
Abstract
Alkaloids are derived mainly from amino acids, and their biosynthesis includes multistep reactions. Benzylisoquinoline alkaloids (BIA), which are secondary metabolites, are produced by the medically and economically important opium poppy (Papaver somniferum L.). The latex of poppy capsule is the main source of BIA, including mainly opium alkaloids such as codeine, papaverine, thebaine, and noscapine. To analyze BIA-related gene expression at the molecular level, genome-wide transcriptome analyses were carried out in opium-poppy capsule tissues, using 12 × 135 K (12 × 135,000 features) NimbleGen microarrays of opium poppy at 0, 3, and 12 h. Fungal elicitor methyl jasmonate (MeJa) treatment was applied to the surface of the capsules of opium poppy at 0, 3, and 12 h. The measurement of specific BIA showed that the treated capsules were especially rich in morphine and noscapine. The differential expression of genes related to BIA biosynthesis was detected from 3 to 12 h after MeJa treatment. Overall, 2,459 out of the 95,608 probes identified specific differential gene expression. Many altered transcripts (629) were detected after 12 h of treatment. Altered levels of gene expression were also detected in BIA biosynthesis-pathway genes, such as NCS, CNMT, SalR, 16TOMT, 6OMT, COR and StySyn. Gene ontology analyses revealed that the differentially expressed genes were involved in many processes and functions. Upon induction with MeJa, response to stimuli- and stress-related transcripts were induced significantly in opium poppies. In addition, transcripts related to specific metabolic processes were well represented. Pathway analysis revealed that BIA biosynthesis, cysteine and methionine metabolism, sucrose and starch metabolism, as well as purine metabolism pathways were affected significantly. This study provides insights into the molecular mechanism of opium-poppy defense upon MeJa treatment, facilitating a better understanding of processes underlying the BIA biosynthesis network.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Opium poppy (Papaver somniferum L.) produces important pharmaceutical benzylisoquinoline alkaloids (BIA), including the narcotic analgesic morphine, the anti-tussive drug codeine, the vasodilator papaverine and anti-tumor agent noscapine (Ye et al. 1998; Desgagné-Penix et al. 2010; Winzer et al. 2012). Since opium poppy is the primary source for opiate production, it is an agronomically and economically important plant (Allen et al. 2004; Unver et al. 2010). In Turkey, opium poppy is grown annually on approximately 13,500 ha; this is the largest legal cultivation area in the world according to Narcotics Drugs Technical reports 2012; http://www.incb.org/documents/Narcotic-Drugs/Technical-Publications/2012/Narcotic_Drugs_Report_2012.pdf.
BIA biosynthesis is a complex and not completely understood pathway in opium poppy. The first step of BIA biosynthesis is the condensation of two l-tyrosine derivatives, 4-hydroxyphenylacetaldehyde (4-HPAA) and dopamine, catalyzed by tyrosine aminotransferase (TyrAT) and tyrosine/DOPA decarboxylase (TYDC) to generate (S)-norcoclourine (Facchini and De Luca 1994; Lee and Facchini 2011). To form the central intermediate of opium alkaloid branch pathway (S)-reticuline, a series of enzymes including norcoclaurine synthase (NCS) (Lee and Facchini 2010), norcoclaurine 6-O-methyltransferase (6OMT) (Morishige et al. 2000), coclaurine N-methyltransferase (CNMT) (Choi et al. 2002), and 4′-O-methyltransferase (4OMT) (Morishige et al. 2000) are involved. The morphinan branch requires the epimerization of (S)-reticuline to (R)-reticuline (De-Eknamkul and Zenk 1992). (R)-reticuline is converted to salutaridine by salutaridine synthase (SalSyn), which is then reduced by salutaridine reductase to yield salutaridinol (Ziegler et al. 2006). In the last step of morphine biosynthesis, conversion of thebaine to morphine occurs via codeine or oripavine produced by demethylases (Hagel and Facchini 2010; Unterlinner et al. 1999). Although several cell types participate in BIA biosynthesis, alkaloid accumulation occurs in large vesicles contained in the articulated laticifers adjacent to the vascular bundle throughout the plant (Facchini and Park 2003).
It is well documented that exogenous application of methyl jasmonate (MeJA) is effective at controlling plant diseases by resulting in the expression of a set of defense genes and inducing resistance of host against pathogens and secondary metabolites (Chen et al. 2014). Plants produce jasmonates (JA) from a group of oxylipins (oxygenated fatty acids) via the octadecanoid pathway (Schaller and Stintzi 2009; Wasternack and Kombrink 2009). The class of cyclopentanone compounds, jasmonic acid and its volatile methyl ester MeJA, are indicated as endogenous regulators playing important roles in stress response, plant growth and development (Creelman and Mullet 1997), and are used extensively in biological experiments. MeJa acts as an elicitor to allow alkaloid accumulation in opium poppy seedlings and cell culture (Holková et al. 2010; Huang and Kutchan 2000; Mishra et al. 2013a). In such studies, transcriptome analysis have been performed either by RNA sequencing or by macroarray analyses. However, no comprehensive genome-wide microarray and transcriptome analysis has been designed for opium poppy capsule. We aim to enhance BIA synthesis in opium poppy capsule with exogenous application of MeJa and to carry out transcriptome profiling at the genome-wide level. For this purpose, we used the Nimblegen microarray platform to analyze gene expression in opium poppy capsule; opium poppy capsule has shown to be a useful tool for analyzing plant gene expression profiles under a variety of conditions (Hagel and Facchini 2010).
Materials and Methods
Plant Materials, Growth Conditions and MeJa Treatments
Papaver somniferum cultivar Ofis 95 obtained from Toprak Mahsulleri Ofisi (TMO) (Ankara, Turkey) was used for transcriptome analysis. Seeds of opium poppy were germinated in fruit trays then were transplanted to pots. All plants were grown with day/night cycles of 16/8 h at 20 °C/18 °C for summer and 10/14 h at 10 °C/4 °C for winter season in greenhouse conditions for 8 months. Before 100 μM MeJa was sprayed on leaves, the stem and capsule were wounded with sterile pins with an average wounding site of approximately one per square millimeter avoiding major injures (Mishra et al. 2013b). MeJa solution was prepared in ethanol using 0.1 % triton X-100. Control plants were treated with equal volumes of 0.1 % triton X-100 dissolved in ethanol. Capsule, leaf, stem and root tissues of treated and control plants were harvested at 0, 3, and 12 h after MeJa treatment. Tissues were frozen in liquid nitrogen and stored at −80 ° C until use.
ToF LC-MS Analyses
HPLC analyses were carried out using the 1260 infinity LC, 6210 ToF-MS Agilent platform (http://www.chem.agilent.com). The plant materials were dried at 28 °C for 2 days. Dried samples (0.1 g) were soaked in methanol (HPLC grade, Merck, Darmstadt, German) at room temperature for 1 day with shaking followed by filtration to separate the marc and evaporated. The extracts were solved in 2,000 ppm and diluted to 1/200 ppm from this stock. To analyze the HPLC ToF MS samples were filtered through 0.45 μm membranes. Morphine, codeine, thebaine, papaverine, noscapine and laudanosine were detected.
RNA Isolation, Labeling and Hybridization
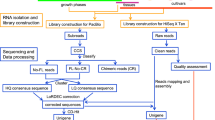
Three biological replicates of each time point were used for the analysis. Frozen capsule tissues were ground to a fine powder in liquid nitrogen for RNA isolation using TRIzol® Reagent (Cat No. 15596–026, Invitrogen, Carlsbad, CA) following the manufacturer’s protocol. RNA was quantified using a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Lenexa, KS) and RNA quality was checked on 1.5 % agarose gels. From 10 μg total RNAs, double stranded cDNA was synthesized using the SuperScript Double-Stranded cDNA Synthesis Kit (Invitrogen) and labeled with Cy3 random nonamers with the One- Color DNA Labeling Kit (Roche NimbleGen, Madison, WI) and the product was precipitated using NimbleGen kits according to the NimbleGen Gene Expression protocol for microarrays. As described in previous report (Turktas et al. 2013), 4 μg labeled products was loaded onto the custom 12X135K array, incubated at 42 °C overnight in a hybridization system, and washed in the NimbleGen Wash Buffer Kit following the NimbleGen protocol. The microarray slide was scanned at 2 μm resolution using a MS 200 Microarray Scanner, generating the corresponding 532 nm TIFF images. The data were then imported into DEVA software (http://www.nimblegen.com) to quantify the signal intensities of the spots on the image.
Microarray Design and Data Analysis
Experiments were repeated with three independent biological replicates per time point. Microarray analysis was performed using the Nimblegen 12X135K expression array. The microarray was constructed with 95,608 60-mer oligonucletide probes corresponding to 23,902 transcripts selected from P. somniferum accession number SRX012997. The MeJa-induced transcriptome profiling was detected over a time-course with untreated plants for controls and at 0, 3, and 12 h after MeJa treatment.
The array data were normalized according to the quantile method for standardization (Bolstad et al. 2003) and the Robust Multichip Average (RMA) algorithm (Irizarry et al. 2003) with DEVA software as indicated previously. The signal intensities of the samples were transformed into log2-ratio data. The dye-normalized and background-subtracted intensity data were exported into ArrayStar software (DNAStar, Madison, WI) to perform gene expression analyses. Student’s t-test was used to identify differentially expressed genes. P ≤ 0.05 and 1.5 fold change were considered to indicate differential expression of genes between two data sets. The basic local alignment search tool (BLAST) algorithm and BLAST to Gene Ontology (Blast2GO; http://www.blast2go.com) tool were used against the National Center for Biotechnology Information (NCBI) database to annotate the genes corresponding to the hybridized cDNA signals. Gene ontology (GO) enrichment analysis was done using Gramene. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed with default parameters for the predicted target genes, to improve the elucidation of the biological functions of the genes. Putative mRNA sequences were used as queries against the KEGG database (http://www.genome.jp/kegg).
qRT-PCR Analysis
In order to validate our microarray data analysis, 11 randomly selected genes were measured by quantitative reverse-transcription polymerase chain reaction (qRT-PCR). Gene-specific primers of selected genes were designed using the Primer3Plus software version 2.3.3 (http://primer3plus.com) (Untergasser et al. 2012) for PCR analysis (Table S4). The 18S rRNA (GenBank accession no: X16077.1) (forward primer: 5′-TTTGACTCAACACGGGGAAA-3′; reverse primer: 5′-CAGACAAATCGCTCCACCAA-3′) served as reference gene. The reverse transcription reaction was carried out with a Fermentas First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol. The qRT-PCR experiments were performed as previously reported (Unver and Budak 2009). cDNA (2 μl) was amplified with 0.1 μl specific primers in a total volume of 18 μl, using a LightCycler 480 Real-Time PCR System with SYBR Green I Master (Roche Applied Science, Penzberg, Germany). The synthesized cDNAs were used as templates for PCR amplification under the following conditions: preheating at 95 °C for 5 min; and 41 cycles (95 °C for 10 s; 53 °C, 59 °C or 61 °C for 20 s depending on the cDNA annealing temperature; and final extension 72 °C for 10s). Three replicates were performed for each sample. Gene expression levels were calculated according to the ddCt algorithm.
Results
Measurement of BIA in MeJa-treated Opium Poppy
To study the response of opium poppy to MeJa treatment, capsule metabolite analysis was carried out in mature opium poppy plants at post anthesis stage, 2–3 days after petal fall. Morphine, codeine, thebaine, noscapine, laudanosine and papaverine levels were analyzed with HPLC-ToF MS (Fig. 1). Morphine and noscapine were found to be the most abundant alkaloids in capsules. The morphine level increased gradually in MeJa-treated plants with increasing treatment time. The morphine content reached the highest level at 12 h following MeJa treatment (Fig. 1a). The same pattern was observed for noscapine (Fig. 1b). The codeine level was up-regulated at 3 h of treatment. The concentration then decreased at 12 h after MeJa application (Fig. 1c). Additionally, an increased concentration of thebaine was observed upon MeJa treatment at 0, 3, and 12 h (Fig. 1d). However, the level of laudanosine and papaverine was very low; papaverine was hard to detect (Fig. 1e) and its level is almost ten-fold lower than that of the other BIAs. The laudanosine content was increased by MeJa treatment (Fig. 1f).

Alkolaoid levels in methyl jasmonate (MeJA)-treated and untreated opium poppy capsule samples measured with HPLC-ToF. Units are milligrams of metabolite per gram of capsule with standard deviations. Control samples represent a mixture of all control time points. a Morphine, b noscapine, c codeine, d thebaine, e laudanosine, f papaverine
Assessment of Microarray Data
All probes produced detectable signals (Table S1). To investigate changes in gene expression levels in the capsule, six combinations of time points were evaluated [0 h vs control, 3 h vs control, 12 h vs control, 3 h vs 0 h, 12 h vs 0 h, 12 h vs 3 h (i.e., 0 h vs control indicates 0 h compared to control)]. Scatter plots were used to indicate the gene expression differences between the two datasets in microarray analyses (Fig. 2). The results revealed that there were substantial numbers of differentially expressed genes among the time points. The Pearson’s coefficient correlation between the time point data sets showed that low R 2 value indicated high number of differentially expressed genes between the time points. The microarray data is available at Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) with accession number: GSE55196.

Scatter plots indicating gene expression differences between the two datasets in microarray analyses (Pearson’s coefficient correlation in parentheses). a 0 h vs Control (0.015) b 3 h vs Control (0.010) c 12 h vs Control (0.004) d 3 h vs 0 h (0.010) e 12 h vs 0 h (0.0004) f 12 h vs 3 h (0.007)
Transcriptome Profiling
Transcripts with P ≤ 0.05 and 1.5 fold change were considered as differentially expressed (Fig. 3 and Table S2). A total of 2,459 unique transcripts were found as differentially expressed in at least one comparison. Differentially regulated genes are shown in Venn diagrams in Fig. 4; 14 transcripts were down-regulated in the control samples compared to any other time points (Fig. 4a). Among them, only two were annotated [light harvesting complex II protein ıhcb1 (SRR030261.67895) and alanine:glyoxylate aminotransferase (SRR030261.99439)], and the rest were unknown transcripts. The data obtained from scatter plot analysis revealed that Pearson’s coefficient correlation ranged from 0.0004 to 0.015 for the six comparisons.

a Heatmap map representing normalized signal intensities of all probes. b Heat map representing normalized signal intensities of differentially expressed 2,459 genes. Gene expression level: red high, blue low

Venn diagrams showing the number of differentially expressed transcripts among time points. Red arrows up-regulation, blue arrows down-regulation. a Number of transcripts within 0 h, 3 h, 12 h and control. b Number of transcripts within 0 h, 3 h and 12 h
The microarray analysis indicated that 460 transcripts were differentially regulated between 0 h and control samples, of which 233 transcripts were down-regulated, and 227 were up-regulated. About 75 % of those remained unannotated (Table S2). Pearson’s coefficient correlation was R 2 = 0.015. A broad range of expression difference was detected from −4.29 to 3.16. In total, 470 transcripts, of which only 23 % were annotated, were found to be differentially expressed between 3 h treated samples and controls. While 231 transcripts were up-regulated, 239 were down-regulated (Fig. 4a). After 12 h treatment, MeJa induced the highest number (515) of differentially expressed transcripts; however, only 20 % were annotated and the rest were genes with known functions. Among the, 249 were down-regulated and 266 were up- regulated (Fig. 4a).
At 12 h of MeJa treatment, 629 transcripts with differential expression were induced, which was the highest number among all datasets. The numbers of down- and up-regulated transcripts were 320 and 309, respectively. The fold change of the transcripts ranged from +3.94 to −2.99. Out of 629, 21 % were annotated. The Pearson’s coefficient correlation was R 2 = 0.0004 (Fig. 4b).
The results of the 3 h treatment vs 0 h time comparison indicated that 496 transcripts were differentially regulated. Of these, 245 were positively regulated and 251 were negatively regulated, and the fold change ranged from +3.21 to −2.79. Almost a quarter of the transcripts were found to be annotated (Fig. 4b). Tthe 12 h vs 3 h time comparison revealed 467 differentially expressed transcripts, of which 252 showed down-regulation and up-regulation. The highest fold change among up-regulated transcripts was found to be 2.78, while the lowest was 3.97 among down-regulated transcripts (Fig. 4b).
GO Enrichment and Pathway Analyses
To identify and to annotate the GO categories of the genes altered by MeJa treatment, we performed GO enrichment analysis in terms of biological processes, molecular function and cellular component. Transcripts were classified into the categories using the Gramene gene ontology tools (Fig. 5).

Gene ontology (GO) categories of ANNOTATED differentially expressed transcripts for a biological processes, b molecular functions and c cellular components. The figure shows the percentage distributions of GO terms
In GO of the 0 h vs control assessment, 115 altered expressed transcripts were identified (Fig. 6). The majority of these were involved in metabolic process, response to stimuli and stress processes. Interestingly, among the stimuli-responsive transcripts, 47 % belong to cadmium ion response, and 60 % of these had higher expression at 0 h compared to control plants. In addition, 65 % of transcripts involving in metabolic processes were up-regulated. Among them, 3-deoxy-d-arabino-heptulosonate 7-phosphate synthase (SRR030261.89386), the precursor of secondary metabolites involving in shikimic acid biosynthesis, was one of the most up-regulated transcripts at 2.16 fold (Table S2). GO analysis showed that 52 % of the transcripts were located in chloroplasts, followed by membranes and vacuoles. The predominant pathways in this time comparison were cysteine and methionine metabolism and isoquinoline alkaloid biosynthesis (Fig. S1a).

Comparative functional gene ontology (GO) analysis for the time courses followed here. The charts show the number of genes that fall into a particular GO term for biological process and molecular function category in up-regulated genes are represented above the x-axis and down-regulated genes are represented belove the x-axis
For the 3 h treatment vs control comparison, the highest number of transcripts was involved in response to stimuli and stress, metabolic process and cellular organization. Similar to 0 h vs control, the largest number of transcripts was involved in the cadmium ion response but 69 % was found to be down regulated. In this assessment, cysteine and methionine metabolism and oxidative phosphorylation were the most detected pathways (Fig. S1b).
The majority of the differentially expressed genes in 12 h MeJa-treated samples were associated with metabolic processes, response to stimuli and stress, development and hormonal regulation. Similar to the other time points, the largest number of transcripts (20 %) belonged to the cadmium ion response, and all of them were up-regulated. The rest (80 %) of the transcripts belonging to hormonal regulation were also up-regulated. The transcript responding to cytokinin and ABA had the highest fold change (2.66 and 2.50, respectively); 42 % of the transcripts were JA and SA related and all of them were up-regulated. The majority of the transcripts had ion binding, hydrolase and oxidoreductase activities, and were located in the chloroplast, plasma membrane and nucleus. A notable number of transcripts at this time point were involved in the purine metabolism pathway (Fig. S1c).
The differentially expressed genes in 3 h MeJa-treated samples were related to response to stimuli and stress, development and metabolic process. Among the transcripts responding to stimuli, 16 % were associated with response to cadmium ion; 69 % of these were up-regulated. Additionally, 62 % of the transcripts in hormonal regulation were down-regulated. Starch and sucrose metabolism, purine metabolism, isoquinoline alkaloid biosynthesis were common pathways for genes differentially expressed in MeJa-treated samples after 3 h exposure (Fig. S1d).
At the 12 h vs 0 h time point, analysis found that metabolic process, response to stimuli and stress and development-related transcripts were found predominantly. Similar to the other time point comparisons, response to the cadmium ion contained the highest number of transcripts with 25 %. Transcripts related to hormonal regulation (74 %) were down-regulated. On the contrary, nine out of ten RNA binding activity transcripts were up-regulated. Cysteine and methionine metabolism, as well as purine metabolism pathways were mainly induced by MeJa treatment (Fig. S1e).
Comparison of 12 h vs 3 h indicated that metabolic process, response to stimuli and stress and development related genes have the highest number of transcripts. Likewise, 12 h vs 0 h timepoint assessment revealed a number of transcripts involved in cysteine and methionine metabolism and nitrogen metabolism (Fig. S1f).
Genes Involved in BIA Biosynthesis are Differentially Expressed Upon MeJa Treatment
Several genes differentially expressed after MeJa treatment were involved in the BIA pathway. These genes included 6OMT, CNMT, 4OMT, Codeinone reductase (COR), NCS, Aspartate aminotransferase, SalSyn, SalR (Table 1). In the 0 h treatment compared to the control, expression of NCS (SRR030261.36518) yielding (S)- norcoclaurine and SalR (SRR030261.37318) converting salutaridine to salutaridinol was induced, with 1.82- and 2.39-fold changes, respectively. COR (SRR030261.27166), which catalyzes the reduction of codeinone to codeine, was 1.52-fold down-regulated in the 0 h vs the control comparison. O-methyl transferases were found to be induced continuously with the application of MeJa with the exception of norcoclaurine 6-o-methyltransferase (SRR030261.23214). On the contrary, the opposite pattern was observed for 16-hydroxytabersonine o-methyltransferase (SRR030261.35138) (Table 1).
Stress and Defense Responsive Genes
Ontology analyses showed that a total of 157 differentially expressed transcripts responded to stimulus; 43 % of them were related to response to cadmium ion. In response to stimuli, Major Latex Protein (MLP) transcripts were expressed in all time points except 0 and 3 h after treatment. MLP encoding transcripts (SRR030261.533409) were down-regulated (1.81 and 2.57) upon MeJa treatment at 0 h and at 3 h, respectively. MLP was also detected as the most differentially expressed gene among all the stimuli-regulated transcripts. However, after 3 h of MeJa treatment, the expression level of MLP decreased; after 12 h treatment, the expression level was down by 1.61-fold compared with the time point at 3 h.
Transcripts corresponding to HSP 70 (SRR030261.59447) and HSP 90 (FG609084) were up-regulated in 3 h compared to 0 h (2.08 and 1.66 fold), whereas they were down-regulated at 12 h compared to 3 h (1.87-fold and 1.81-fold).
MeJa treatment altered the transcripts levels of pathogen related proteins (PRs) Class I Chitinase, Chitinase-like protein 1 and Chitinase-like protein 2. Chitinase-like protein 1 and 2 were detected only in 3 h vs control with reverse pattern. Besides, Class I Chitinase transcript was detected in 3 h vs 0 h with the fold change of 1.59.
Among all stimuli response transcripts, 80 % were related to stress responsive, such as uridine diphosphate glycosyltransferase 74e2 (UGT74E2) (SRR030261.35893). Compared to the control, a rapid increase of UGT74E2 occurred immediately after MeJa application (0 h) with a fold change of 2.50; however, its expression was down-regulated (2.17 fold) after 3 h. Moreover, it was down-regulated (2.50 fold) at 12 h. The expression pattern of UGT74E2 was consistent with that of morphine content at all time points (Table S2).
KEGG Pathway Enrichment
KEGG pathway enrichment analyses suggest that the transcripts especially morphinan branch genes, such as 6OMT, CNMT, 4OMT, COR, NCS, Aspartate aminotransferase, SalSyn, and SalR were involved in BIA biosynthesis (Fig. 7). In microarray experiments, the most abundant transcripts detected in BIA biosynthesis were cysteine and methionine metabolism, oxidative phosphorylation, purine metabolism, and starch and sucrose metabolism pathways. As many as 83 % of the transcripts involved in cysteine and methionine metabolism were down-regulated at the early treatment (0 h and 3 h). However, the transcripts belonging to cysteine and methionine metabolism displayed up-regulation at 12 h. In the oxidative phosphorylation pathway, 80 % of the transcripts were up-regulated at 3 h. Starch and sucrose metabolism transcripts were up-regulated at 0 h, whereas 67 % of the transcripts were down-regulated at 3 h; 60 % of the transcripts were related to purine metabolism, and were up-regulated at 12 h (Table S3).

Transcripts (colored boxes) involved in benzylisoquinoline alkaloids (BIA) synthesis pathways
Validation of Microarray Data
qRT-PCR was performed in order to validate the microarray results, and to determine the correlation between the two expression platforms. Ten genes, three of them belonging to the BIA biosynthesis pathway and the others belonging to different metabolic mechanisms, were selected for validation. Comparison of the results obtained from qRT-PCR with those from microarray data showed almost similar patterns with some exceptions. The qRT-PCR data on expression of seven genes (methyltransferase-like protein 1-like, calvin cycle protein cp12, vacuolar amino acid transporter 1-like protein, flavanol synthase flavon 3, norcoclaurine synthase, (+) neomentholdehydrogenase and 4OMT) were highly correlated with the microarray data. For example; (+)-neomenthol dehydrogenase was up-regulated by 2.39-fold by microarray and 8-fold by qRT-PCR, respectively. However, some inconsistencies between the microarray and qRT-PCR results were also observed. For example, SRR030261.74271 was down-regulated in 12 h vs 0 h and 12 h vs 3 h in microarray analysis, whereas it was found to be the opposite in qRT-PCR; however, comparison of 0 h-C and 3 h-C was consistent with qRT-PCR results. Taken together, the majority of the data provide an indication of the validity and reliability of our microarray results (Table 2, Fig. 8).

Expression levels of seven selected genes calculated by qRT-PCR. Standard error bars are indicated on each column. Expression was normalized to 18S rRNA
Discussion
With advanced genomic approaches, metabolic and gene expression profiling for plant–pathogen interactions are widely required to understand plant defense mechanisms against various environmental stresses (Figueiredo et al. 2008). MeJa treatment induces secondary metabolite production in plant tissues and cell cultures (D’Onofrio et al. 2009; Ku et al. 2014; Mishra et al. 2013a). Here, HPLC-ToF MS and microarray analyses were performed to identify changes in the level of six alkaloids as well as gene expression in opium poppy capsules after MeJa treatment.
MeJa Exposure Altered Alkaloid Biosynthesis
The opium poppy capsule is rich in latex alkaloids (Weid et al. 2004). The results of our metabolite analysis showed that all of the alkaloid levels were induced after 3 h MeJa treatment, and increased continuously to 12 h after treatment. The most abundant alkaloids in capsule tissue were morphine and noscapine. Morimoto et al. (2001) suggested that mechanical damage resulted in the rapid conversion of morphine to bismorphine, and incorporation of bismorphine into the cell wall had a defensive function. It is known that wounding is a stress factor, and the gradual induction in the morphine concentration upon MeJa treatment indicates a response to stress (Morimoto et al. 2001; Bonilla et al. 2014).
Morphine and noscapine are usually branched after (S)-reticuline formation. However, our analyses revealed that morphine and noscapine existed at all time points. This result suggests that genes upstream of (S)-reticuline might be the key players in noscapine accumulation rather than the downstream genes. Alternatively, there might be unknown steps and/or regulators for noscapine biosynthesis (Bonilla et al. 2014; Ziska et al. 2008; Lachman et al. 2006).
Gene Associated with BIA Biosynthesis Pathway
Microarray analysis showed that 6OMT, CNMT, 4OMT, COR, NCS, Aspartate aminotransferase, SalSyn, SalR transcripts involved in BIA pathway were aberrantly expressed at various time points. Cell- or tissue-specific accumulation of plant secondary metabolites generally correlates with expression of the corresponding biosynthetic genes (Ziegler et al. 2009). NCS and SalR transcript levels showed a correlation between most of the alkaloid accumulation in capsule. Previously, the NCS enzyme catalyzing the first committed step in BIA biosynthesis was shown to be a member of PR10/Betv1 family (Samanani et al. 2004; Liscombe et al. 2005), and Ziegler et al. (2009) suggested that SalR might play a role in pathogen defense due to exhibiting amino acid similarity with the neomenthol reductases that participate in the protection of plants against pathogens. Taken together, these results indicated that NCS and SalR genes were induced under MeJa treatment. Additionally, alkaloid accumulation as a defense response and the decrease of alkaloid levels in comparison to 0 h might be one reason for the repressed levels of BIA pathway gene expression. NCS and SalR might act together to regulate the secondary metabolites produced upon pathogen attack (Table 1).
Plant Growth Regulators Involved in BIA Biosynthesis
It is well known that the phytohormones jasmonic acid (JA), salicyclic acid (SA), abscisic acid (ABA) and ethylene (ET) play important roles in plants to cope with pathogen stress (Pieterse et al. 2009; Denancé et al. 2013). Similarly, the most abundant transcripts were found to be related with ABA, auxin, JA and SA hormone regulations under MeJa treatment. ABA-related transcripts represented 28.4 % of all probes in hormone regulation, suggesting involvement of ABA in response to pathogen stress. Moreover, brassinosteroid (BR) biosynthesis genes were detected in opium poppy capsule. The majority of the BR-related transcripts were found at 3 h after MeJa treatment; 80 % of them were negatively regulated (Table S2).
Genes Responding to Stress
The large number of biotic and abiotic stress-related genes identified in this study suggests that our opium poppy transcriptome data reveal extensive changes in response to MeJa treatment. The number of differentially expressed transcripts and alkaloid concentration consistently reached their highest levels at 12 h after treatment. Opium poppy latex contains abundant, low molecular mass major latex proteins (Nessler et al. 1985). Although the function of the major latex proteins is not completely understood, the opium poppy transcriptome has been shown to be affected by MeJa treatment. Facchini and Park (2003) reported that there is no induction of MLP transcripts after elicitation of cell cultures of opium poppy from 0 to 80 h, in contrast our results, which indicated alterations at all time points.
Heat shock proteins (HSPs) play important roles in protecting plants against stress by re-establishing normal protein configuration, thus helping to maintain cellular homeostasis. Hsp90 family members are also involved in protein folding, but they also play a key role in cellular signal regulation, and seem to be necessary for functionality of resistance (R) protein in A. thaliana (Hubert et al. 2003). Previously, it was reported that elicitor treatment did not substantially increase HSP70 protein levels in opium poppy cell cultures (Zulak et al. 2009). However, Decker et al. (2000) noticed that some defined proteins found in capsule latex might be related to chaperones; this was also the case in California poppy (Eschscholzia californica)—a species closely related to P. somniferum. HSP70 is one of the most up-regulated proteins in response to both low and high-elicitor treatment in root-derived cell cultures of California poppy (Angelova et al. 2010). Consistently, our results suggest that HSP 70 and HSP 90 might work together to promote protection and the maintenance of homeostasis within 3 h in opium poppy.
Chitinase, which belongs to the diverse group of PR genes, has the ability to degrade fungal cell walls in order to enhance the ability of a plant to resist fungal pathogens. Being an early responsive gene, chitinase was induced 3 h after MeJa treatment.
It was reported that uridine diphosphate glycosyltransferases (UGTs) transfer glycosyl residues from activated nucleotide sugars to a wide range of acceptor molecules, such as secondary metabolites including phytoalexins, cell wall precursors and plant hormones like SA (Ross et al. 2001; Bricchi et al. 2012; Glombitza et al. 2004). However, to date, no data is available about whether UGT 74e2 plays a role in biosynthesizing alkaloids. Here, the results indicated that UGT 74e2 transcript levels were altered in time courses under MeJa treatment (Table S2).
Metal Ions Might Regulate Defense and Secondary Metabolite Production in Opium Poppy
Further investigations have reported that high levels of Ni, Zn, Cd or Se provided effective protection against fungi, or even snails and viruses (Franceschi et al. 2005). Cadmium-induced resistance against fusarium infection has been related to metal-induced proteins in wheat (Mittra et al. 2004), and different cadmium treatments in Catharanthus roseus also enhanced the production of ajmalicine alkaloid (Zheng and Wu 2004). In this study, a huge number of transcripts involved in response to cadmium ion were represented in all time courses. Lachman et al. (2006) reported that the higher Cd content in seeds and capsules of opium poppy caused an increase and activation of noscapine and morphine biosynthesis (Bonilla et al. 2014; Ziska et al. 2008; Lachman et al. 2006). This provides a strong indication for a link between cadmium ion and production of secondary metabolites and defense mechanisms in opium poppy.
Induced KEGG Pathways
In this study, transcripts belonging to cysteine (Cys) and methionine (Met) metabolism altered under MeJa treatment. Amino acids Cys and Met are essential to all living organisms because of their prominent functions in primary and secondary metabolism (Leustek et al. 2000; Droux 2004; Komarnisky et al. 2003). S-Adenosyl-l-methionine (AdoMet) functions as a donor of methyl groups in different reactions catalyzed by methlytransferase enzymes (Tabor and Tabor 1985), and it was reported that fungal elicitors activate AdoMet transcription (Kawalleck et al. 1992; Liscombe and Facchini 2007). In the present study, starch and sucrose metabolism related transcript levels showed alterations, especially within 3 h (Table S3). These findings support previous results showing activation of carbohydrate metabolism upon pathogen infection and wounding (Ehness et al. 1997).
Exogenous application of 50 μM MeJa induced only 6OMT, 7OMT, BBE, COR, and T6ODM transcripts in poppy seedlings (Mishra et al. 2013a). Cellular or tissue localization of morphine alkaloid biosynthesis is not completely understood, but it was reported that the distribution of expression levels differs according to tissue types or plant growth stages (Huang and Kutchan 2000). We detected some transcripts playing roles in BIA synthesis pathways as indicated in Fig. 7 (blue boxes) by microarray analyses. Therefore, differentially expressed genes related to BIA synthesis are regulated in elicitor-treated opium poppy at different time points.
Conclusions
The genome-wide transcriptome profiling of opium poppy capsules induced with MeJa described the relationship between stress response and BIA biosynthesis. The majority of the differentially expressed genes are involved in metal ion binding, response to stimuli, and plant hormone regulation. On the other hand, several genes related to the BIA pathway were also expressed differentially. The KEGG pathway enrichment analyses showed that genes involved in BIA biosynthesis are possibly linked with MeJa induction. However, many transcripts were unannotated and still need to be investigated. Our results suggest that there might be other pathways connected to BIA production. The results of this study provide new insights into out understanding of transcriptome rearrangements for BIA synthesis.
References
Allen RS, Millgate AG, Chitty JA, Thisleton J, Miller JA, Fist AJ, Gerlach WL, Larkin PJ (2004) RNAi-mediated replacement of morphine with the nonnarcotic alkaloid reticuline in opium poppy. Nat Biotechnol 22(12):1559–1566. doi:10.1038/nbt1033
Angelova S, Buchheim M, Frowitter D, Schierhorn A, Roos W (2010) Overproduction of alkaloid phytoalexins in California poppy cells is associated with the co-expression of biosynthetic and stress-protective enzymes. Mol Plant 3(5):927–939
Bolstad BM, Irizarry RA, Åstrand M, Speed TP (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and BIA. Bioinformatics 19(2):185–193
Bonilla A, Sarria A, Algar E, Muñoz Ledesma F, Ramos Solano B, Fernandes J, Gutierrez Mañero F (2014) Microbe associated molecular patterns from rhizosphere bacteria trigger germination and Papaver somniferum metabolism under greenhouse conditions. Plant Physiol Biochem 74:133–140
Bricchi I, Bertea CM, Occhipinti A, Paponov IA, Maffei ME (2012) Dynamics of membrane potential variation and gene expression induced by Spodoptera littoralis, Myzus persicae, and Pseudomonas syringae in Arabidopsis. PLoS One 7(10):e46673
Chen J, Zou X, Liu Q, Wang F, Feng W, Wan N (2014) Combination effect of chitosan and methyl jasmonate on controlling Alternaria alternata and enhancing activity of cherry tomato fruit defense mechanisms. Crop Prot 56:31–36
Choi K-B, Morishige T, Shitan N, Yazaki K, Sato F (2002) Molecular cloning and characterization of coclaurinen-methyltransferase from cultured cells of Coptis japonica. J Biol Chem 277(1):830–835
Creelman RA, Mullet JE (1997) Biosynthesis and action of jasmonates in plants. Annu Rev Plant Biol 48(1):355–381
D’Onofrio C, Cox A, Davies C, Boss PK (2009) Induction of secondary metabolism in grape cell cultures by jasmonates. Funct Plant Biol 36(4):323–338
De-Eknamkul W, Zenk MH (1992) Purification and properties of 1, 2-dehydroreticuline reductase from Papaver somniferum seedlings. Phytochemistry 31(3):813–821
Decker G, Wanner G, Zenk MH, Lottspeich F (2000) Characterization of proteins in latex of the opium poppy (Papaver somniferum) using two-dimensional gel electrophoresis and microsequencing.Electrophoresis 21:3500–3516
Denancé N, Sánchez-Vallet A, Goffner D, Molina A (2013) Disease resistance or growth: the role of plant hormones in balancing immune responses and fitness costs. Front Plant Sci 4:155
Desgagné-Penix I, Khan M, Schriemer D, Cram D, Nowak J, Facchini P (2010) Integration of deep transcriptome and proteome analyses reveals the components of alkaloid metabolism in opium poppy cell cultures. BMC Plant Biol 10(1):252
Droux M (2004) Sulfur assimilation and the role of sulfur in plant metabolism: a survey. Photosynth Res 79(3):331–348
Ehness R, Ecker M, Godt DE, Roitsch T (1997) Glucose and stress independently regulate source and sink metabolism and defense mechanisms via signal transduction pathways involving protein phosphorylation. Plant Cell Online 9(10):1825–1841
Facchini PJ, De Luca V (1994) Differential and tissue-specific expression of a gene family for tyrosine/dopa decarboxylase in opium poppy. J Biol Chem 269(43):26684–26690
Facchini PJ, Park S-U (2003) Developmental and inducible accumulation of gene transcripts involved in alkaloid biosynthesis in opium poppy. Phytochemistry 64(1):177–186
Figueiredo A, Fortes AM, Ferreira S, Sebastiana M, Choi YH, Sousa L, Acioli-Santos B, Pessoa F, Verpoorte R, Pais MS (2008) Transcriptional and metabolic profiling of grape (Vitis vinifera L.) leaves unravel possible innate resistance against pathogenic fungi. J Exp Bot 59(12):3371–3381
Franceschi VR, Krokene P, Christiansen E, Krekling T (2005) Anatomical and chemical defenses of conifer bark against bark beetles and other pests. New Phytol 167(2):353–376
Glombitza S, Dubuis P-H, Thulke O, Welzl G, Bovet L, Götz M, Affenzeller M, Geist B, Hehn A, Asnaghi C (2004) Crosstalk and differential response to abiotic and biotic stressors reflected at the transcriptional level of effector genes from secondary metabolism. Plant Mol Biol 54(6):817–835
Hagel JM, Facchini PJ (2010) Dioxygenases catalyze the O-demethylation steps of morphine biosynthesis in opium poppy. Nat Chem Biol 6(4):273–275
Holková I, Bezáková L, Bilka F, Balažová A, Vanko M, Blanáriková V (2010) Involvement of lipoxygenase in elicitor-stimulated sanguinarine accumulation in Papaver somniferum suspension cultures. Plant Physiol Biochem 48(10):887–892
Huang F-C, Kutchan TM (2000) Distribution of morphinan and benzo [c] phenanthridine alkaloid gene transcript accumulation in Papaver somniferum. Phytochemistry 53(5):555–564
Hubert DA, Tornero P, Belkhadir Y, Krishna P, Takahashi A, Shirasu K, Dangl JL (2003) Cytosolic HSP90 associates with and modulates the Arabidopsis RPM1 disease resistance protein. EMBO J 22(21):5679–5689
Irizarry RA, Hobbs B, Collin F, Beazer‐Barclay YD, Antonellis KJ, Scherf U, Speed TP (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4(2):249–264
Kawalleck P, Plesch G, Hahlbrock K, Somssich IE (1992) Induction by fungal elicitor of S-adenosyl-L-methionine synthetase and S-adenosyl-L-homocysteine hydrolase mRNAs in cultured cells and leaves of Petroselinum crispum. Proc Natl Acad Sci USA 89(10):4713–4717
Komarnisky LA, Christopherson RJ, Basu TK (2003) Sulfur: its clinical and toxicologic aspects. Nutrition 19(1):54–61
Ku KM, Jeffery EH, Juvik JA (2014) Optimization of methyl jasmonate application to broccoli florets to enhance health‐promoting phytochemical content. J Sci Food Agric 94:2090–2096
Lachman J, Hejtmankova A, Miholova D, Kolihova D, Tluka P (2006) Relations among alkaloids, cadmium and zinc contents in opium poppy (Papaver somniferum L.). Plant Soil Environ 52(6):282
Lee E-J, Facchini P (2010) Norcoclaurine synthase is a member of the pathogenesis-related 10/Bet v1 protein family. Plant Cell Online 22(10):3489–3503
Lee E-J, Facchini PJ (2011) Tyrosine aminotransferase contributes to benzylisoquinoline alkaloid biosynthesis in opium poppy. Plant Physiol 157(3):1067–1078
Leustek T, Martin MN, Bick J-A, Davies JP (2000) Pathways and regulation of sulfur metabolism revealed through molecular and genetic studies. Annu Rev Plant Biol 51(1):141–165
Liscombe DK, Facchini PJ (2007) Molecular cloning and characterization of tetrahydroprotoberberine cis-N-methyltransferase, an enzyme involved in alkaloid biosynthesis in opium poppy. J Biol Chem 282(20):14741–14751
Liscombe DK, MacLeod BP, Loukanina N, Nandi OI, Facchini PJ (2005) Evidence for the monophyletic evolution of benzylisoquinoline alkaloid biosynthesis in angiosperms. Phytochemistry 66(11):1374–1393
Mishra S, Triptahi V, Singh S, Phukan UJ, Gupta M, Shanker K, Shukla RK (2013a) Wound ınduced tanscriptional regulation of benzylisoquinoline pathway and characterization of wound ınducible PsWRKY transcription factor from Papaver somniferum. PLoS One 8(1):e52784
Mishra S, Triptahi V, Singh S, Phukan UJ, Gupta MM, Shanker K, Shukla RK (2013b) Wound induced tanscriptional regulation of benzylisoquinoline pathway and characterization of wound inducible PsWRKY transcription factor from Papaver somniferum. PLoS One 8(1):e52784. doi:10.1371/journal.pone.0052784
Mittra B, Ghosh P, Henry S, Mishra J, Das T, Ghosh S, Babu C, Mohanty P (2004) Novel mode of resistance to Fusarium infection by a mild dose pre-exposure of cadmium in wheat. Plant Physiol Biochem 42(10):781–787
Morimoto S, Suemori K, Moriwaki J, Taura F, Tanaka H, Aso M, Tanaka M, Suemune H, Shimohigashi Y, Shoyama Y (2001) Morphine metabolism in the opium poppy and ıts possible physiological function bıochemıcal characterızatıon of the morphıne metabolite, bısmorphıne. J Biol Chem 276(41):38179–38184
Morishige T, Tsujita T, Yamada Y, Sato F (2000) Molecular characterization of the S-adenosyl-l-methionine: 3′-hydroxy-N-methylcoclaurine 4′-O-methyltransferase ınvolved in ısoquinoline alkaloid biosynthesis in Coptis japonica. J Biol Chem 275(30):23398–23405
Nessler CL, Allen RD, Galewsky S (1985) Identification and characterization of latex-specific proteins in opium poppy. Plant Physiol 79(2):499–504
Pieterse CM, Leon-Reyes A, Van der Ent S, Van Wees SC (2009) Networking by small-molecule hormones in plant immunity. Nat Chem Biol 5(5):308–316
Ross J, Li Y, Lim E-K, Bowles DJ (2001) Higher plant glycosyltransferases. Genome Biol 2(2):3004.3001–3004.3006
Samanani N, Liscombe DK, Facchini PJ (2004) Molecular cloning and characterization of norcoclaurine synthase, an enzyme catalyzing the first committed step in benzylisoquinoline alkaloid biosynthesis. Plant J 40(2):302–313
Schaller A, Stintzi A (2009) Enzymes in jasmonate biosynthesis–structure, function, regulation. Phytochemistry 70(13):1532–1538
Tabor CW, Tabor H (1985) Polyamines in microorganisms. Microbiol Rev 49(1):81
Turktas M, Inal B, Okay S, Erkilic EG, Dundar E, Hernandez P, Dorado G, Unver T (2013) Nutrition metabolism plays an Important role in the alternate bearing of the olive tree (Olea europaea L.). PLoS One 8(3):e59876
Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG (2012) Primer3—new capabilities and interfaces. Nucleic Acids Res 40(15):e115
Unterlinner B, Lenz R, Kutchan TM (1999) Molecular cloning and functional expression of codeinone reductase: the penultimate enzyme in morphine biosynthesis in the opium poppy Papaver somniferum. Plant J 18(5):465–475
Unver T, Budak H (2009) Conserved microRNAs and their targets in model grass species Brachypodium distachyon. Planta 230(4):659–669
Unver T, Parmaksiz I, Dundar E (2010) Identification of conserved micro-RNAs and their target transcripts in opium poppy (Papaver somniferum L.). Plant Cell Rep 29(7):757–769. doi:10.1007/s00299-010-0862-4
Wasternack C, Kombrink E (2009) Jasmonates: structural requirements for lipid-derived signals active in plant stress responses and development. ACS Chem Biol 5(1):63–77
Weid M, Ziegler J, Kutchan TM (2004) The roles of latex and the vascular bundle in morphine biosynthesis in the opium poppy, Papaver somniferum. Proc Natl Acad Sci USA 101(38):13957–13962
Winzer T, Gazda V, He Z, Kaminski F, Kern M, Larson TR, Li Y, Meade F, Teodor R, Vaistij FE (2012) A Papaver somniferum 10-gene cluster for synthesis of the anticancer alkaloid noscapine. Science 336(6089):1704–1708
Ye K, Ke Y, Keshava N, Shanks J, Kapp JA, Tekmal RR, Petros J, Joshi HC (1998) Opium alkaloid noscapine is an antitumor agent that arrests metaphase and induces apoptosis in dividing cells. Proc Natl Acad Sci USA 95(4):1601–1606
Zheng Z, Wu M (2004) Cadmium treatment enhances the production of alkaloid secondary metabolites in Catharanthus roseus. Plant Sci 166(2):507–514
Ziegler J, Facchini PJ, Geißler R, Schmidt J, Ammer C, Kramell R, Voigtländer S, Gesell A, Pienkny S, Brandt W (2009) Evolution of morphine biosynthesis in opium poppy. Phytochemistry 70(15):1696–1707
Ziegler J, Voigtländer S, Schmidt J, Kramell R, Miersch O, Ammer C, Gesell A, Kutchan TM (2006) Comparative transcript and alkaloid profiling in Papaver species identifies a short chain dehydrogenase/reductase involved in morphine biosynthesis. Plant J 48(2):177–192
Ziska LH, Panicker S, Wojno HL (2008) Recent and projected increases in atmospheric carbon dioxide and the potential impacts on growth and alkaloid production in wild poppy (Papaver setigerum DC.). Clim Chang 91(3–4):395–403
Zulak KG, Khan MF, Alcantara J, Schriemer DC, Facchini PJ (2009) Plant defense responses in opium poppy cell cultures revealed by liquid chromatography-tandem mass spectrometry proteomics. Mol Cell Proteomics 8(1):86–98
Acknowledgments
This study was supported by Gaziosmanpaşa University (grant no: 212/47), the Ministry of Development (grant no: DPT2010K120720), and TUBITAK (grant no: 109O661 and 111O036).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Figure S1a
(XLSX 74 kb)
Figure S1b
(XLSX 81 kb)
Figure S1c
(XLSX 66 kb)
Figure S1d
(XLSX 58 kb)
Figure S1e
(PDF 1104 kb)
Figure S1f
(XLSX 70 kb)
Table S1
(XLSX 173 kb)
Table S2
(XLSX 119 kb)
Table S3
(XLSX 16 kb)
Table S4
(DOC 31 kb)
Rights and permissions
About this article

Cite this article
Gurkok, T., Turktas, M., Parmaksiz, I. et al. Transcriptome Profiling of Alkaloid Biosynthesis in Elicitor Induced Opium Poppy. Plant Mol Biol Rep 33, 673–688 (2015). https://doi.org/10.1007/s11105-014-0772-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-014-0772-7