_g^2}{\sigma_g^2-\raisebox{1ex}{${\sigma}_e^2$}\!\left/ \!\raisebox{-1ex}{$ y$}\right.} \), where σ 2 g is the genetic variance of progeny lines, σ 2 e is the error variance, and y is the number of years.
The statistical analyses were performed using the software R 2.15.0 [R Development Core Team (2012); R: a language and environment for statistical computing; R Foundation for Statistical Computing, Vienna, Austria; ISBN 3-900051-07-0, URL http://www.R-project.org/].
Linkage Map and QTL Analysis
Both mapping populations used in this study were obtained from a cross-pollination of heterozygous parents. The development of RosBREED cherry 6K SNP array v1 and RosCOS markers for genotyping sweet cherry progenies opens the possibility to directly ascertain the position of markers on the Peach Genome v1.0. SNP markers from the RosBREED cherry 6K SNP array v1 (Peace et al. 2012) were used to genotype both families, using the mapping protocol previously described by Klagges et al. (2013). Additionally, a set of RosCOS (Cabrera et al. 2012) markers, not included in the RosBREED cherry 6K SNP array v1, was also used for genotyping using Sequenom®. R × L progeny was previously used for genetic map construction using SSRs (Dirlewanger et al. 2004; Dirlewanger et al. 2012) and SNPs (Cabrera et al. 2012; Klagges et al. 2013), and R × G genetic map has been recently published (Castède et al. 2014) (Online Resource 1). Parental maps were built for QTL analysis using markers segregating only for one parent. Markers situated closer than 2 cM were deleted to ease computing analysis. In addition, the positions of the 23 CNRs were shown using in silico mapping based on the synteny between peach and sweet cherry (Klagges et al. 2013). A linear interpolation was used between the physical peach position of the closest markers, their position in centimorgan and the physical position of the PpCNR (De Franceschi et al. 2013).
QTL mapping was carried out using MultiQTL V2.6 software. The multiple interval mapping (MIM) approach was used (Haifa, Israel, 2005; http://www.multiQTL.com).
For both firmness and fruit weight, each year (by-year analysis) was analyzed independently in order to examine the stability of the QTL. Analysis combining all years together was performed using the multiple environment option available, increasing the accuracy of the QTL detection. QTL analyses were performed according to Castède et al. (2014).
The graphical presentation of linkage maps and QTL was obtained using the MapChart software version 2.2 (Voorrips 2002).
In Silico CG Research
Chromosomal regions for in silico CG analysis were initially selected based on the location of the QTLs associated with firmness and fruit weight. Predicted peach protein sequences derived from scaffolds underlying the major QTLs were downloaded from the Genome Database for Rosaceae (http://www.rosaceae.org/node/355) and blasted against the NCBI nr database using BLASTP in the program Blast2GO (Conesa et al. 2005) with an E-value cutoff of 0.001. Blast2GO was then used for the Gene Ontology annotation.
Results
Distribution of Traits, Genetic Variation and Broad-Sense Heritability of Traits
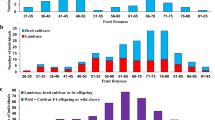
The mean values for fruit firmness and weight were calculated for each year in both populations (Online Resource 2). Variability for both traits was observed between growing seasons for both families. R × L progeny showed higher values of firmness but lower fruit weight in comparison with R × G throughout the years of evaluation. During the year 2006, the intra-family variation for R × L, in terms of coefficient variation, was significantly higher than that during the remaining years. The analysis of firmness and fruit weight revealed a normal distribution, for both populations across most of the years phenotyped (Online Resource 2, Online Resource 6).
For both progenies, the genetic variation among the progeny individuals was highly significant (Table 1). The broad-sense heritability for fruit firmness was 0.85 (R × L) and 0.78 (R × G), and 0.88 (R × L) and 0.76 (R × G) for fruit weight. The highest value of heritability (0.88) was found for fruit weight in R × L progeny.
Correlation of Traits
Correlation coefficients were calculated for each phenotypic trait measured in different years and between fruit firmness and weight for the same year (Table 2). For fruit firmness, significant correlations (p < 0.05) between years were found for most of the comparisons, both for R × L (0.26–0.72) and R × G (0.48–0.67). Similarly, highly significant correlations were also observed for fruit weight: R × L (0.40–0.74) and R × G (0.40–0.59). In addition, significant negative correlations (p < 0.05) were detected between fruit firmness and weight, being higher in R × L (0.45–0.64) than in R × G (0.29–0.40).
Linkage Map and QTL Analysis
The final maps, used for QTL analysis, for R × L consisted of 136 and 127 SNP markers over 8 LGs, named R1 to R8 for Regina and L1 to L8 for Lapins, covering 712.4 and 710.4 cM, respectively (Fig. 1, Online Resource 1). For R × G, parental maps consisted of 142 and 137 markers over 8 LGs, named G1 to G8 for Garnet, and covering 657.6 and 823.6 cM for Regina and Garnet, respectively (Fig. 1, Online Resource 1).

QTL analysis in two progenies “Regina” × “Lapins” (left) and “Regina” × “Garnet” (right) for fruit firmness (Ff) (in orange) and fruit weight (Fw) (in red). Only results using multiyear analyses from MultiQTL are indicated. QTLs are indicated by bars (orange bars, Ff; red bars, Fw). Distance between markers is represented in centimorgan. CNR genes are mapped in silico using the peach genome sequence
Results of by-year and multiyear QTL detection for fruit firmness and weight were calculated for the two progenies (Table 3). For the R × L progeny, 39 fruit firmness QTLs were detected on seven of the eight Prunus LGs (except for LG7) along the 6 years of analysis. Among these QTLs, maximum explained variation (EV) was found on L2. Only the QTLs found on L2, R1, and R5 were detected at least throughout 3 of the 6 years of the study. Using the multiyear analysis, mean explained variation (MEV) ranged from 4.1 to 20.0, and the highest values were obtained for the QTLs for L2 and R2 (20.0 and 12.5, respectively), R1 (14.0), and L6 (12.0) and L5 (11.8). The multiyear analysis detected a total of 23 QTLs, including a second QTL on the top of L8 and two in the middle of R6, not previously detected by the by-year analysis.
Regarding fruit weight, 53 QTLs were detected considering the analysis of all 7 years. Maximum EV was found on L2 in 2009 (39.9 %), R2 in 2012 (37.5 %), and R6 in 2010 (31.9 %). Only the QTLs on R2 were detected every year. With the multiyear analysis, MEV values ranged from 6.5 to 20.2, and the highest values were obtained for L2 and R2 (20.2 and 18.3, respectively), R6 (16.7), and R3 (11.1). The multiyear analysis detected a total of 21 QTLs, including two QTLs not previously found by the by-year analysis: the first on L3 and the second on the top extreme of L5.
In the R × G progeny, 18 fruit firmness QTLs were detected on six of the eight Prunus LGs (except for LG3 and LG4) considering the analysis of 4 years. Maximum EV was found on G2 in 2012 (29.8 %), R5 in 2011 (25.9 %), and G1 in 2011 (20.4 %). The QTL on R5 was detected every year. The multiyear analysis showed MEV values of 16.1, 10.1, and 24.1 for G2, G5, and R5, respectively. The multiyear analysis detected a total of 11 QTLs including one QTL on G3 and one on the middle of R5 not found previously by the by-year analysis. In addition, the confidence interval of the QTL situated at 67.7 cM on R5 was not confirmed by MultiQTL 2.5 due to the low MV (<2 %) (m value of bootstrapping).
Regarding fruit weight, 32 QTLs were detected on all Prunus LGs as previously shown for the R × L progeny. Maximum EV in R × G was found on G8 in 2010 (31.2 %), R2 in 2012 (31.0 %), and R2 in 2010 (30.7 %). With the multiyear analysis, MEV values ranged from 6.1 to 18.3, the highest values observed for R2 (18.2), G8 (14.0), R5 (13.8), and G6 and R6 (11.3 and 10.1 respectively). The multiyear analysis revealed a total of 15 QTLs.
Most of the detected QTLs colocalized in similar regions in R × L and R × G for both firmness and weight (Fig. 1). This was especially evident for the colocalizations of QTLs with small confidence intervals at the bottom of LG5 both in R × L and R × G and in the middle of LG6 in R × L, when considering fruit weight QTLs on Regina and fruit firmness QTLs on Lapins (Fig. 1). In addition, a colocalization between fruit firmness and fruit weight QTLs was found throughout the genome of sweet cherry for each progeny (Fig. 1). Some of the clustered QTLs overlapped with PpCNR in silico position previously described by De Franceschi et al. (2013): PpCNR09, PpCNR10, PpCNR11 on LG1; PpCNR12 on the proximal part of LG2; PpCNR16 on LG3; PpCNR18 and PpCNR19 on the bottom of LG5; and PpCNR22 on LG7 (Fig. 1).
Haplotype Construction
Given that parental maps for QTL analysis were built using markers segregating only for one parent, segregation of a maximum of two different alleles is expected for each QTL in our diploid cross-pollination. Two fruit firmness QTLs were found in R5 (R × G population) (Online Resource 3), named ff5.1 and ff5.2. The ff5.2 was chosen for haplotype construction according to the QTL stability (found in every year of the study) and shorter confidence interval (Online Resource 3). The multiyear analysis detected a peak position for ff5.2 at 67.7 cM, very similar to the QTL peaks found by the by-year analysis. For the ff5.2, markers Rsweet_5_16416089 (65.1 cM) and Rsweet_5_16741368 (67.8 cM) were selected for haplotype construction (Online Resource 3). Only haplotypes that did not have a recombination between the parental markers were used to examine the effects of the allele of fruit firmness QTL on R5. Thus, recombinant seedlings for the QTL region were not included in the analysis. Mean fruit firmness and weight values were calculated for both haplotypes (Online Resource 3). For all 4 years, those progeny individuals that received the Regina’s “a” haplotype consistently had firmer fruits than those that received the Regina’s “b” haplotype (Online Resource 3). On the contrary, those progeny individuals that received the a haplotype consistently had lighter fruits than those that received the b haplotype.
In silico CG Analysis
Our QTL results and CG analysis validated the CG CNR12 previously found in LG2 (De Franceschi et al. 2013). In addition, in silico CG analysis was performed within small confidence intervals colocalizing for fruit firmness and weight found for the QTLs on LG5 and LG6. The region covered by the confidence intervals was shortened thanks to the high number of phenotyping years and to the high heritability of the traits. The high degree of synteny and colinearity conservation found between peach and cherry (Dirlewanger et al. 2004; Klagges et al. 2013) made it possible to use the peach genome sequence as a reference for CG investigation. The intervals for fruit weight (Lapins) and firmness (Regina) expressed in base pairs (bp) of the peach physical position, considerably overlapped on LG6: 14,770,602–17,706,516 bp (fw6.1) and 19,939,133–20,820,863 bp (fw6.2) overlapped with 15,179,474–16,172,722 bp (ff6.1) and 17,018,687–20,986,380 bp (ff6.2) (Fig. 1). In order to include both fruit weight (fw6.1 and fw6.2) and firmness (ff6.1 and ff6.2) QTLs, 6,215,778 bp from 14,770,602 bp of the scaffold 6 were screened. In this region, 634 genes were predicted on the peach dihaploid “Lovell” genome from which 25.2 % had Blast hits with unknown proteins. On LG5, ff5.2 interval (16,489,338–17,743,459 bp) detected in R × L overlapping the fruit weight interval found for R × G has been screened for CG. This interval was chosen according to the ff5.2 observed in Lapins and contained the Regina’s (R × L) ff5.2 located at 67.7 cM (Table 3). In the ff5.2 region, 254 genes were predicted from which 12.6 % had Blast hits with unknown proteins (Online Resource 4). Data mining on the two loci based on predicted protein function, gene ontology annotation, and their potential involvement in fruit weight and firmness resulted in the selection of 43 CGs, 15 on LG5 (covering fw5.2 and ff5.2), and 29 on LG6 (covering fw6.1, fw6.2, ff6.1, and ff6.2) (Online Resource 5). Among the 15 CGs found on LG5, three were selected for their potential involvement in fruit weight control: cytochrome p450 78A3-like, CNR1, and plac8 family protein (Blast2GO annotation) corresponding to PpCNR18 and PpCNR19, respectively (peach CNR gene names based on their order on the peach genome scaffolds) (De Franceschi et al. 2013). The 12 other CGs were selected for their potential involvement in fruit firmness via cell wall biogenesis, modifications, or degradations. On LG6, three CGs were selected for their potential involvement in fruit weight control: cyclin-dependant kinase c-1-like, cyclin-d-binding myb-like transcription factor 1-like and cyclin-d5-1-like. The cyclin kinase and the cyclin-d5-1-like were selected because they are adjacent to the boundaries of the fruit weight QTLs intervals, even if they are not comprised in these intervals. The 26 other CGs on LG6 were selected for their potential involvement in fruit firmness via cell wall biogenesis, modifications, or degradations as in LG5.
Discussion
Both populations exhibited similar patterns of data distribution for fruit firmness and weight for the different years of study, showing the expected continuous distribution, characteristic of quantitative traits (Fig. 1). Similar distributions for fruit weight were observed in other Prunus: sweet cherry (Lamb 1953; Zhang et al. 2010), sour cherry (Wang et al. 2000), and peach (Dirlewanger et al. 1999). In all these studies, the mean fruit weight for the F1 progeny was lower than the parental midpoint, showing an apparent dominance of small-fruited alleles. This fact highlights the importance of QTL identification and CG search for fast breeding for fruit weight in Prunus species.
Broad-sense heritability (H BS) has been used as an index of reliability of phenotypic selection for genetic characteristics (Holland et al. 2003), and the detection of QTLs is easier for traits when numbers of contributing loci are low and heritability is high (Li et al. 2011). In both R × L and R × G mapping populations, H BS was sufficiently high (>0.8) to enable genetic analysis of fruit firmness and weight (Table 1). Nonetheless, the higher heritability found for R × L, for both traits, could be associated to the higher number of QTLs found compared to R × G (Table 1). Previous studies on fruit weight heritability have also shown high values: 0.76 in a 3-year study on a population derived from a domesticated and a wild cherry (Zhang et al. 2010) and 0.88 in sour cherry (Wang et al. 2000).
The negative correlation between fruit firmness and weight found in this study is not a constant within the Rosaceae fruit species. For example, negative correlations between these traits have previously been described in apple (Johnson 1994), whereas no correlation has been found in apricot (Badenes et al. 1998; Ruiz and Egea 2008) or strawberry (Lerceteau-Kohler et al. 2012). On the contrary, a positive correlation was found in peach and nectarine (Cantin et al. 2010b). These results may suggest a different interaction between characters depending on the species. Indeed, the differences in the negative correlation values between fruit firmness and size found in R × L (from −0.49 to −0.72) and R × G (from −0.29 to −0.40) suggest a strong genotype effect in the correlation between these two traits within sweet cherry.
The QTLs for fruit weight (fw5.2) and fruit firmness (ff5.2), colocalizing at the bottom of LG5, have not been described before in sweet cherry. Also, fw5.2 and ff5.2 colocalize with the LD block around the marker EMPAS14 (15,142,021 bp in the peach genome v1.0) in cultivated sweet cherry not found in wild cherry (Arunyawat et al. 2012), suggesting a selection during domestication. It should be noted that Arunyawat et al. (2012) used only 35 SSRs for the eight LGs, so the colocalization of disequilibrium blocks should be considered with caution.
Although no fruit firmness QTL study has been reported for sweet cherry, several QTL regions found in our study colocalize with QTL regions previously identified for other Rosaceae species. Cevik et al. (2010) found an association between MdMADS2.1 gene and fruit firmness in apple LG5. A likely orthologous of the Malus MdMADS2.1 (also known as MdMADS2) gene in Prunus is EST PrpAP1 [BU039475; (Silva et al. 2005)]. The PrpAP1, mapped just 1 cM away from the marker AG108 of the LG5 of Prunus reference map, which colocalized with sweet cherry QTL ff5.2 described in R × G. Thus, a colocalization for fruit firmness genes found using an association genetics study between landraces and modern cultivars of apple (Cevik et al. 2010) and in this QTL study in a cross of modern cultivars was revealed. Remarkably, this region also colocalizes with fruit weight QTL found in different species. For example, a QTL for fruit weight was also found at the bottom of LG5 in the cross of a wild species and a modern nectarine P. davidiana × P. persica (Quilot et al. 2004). This QTL was found to be associated again to AG108, situated just below BPPCT014, in which physical position in the peach genome is 16.6 Mb and is colocalizing with the fw5.2 found in this study. Thus, the ff5.2 in sweet cherry colocalizes with the fw5.2 found in crosses between wild species × modern cultivar of peach.
Zhang et al. (2010) reported fruit weight QTLs on LG2 and LG6 in a population derived from a domesticated and a wild sweet cherry. Later on, Rosyara et al. (2013), conducting pedigree-based QTL mapping on 23 founders and 424 progeny individuals from four full-sib families, one of which was Regina × Lapins, found one QTL in the middle of LG1 (17–27-Mb region of peach genome v1.0), three QTLs on LG2 (15–23-Mb region on peach), one in the middle of LG3 (surrounding 10 Mb on peach), and one on the lower half of LG6 (ca. 19 Mb on peach). In our study, we found two QTLs per LG in the LG1, LG3, and LG6, which means that three new QTLs were reported in those LGs compared to the study of Rosyara et al. (2013).
The QTL for fruit weight located on the top of LG2 was found in all 7 years of study in the R × L population and colocalizes with CNR copy PavCNR12 (De Franceschi et al. 2013). In agreement with these results, Arunyawat et al. (2012) found an LD block in the area of this QTL (between EMPA017—10.5 Mb—and BPPCT002—16.5 Mb—in the peach genome v1.0) in cultivated sweet cherry but not in wild cherry. Also, De Franceschi et al. (2013) showed the association of PavCNR12 haplotypes with the QTL effects, strongly supporting the hypothesis that both PavCNR12 and PcrCNR12 control fruit size in sweet and sour cherry, respectively. The second QTL found in LG2 colocalizes with both the QTL in the distal part of LG2 found in multiple pedigreed populations (Rosyara et al. 2013) and the LD blocks found in modern varieties in sweet cherry (Arunyawat et al. 2012), suggesting a selection of genes in this part of the genome.
Regarding the fruit weight QTL on LG6, fw6.1 colocalizes with the QTL found in multiple pedigreed populations (Rosyara et al. 2013). Interestingly, an LD block was found for the fw6.1 and the fw6.2 (EMPA004 and UDP98021) markers situated at 14,794,983 and 22,785,018 bp, respectively; in the peach genome v1.0 in both cultivated and wild sweet cherry (Arunyawat et al. 2012). This LD block in wild cherry could be associated to the incompatibility gene S located a few megabase downstream the QTL. As for the fw6.2, no QTL colocalization was found on LG6 for PpCNR20 in sweet cherry as expected. De Franceschi et al. (2013) suggested that the small fruit allele of PpCNR20 was unique to the wild mazzard “New York 54,” after studying 16 other sweet cherry cultivars, including Regina and Lapins. In the light of previous results, fw6.2 might correspond to a QTL for pit size, since it is located in the same region as a major QTL detected for stone weight in peach (Quilot et al. 2004) and for length and width pit size in sweet cherry (Zhang et al. 2010).
Comparing our results and those of Zhang et al. (2010), a colocalization between QTL for fruit weight in Rosaceae crosses between wild species × modern cultivar and modern cultivar × modern cultivar on LG2 and LG6 has been observed, as described above for fruit firmness within the Rosaceae.
A significant interaction between QTLs for both fruit firmness and weight and year was detected. Indeed, considering both progenies, QTLs were detected during all years only for fruit firmness on R5 (R × G) and for fruit weight on R2 (R × L). This highlights the need of conducting this type of study during a large number of years, to account for between-year climatic variations. These variations may not affect significantly overall heritability values, as well as the detection of major QTLs, but they can be very important for the detection of minor QTLs (Dirlewanger et al. 2012). Thus, a precise understanding of these complex and critical traits for fruit trees requires multiyear approaches.
In Silico CG Analysis
In the present study, the reduction of QTLs confidence intervals allowed us to use the CG approach in exploring two small regions with a high effect on fruit firmness and weight. These regions complement the candidate genes found on LG2 PpCNR12 and LG6 PpCNR20 in cherry (De Franceschi et al. 2013).
Concerning the colocalization of QTLs for fruit firmness and weight on LG5, the CG analysis has identified a conserved region in peach and tomato including a member of the cytochrome P450 superfamily and several Cobra genes (Fig. 2). The peach cytochrome P450 protein (P450 78A subfamily) contains the KLUH/CYP78A5 protein. The closest peach ortholog of Arabidopsis thaliana KLUH is the P450 gene found on the LG5. KLUH controls plant organ size by cell proliferation regulation (Anastasiou et al. 2007). In tomato, KLUH has been identified as the gene underlying the fw3.2 locus for fruit weight QTL (Chakrabarti et al. 2013; Zhang et al. 2012). Furthermore, in the same sweet cherry locus, two CNRs have been identified (De Franceschi et al. 2013). To date, only two genes underlying fruit weight QTLs are known: KLUH and CNR underlying the fw3.2 and the fw2.2 loci, respectively, in tomato, located in two different LGs. The homolog of KLUH and two homologs of CNR genes were found within the confidence interval of the fruit weight and firmness sweet cherry QTL. Additionally, this region has been shown to coincide with a peak of high LD in peach (Verde et al. 2013) which may result from selective sweeps related to domestication, diversification, and breeding.

Comparison of fw3.2 genomic regions in tomato (Solanum lycopersicon) and peach (Prunus persica). ABC ABC transporter, COBRA COBRA-like protein, mTERF mitochondrial transcription termination factor, PPR pentatricopeptide repeat-containing protein, CSF cleavage stimulation factor, S/T kinase G-type lectin S-receptor-like serine/threonine-protein kinase, Rhomboid rhomboid-like protein, KELCH KELCH repeat protein, PsbP photosystem II oxygen-evolving enhancer protein 2
An unexpected finding resulting from the CG analysis is the conservation of several genes surrounding the KLUH/CYP78 gene between peach and tomato (Zhang et al. 2012) (Fig. 2). Among them, the Cobra genes may play a role in fruit firmness. Indeed, a previous study in tomato suggests that SlCOBRA-like plays an important role in cell wall architecture (Cao et al. 2012b), which is a key factor determining the fruit firmness. Furthermore, a MIKC MADS BOX transcription factor, homolog of the apple MdMadS2 (Cevik et al. 2010), has been found in the near vicinity of the COBRA genes, and a significant association between this gene and fruit flesh firmness has been described in apple (Cevik et al. 2010). All these CGs (COBRA, KLUH, MdMadS2, and CNR) could be found on the apple genome in a very similar disposition but dispatched on Malus chromosomes 6 and 14, which are homeologous chromosomes orthologous of peach LG5 (Jung et al. 2012). This colocalization could explain the correlation between fruit size and firmness found in non-related species.
Most of the other CGs found in the LG5 QTL confidence interval belong to the cell wall modifying/synthesis/degradation pathways and could affect the fruit firmness by combinatorial effects: galacturonosyltransferase synthesizes homogalacturonan (Doong and Mohnen 1998), endoglucanase, and beta-glucosidase are implicated in cell wall loosening (Cosgrove 2005; Minic and Jouanin 2006) and ethylene responsive factor has been identified as the product of a gene underlying a tomato fruit firmness QTL (Chapman et al. 2012).
The last CG on the QTL confidence interval on LG5, WUSCHEL-related homeobox, has been identified as one of the two genes underlying the tomato locule number (lc) QTL controlling locule number and fruit weight (Munos et al. 2011).
The analysis of the LG6 QTL confidence interval highlighted several CGs potentially involved in fruit firmness by acting on the cell wall structure: beta-glucosidases, glycosyltransferases, pectine esterases and one pectine esterase inhibitor, ERF transcription factors, and rhamnosyltransferases. Additionally, an endopolygalacturonase (endoPG) homolog is found in this region. This is probably the best CG for fruit firmness control. Indeed, pectinase endoPG is implicated in fruit softening by cell wall disassembly. Silencing and downregulation of Fragaria (a non-climacteric fruit like sweet cherry) FaPG1 significantly improved fruit firmness (Pose et al. 2013; Quesada et al. 2009). Correlations between fruit softening and cell wall hydrolase endoPG1 have been also described in apple (Costa et al. 2010; Longhi et al. 2012), and reduced levels of PG1 expression have been correlated with firmer fruit (Atkinson et al. 2012; Mann et al. 2008; Wakasa et al. 2006). One CG or a complex network interaction of these multiple CGs may be the predominant factor explaining the effect of this QTL on the sweet cherry fruit firmness control.
Three CGs found in the LG6 QTL confidence interval could be involved in the fruit weight control by regulating cell proliferation. These three CGs (cyclin-d5-1-like, cyclin-d-binding myb-like transcription factor 1-like, and cyclin-dependent kinase c-1-like) are related to the cyclin D family which are important regulators of cell division (Cui et al. 2014). An action (repression or activation) of KLUH and/or CNR on these CycD genes leading to a balanced control of cell proliferation and fruit weight could be hypothesized.
The suggested conserved control of fruit firmness and fruit size across species and the colocalization of QTLs described may give some clues about the domestication in Rosaceae. In the case of plant domestication, relevant crosses for QTL studies would be between individuals from domesticated plants and their closest wild relatives or potentially between a landrace and an elite cultivar (Miller and Gross 2011). However, the described colocalizations for fruit weight and firmness in LG2, LG5, and LG6 in QTL studies in wild species × modern cultivar and modern cultivar × modern cultivar crosses may indicate that fruit firmness and fruit weight alleles are not fixed in sweet cherry modern cultivars. Like sweet cherry, many perennial fruit species are long-lived fruit tree species, with a relatively low history-breeding profile, long juvenile phase, self-incompatibility system, high rates of hybridization, clonal propagation, extensive population genetic variation, and relatively limited population structure in comparison to annual crops. These characteristics may explain the absence of fixation of alleles in traits traditionally associated to domestication in a fruit perennial domesticated species as sweet cherry. Domestication genes have been proposed to meet three criteria: characterized function underlying the trait, evidence of positive selection at that locus, and complete or near-complete fixation of the mutation in all lineages from a domestication event (Meyer and Purugganan 2013). The lack of genomic fixation in these genomic regions controlling fruit firmness and size in wild, landraces, and modern cultivars of different Rosaceae species may suggest that they could be considered as diversification traits in fruit species within the Rosaceae family. This would be in agreement with previous studies in the Solanaceae. In this family, fw2.2, a gene controlling fruit size, has been erroneously inferred to be a domestication locus and is instead important in more recent diversification of domesticated species (Meyer and Purugganan 2013). Furthermore, the conservation of genes controlling the same character not only within Rosaceae but also in Solanaceae (pepper and tomato) may suggest a parallel selection of these traits in different species during diversification process.
Further studies are necessary to analyze the functional role of the aforementioned genes to understand fruit firmness and weight control. This will facilitate the extension of domestication and diversification research beyond the cereal crops and to design more efficient breeding strategies specifically tailoring new varieties to consumer preferences. A preliminary step will be to study the polymorphism within these CGs between the wild, landraces, and modern cultivars, in order to confirm the QTL/CG in silico colocalizations.
References
Anastasiou E, Kenz S, Gerstung M, MacLean D, Timmer J, Fleck C, Lenhard M (2007) Control of plant organ size by KLUH/CYP78A5-dependent intercellular signaling. Dev Cell 13:843–856
Arunyawat U, Capdeville G, Decroocq V, Mariette S (2012) Linkage disequilibrium in French wild cherry germplasm and worldwide sweet cherry germplasm. Tree Genet Genome 8:737–755. doi:10.1007/s11295-011-0460-9
Atkinson RG et al (2012) Down-regulation of POLYGALACTURONASE1 alters firmness, tensile strength and water loss in apple (Malus x domestica) fruit. BMC Plant Biol 12
Badenes ML, Martinez-Calvo J, Llacer G (1998) Analysis of apricot germplasm from the European ecogeographical group. Euphytica 102:93–99. doi:10.1023/a:1018332312570
Cabrera A et al (2012) Rosaceae conserved orthologous sequences marker polymorphism in sweet cherry germplasm and construction of a SNP-based map. Tree Genet Genome 8:237–247. doi:10.1007/s11295-011-0436-9
Cantin CM, Crisosto CH, Ogundiwin EA, Gradziel T, Torrents J, Moreno MA, Gogorcena Y (2010a) Chilling injury susceptibility in an intra-specific peach Prunus persica (L.) Batsch progeny. Postharvest Biol Technol 58:79–87
Cantin CM, Gogorcena Y, Moreno AM (2010b) Phenotypic diversity and relationships of fruit quality traits in peach and nectarine Prunus persica (L.) Batsch breeding progenies. Euphytica 171:211–226. doi:10.1007/s10681-009-0023-4
Cao K, Wang L, Zhu G, Fang W, Chen C, Luo J (2012a) Genetic diversity, linkage disequilibrium, and association mapping analyses of peach (Prunus persica) landraces in China. Tree Genet Genome 8:975–990. doi:10.1007/s11295-012-0477-8
Cao Y, Tang X, Giovannoni J, Xiao F, Liu Y (2012b) Functional characterization of a tomato COBRA-like gene functioning in fruit development and ripening BMC Plant Biol 12 doi:10.1186/1471-2229-12-211
Castède S et al (2014) Genetic determinism of phenological traits highly affected by climate change in Prunus avium: flowering date dissected into chilling and heat requirements. New Phytol. doi:10.1111/nph.12658
Cevik V, Ryder CD, Popovich A, Manning K, King GJ, Seymour GB (2010) A FRUITFULL-like gene is associated with genetic variation for fruit flesh firmness in apple (Malus domestica Borkh.). Tree Genet Genome 6:271–279. doi:10.1007/s11295-009-0247-4
Chagne D et al (2012) Genome-wide SNP detection, validation, and development of an 8K SNP array for apple. PLoS ONE 7:e31745–e31745
Chakrabarti M et al (2013) A cytochrome P450 regulates a domestication trait in cultivated tomato. Proc Natl Acad Sci U S A 110:17125–17130
Chapman NH et al (2012) High-resolution mapping of a fruit firmness-related quantitative trait locus in tomato reveals epistatic interactions associated with a complex combinatorial locus. Plant Physiol 159:1644–1657. doi:10.1104/pp. 112.200634
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Cong B, Liu JP, Tanksley SD (2002) Natural alleles at a tomato fruit size quantitative trait locus differ by heterochronic regulatory mutations. Proc Natl Acad Sci U S A 99:13606–13611. doi:10.1073/pnas.172520999
Cosgrove DJ (2005) Growth of the plant cell wall. Nat Rev Mol Cell Biol 6:850–861. doi:10.1038/nrm1746
Costa F et al (2010) QTL dynamics for fruit firmness and softening around an ethylene-dependent polygalacturonase gene in apple (Malusxdomestica Borkh.). J Exp Bot 61:3029–3039. doi:10.1093/jxb/erq130
Cui L, Li J, Zhang T, Guo Q, Xu J, Lou Q, Chen J (2014) Identification and expression analysis of D-type cyclin genes in early developing fruit of cucumber (Cucumis sativus L.). Plant Mol Biol Report 32:209–218. doi:10.1007/s11105-013-0637-5
De Franceschi P et al (2013) Cell number regulator genes in Prunus provide candidate genes for the control of fruit size in sweet and sour cherry. Mol Breed 32:311–326. doi:10.1007/s11032-013-9872-6
Dirlewanger E et al (1999) Mapping QTLs controlling fruit quality in peach (Prunus persica (L.) Batsch). Theor Appl Genet 98:18–31
Dirlewanger E, Graziano E, Joobeur T, Garriga-Caldere F, Cosson P, Howad W, Arus P (2004) Comparative mapping and marker-assisted selection in Rosaceae fruit crops. Proc Natl Acad Sci U S A 101:9891–9896. doi:10.1073/pnas.0307937101
Dirlewanger E et al (2012) Comparison of the genetic determinism of two key phenological traits, flowering and maturity dates, in three Prunus species: peach, apricot and sweet cherry. Heredity 109:280–292
Doebley JF, Gaut BS, Smith BD (2006) The molecular genetics of crop domestication. Cell 127:1309–1321. doi:10.1016/j.cell.2006.12.006
Doganlar S, Frary A, Daunay MC, Lester RN, Tanksley SD (2002) Conservation of gene function in the Solanaceae as revealed by comparative mapping of domestication traits in eggplant. Genetics 161:1713–1726
Doong RL, Mohnen D (1998) Solubilization and characterization of a galacturonosyltransferase that synthesizes the pectic polysaccharide homogalacturonan. Plant J 13:363–374. doi:10.1046/j.1365-313X.1998.00042.x
Eduardo I, Pacheco I, Chietera G, Bassi D, Pozzi C, Vecchietti A, Rossini L (2011) QTL analysis of fruit quality traits in two peach intraspecific populations and importance of maturity date pleiotropic effect. Tree Genet Genome 7:323–335. doi:10.1007/s11295-010-0334-6
Frankel OH, Brown AHD, Burdo JJ (1995) The conservation of plant biodiversity. Cambridge University Press, Cambridge
Frary A et al (2000) fw2.2: a quantitative trait locus key to the evolution of tomato fruit size. Science 289:85–88. doi:10.1126/science.289.5476.85
Goffinet MC, Robinson TL, Lakso AN (1995) A comparison of Empire apple fruit size and anatomy in unthinned and hand-thinned trees. J Hortic Sci 70:375–387
Gross BL, Olsen KM (2010) Genetic perspectives on crop domestication. Trends Plant Sci 15:529–537. doi:10.1016/j.tplants.2010.05.008
Guyer DE, Sinha NK, Chang TS, Cash JN (1993) Physicochemical and sensory characteristics of selected Michigan sweet cherry (Prunus avium L) cultivars. J Food Qual 16:355–370
Holland JB, Nyquist WE, Cervantes-Martinez CT (2003) Estimating and interpreting heritability for plant breeding: an update. Plant Breed Rev 22:9–112
Illa E et al (2011) Saturating the Prunus (stone fruits) genome with candidate genes for fruit quality. Mol Breed 28:667–682. doi:10.1007/s11032-010-9518-x
Johnson DS (1994) Influence of time of flower and fruit thinning on the firmness of Coxs Orange Pippin apples at harvest and after storage. J Hortic Sci 69:197–203
Jung S et al. (2012) Whole genome comparisons of Fragaria, Prunus and Malus reveal different modes of evolution between Rosaceous subfamilies Bmc Genomics 13 doi:10.1186/1471-2164-13-129
Kenis K, Keulemans J, Davey MW (2008) Identification and stability of QTLs for fruit quality traits in apple. Tree Genet Genome 4:647–661. doi:10.1007/s11295-008-0140-6
Klagges C et al. (2013) Construction and comparative analyses of highly dense linkage maps of two sweet cherry intra-specific progenies of commercial cultivars. PLoS ONE 8 doi:10.1371/journal.pone.0054743
Koinange EMK, Singh SP, Gepts P (1996) Genetic control of the domestication syndrome in common bean. Crop Sci 36:1037–1045
Lamb NC (1953) Notes on the inheritance of some characters in the sweet cherry Prunus avium. Proc Am Soc Hortic Sci 61:293–298
Lerceteau-Kohler E, Moing A, Guerin G, Renaud C, Petit A, Rothan C, Denoyes B (2012) Genetic dissection of fruit quality traits in the octoploid cultivated strawberry highlights the role of homoeo-QTL in their control. Theor Appl Genet 124:1059–1077. doi:10.1007/s00122-011-1769-3
Li HH, Bradbury P, Ersoz E, Buckler ES, Wang JK (2011) Joint QTL linkage mapping for multiple-cross mating design sharing one common parent. PLoS ONE 6 doi:10.1371/journal.pone.0017573
Longhi S, Moretto M, Viola R, Velasco R, Costa F (2012) Comprehensive QTL mapping survey dissects the complex fruit texture physiology in apple (Malus x domestica Borkh.). J Exp Bot 63:1107–1121. doi:10.1093/jxb/err326
Mann HS, Alton JJ, Kim S, Tong CBS (2008) Differential expression of cell-wall-modifying genes and novel cDNAs in apple fruit during storage. J Am Soc Hortic Sci 133:152–157
Mariette S, Tavaud M, Arunyawat U, Capdeville G, Millan M, Salin F (2010) Population structure and genetic bottleneck in sweet cherry estimated with SSRs and the gametophytic self-incompatibility locus Bmc Genetics 11 doi:10.1186/1471-2156-11-77
Martinez-Garcia PJ et al (2013) High density SNP mapping and QTL analysis for fruit quality characteristics in peach (Prunus persica L.). Tree Genet Genome 9:19–36. doi:10.1007/s11295-012-0522-7
Meyer RS, Purugganan MD (2013) Evolution of crop species: genetics of domestication and diversification. Nat Rev Genet 14:840–852. doi:10.1038/nrg3605
Miller AJ, Gross BL (2011) From forest to field: perennial fruit crop domestication. Am J Bot 98:1389–1414. doi:10.3732/ajb.1000522
Minic Z, Jouanin L (2006) Plant glycoside hydrolases involved in cell wall polysaccharide degradation. Plant Physiol Biochem 44:435–449. doi:10.1016/j.plaphy.2006.08.001
Munos S et al (2011) Increase in tomato locule number is controlled by two single-nucleotide polymorphisms located near WUSCHEL. Plant Physiol 156:2244–2254. doi:10.1104/pp. 111.173997
Ogundiwin EA, Peace CP, Gradziel TM, Parfitt DE, Bliss FA, Crisosto CH (2009) A fruit quality gene map of Prunus Bmc Genomics 10 doi:10.1186/1471-2164-10-587
Olmstead JW, Lezzoni AF, Whiting MD (2007) Genotypic differences in sweet cherry fruit size are primarily a function of cell number. J Am Soc Hortic Sci 132:697–703
Panda S, Martin JP, Aguinagalde I, Mohanty A (2003) Chloroplast DNA variation in cultivated and wild Prunus avium L: a comparative study. Plant Breed 122:92–94. doi:10.1046/j.1439-0523.2003.00768.x
Paterson AH (2002) What has QTL mapping taught us about plant domestication? New Phytol 154:591–608. doi:10.1046/j.1469-8137.2002.00420.x
Peace CP, Crisosto CH, Gradziel TM (2005) Endopolygalacturonase: a candidate gene for freestone and melting flesh in peach. Mol Breed 16:21–31. doi:10.1007/s11032-005-0828-3
Peace C et al. (2012) Development and evaluation of a genome-wide 6K SNP array for diploid sweet cherry and tetraploid sour cherry. PLoS ONE 7 doi:10.1371/journal.pone.0048305
Pose S, Paniagua C, Cifuentes M, Blanco-Portales R, Quesada MA, Mercado JA (2013) Insights into the effects of polygalacturonase FaPG1 gene silencing on pectin matrix disassembly, enhanced tissue integrity, and firmness in ripe strawberry fruits. J Exp Bot 64:3803–3815
Purugganan MD, Fuller DQ (2009) The nature of selection during plant domestication. Nature 457:843–848. doi:10.1038/nature07895
Quesada MA et al (2009) Antisense down-regulation of the FaPG1 gene reveals an unexpected central role for polygalacturonase in strawberry fruit softening. Plant Physiol 150:1022–1032
Quilot B, Wu BH, Kervella J, Genard M, Foulongne M, Moreau K (2004) QTL analysis of quality traits in an advanced backcross between Prunus persica cultivars and the wild relative species P-davidiana. Theor Appl Genet 109:884–897. doi:10.1007/s00122-004-1703-z
Romano GS, Cittadini ED, Pugh B, Schouten R (2006) Sweet cherry quality in the horticultural production chain. Stewart Postharvest Rev 2:1–9. doi:10.2212/spr.2006.6.2
Rosyara U et al. (2013) Fruit size QTL identification and the prediction of parental QTL genotypes and breeding values in multiple pedigreed populations of sweet cherry Mol Breed:1–13 doi:10.1007/s11032-013-9916-y
Ruess F, Stosser R (1993) Investigations on the intercellular system of apple fruit by digital image-processing methods. Gartenbauwissenschaft 58:197–205
Ruiz D, Egea J (2008) Phenotypic diversity and relationships of fruit quality traits in apricot (Prunus armeniaca L.) germplasm. Euphytica 163:143–158
Sato S et al (2012) The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485:635–641. doi:10.1038/nature11119
Schreckenberg K, Awono A, Degrande A, Mbosso C, Ndoye O, Tchoundjeu Z (2006) Domesticating indigenous fruit trees as a contribution to poverty reduction. Forest Trees Livelihoods 16:35–51
Silva C, Garcia-Mas J, Sanchez AM, Arus P, Oliveira M (2005) Looking into flowering time in almond (Prunus dulcis (Mill) D. A. Webb): the candidate gene approach. Theor Appl Genet 110:959–968
Tavaud M (2000) Diversité génétique du cerisier doux (Prunus avium L.) sur son aire de répartition : comparaison avec ses espèces apparentées (P. cerasus et P. x gondouinii) et son compartiment sauvage., Thèse de Doctorat Ecole Nationale Supérieur Agronomique de Montpellier
Van Tassel DL, DeHaan LR, Cox TS (2010) Missing domesticated plant forms: can artificial selection fill the gap? Evol Appl 3:434–452. doi:10.1111/j.1752-4571.2010.00132.x
Velasco R et al (2010) The genome of the domesticated apple (Malus x domestica Borkh.). Nat Genet 42:833. doi:10.1038/ng.654
Verde I et al. (2012) Development and Evaluation of a 9K SNP Array for Peach by Internationally Coordinated SNP Detection and Validation in Breeding Germplasm PLoS ONE 7 doi:10.1371/journal.pone.0035668
Verde I et al (2013) The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet 45:487–U447
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78. doi:10.1093/jhered/93.1.77
Wakasa Y, Kudo H, Ishikawa R, Akada S, Senda M, Niizeki M, Harada T (2006) Low expression of an endopolygalacturonase gene in apple fruit with long-term storage potential. Postharvest Biol Technol 39:193–198. doi:10.1016/j.postharvbio.2005.10.005
Wang D, Karle R, Iezzoni AF (2000) QTL analysis of flower and fruit traits in sour cherry. Theor Appl Genet 100:535–544. doi:10.1007/s001229900121
Whiting MD, Ophardt D, McFerson JR (2006) Chemical blossom thinners vary in their effect on sweet cherry fruit set, yield, fruit quality, and crop value. Horttechnology 16:66–70
Yamamoto T et al (2001) Characterization of morphological traits based on a genetic linkage map in peach. Breed Sci 51:271–278. doi:10.1270/jsbbs.51.271
Zhang GR, Sebolt AM, Sooriyapathirana SS, Wang DC, Bink M, Olmstead JW, Iezzoni AF (2010) Fruit size QTL analysis of an F-1 population derived from a cross between a domesticated sweet cherry cultivar and a wild forest sweet cherry. Tree Genet Genome 6:25–36. doi:10.1007/s11295-009-0225-x
Zhang N, Brewer MT, van der Knaap E (2012) Fine mapping of fw3.2 controlling fruit weight in tomato. Theor Appl Genet 125:273–284
Acknowledgments
The authors thank the technical team of the department of “Biologie du Fruit et Pathologie” of the “Institut National de Recherche Agronomique,” Jacques Joly and Lydie Fouilhaux, for producing the hybrids and collaborating actively in the phenotyping, and the Experimental Unit of INRA-Toulenne (UEA) for growing the trees.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Online Resource 1
Markers used in the final parental maps. (PDF 336 kb)
Online Resource 2
Mean, s.d., IQR (interquartile range), c.v., value range, number of individuals (n) for the sweet cherry (Prunus avium) populations derived from the crosses between ‘Regina’ x ‘Lapins’ and ‘Regina’ x ‘Garnet’ observed during the different years of evaluation of the fruit firmness and weight. (PDF 101 kb)
Online Resource 3
Fruit firmness (Ff) and weight (Fw) of haplotype combinations for the QTL region on the extreme end of linkage group 5 for ‘Regina’ × ‘Garnet’ progeny. Significant difference between means (P > 0,05) are indicated by a star. (PDF 164 kb)
Online Resource 4
Genes located on the Quantitative Trait Loci for fruit firmness and fruit size in linkage groups 5 and 6. Linkage group, sequence name, sequence description, sequence length, #Hits, min. eValue, mean Similarity, number of gene ontologies, description of gene ontologies, enzyme codes and InterProScan codes are provided for each gene. (PDF 1054 kb)
Online Resource 5
Candidate genes located on the Quantitative Trait Loci for fruit firmness and fruit size in linkage groups 5 and 6. Linkage group, sequence name, sequence description, sequence length, #Hits, min. eValue, mean Similarity, number of gene ontologies, description of gene ontologies, enzyme codes and InterProScan codes are provided for each candidate gene. (PDF 207 kb)
Online Resource 6
Distribution of fruit weight (left) and firmness (right) measurements for two cross-pollination populations derived from ‘Regina’ and ‘Lapins’ (a) and ‘Regina’ and ‘Garnet’ (b) (PDF 214 kb)
Rights and permissions
About this article

Cite this article
Campoy, J.A., Le Dantec, L., Barreneche, T. et al. New Insights into Fruit Firmness and Weight Control in Sweet Cherry. Plant Mol Biol Rep 33, 783–796 (2015). https://doi.org/10.1007/s11105-014-0773-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-014-0773-6