
Abstract
microRNAs (miRNAs), endogenous non-coding RNAs of approximately 21–24 nucleotides, are important regulators of transcriptional and post-transcriptional gene expression. These regulators play a key role during plant growth and development, including embryogenesis, which is crucial to the life cycle of most plant species. However, although embryogenesis-associated miRNAs have been isolated in a few species, the diversity of these regulatory miRNAs remains largely unexplored, especially in radish. In this study, two small RNA libraries were constructed from radish ovules before and after fertilization. Both libraries were sequenced by next generation sequencing (NGS) technology. This analysis identified 144 conserved and 34 non-conserved miRNAs (representing 43 known miRNA families) and 38 novel miRNAs (representing 28 miRNA families). Comparative analysis revealed that 29 known and 10 novel miRNA families were differentially expressed during embryogenesis. QRT-PCR analysis confirmed miRNA expression patterns and revealed tissue-specific and/or developmental stage-dependent expression for some miRNAs. Moreover, potential target predictions indicated that most of these targets were transcription factors involved in regulating plant growth, development and metabolism. Notably, target transcripts such as squamosa promoter-binding protein, auxin response factor and agamous-like MADS-box protein participated in radish embryogenesis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Embryogenesis represents a core stage in the life cycle of more than 80 % of diverse green plants (Zhang and Ogas 2009; Xiang et al. 2011). This program plays an important role in defining many key aspects of seed formation and diversity, and impacts multiple agronomic traits (Goldberg et al. 1994; Braybrook and Harada 2008). Embryogenesis consists of two distinct phases: early morphological events and late maturation (Yazawa et al. 2004; Zhang et al. 2012). Research within the last decade has demonstrated that several key developmental processes condensed in early embryogenesis can be considered a miniature model for plant development (Palovaara et al. 2013). So far, however, the molecular events related to plant early embryogenesis are largely unknown compared with animals. This lag in research is partly due to the poor accessibility and tiny size of embryos in plants (Xiang et al. 2011; Palovaara et al. 2013).
MicroRNAs (miRNAs), approximately 21–24 nucleotides in length, are a class of small, endogenous, non-coding single-stranded RNA molecules. miRNAs are processed from larger RNA hairpin precursors cleaved by the RNase III Dicer-like1 (DCL1) and bind complementary target mRNAs that directly or indirectly regulate specific target messenger RNA in both animals and plants (Voinnet 2009; Chorostecki et al. 2012). Arising from the discovery that miR398 functioned in plant stress tolerance (Sunkar and Zhu 2004; Sunkar et al. 2006), increasing evidence suggests that miRNAs have emerged as effective regulators of numerous physiological and developmental processes during the life cycle of plants, including the development of vegetative organs (Coudert et al. 2010; Li et al. 2011) and reproductive organs (Xue et al. 2009; Gao et al. 2012), hormone regulation (Liu et al. 2007; Meng et al. 2009), nutrient homeostasis (Martin et al. 2010; Wang et al. 2012c), stress response (Sunkar 2010; Xu et al. 2013b; Zhang et al. 2013), and self-regulation in the miRNA biogenesis pathway (Carrington and Ambros 2003; Yan et al. 2012).
Recently, with the development of the next-generation sequencing (NGS) technology, an increasing number of miRNAs, particularly some non-conserved, low-abundance miRNAs were identified in several plant species (Mao et al. 2012; Wang et al. 2012a, c; Yang et al. 2013a). To date, more than 7,000 miRNAs in diverse plant species have been released in the miRBase 20.0 database (http://www.mirbase.org/). Several conserved miRNAs have been reported to participate specifically in embryogenesis. For example, certain miRNA-mediated genes may play a critical role in the morphogenesis stages of embryogenesis in Arabidopsis (Willmann et al. 2011), cotton (Yang et al. 2013b), sweet orange (Wu et al. 2011), longan (Lin and Lai 2013), larch (Zhang et al. 2012), and loblolly pine (Oh et al. 2008). Although previous studies provide valuable information for roles of miRNAs during embryo morphogenesis and maturation, our understanding of the complex miRNA-mediated regulatory networks throughout plant embryogenesis is still in its infancy.
Radish, Raphanus sativus L. (2n = 2x = 18), is a major annual or biennial root vegetable crop of the Brassicaceae family. Recently, a total of 48 conserved miRNAs representing 9 miRNA families were isolated from expressed sequence tags (EST) of R. sativus by a comparative analysis approach (Muvva et al. 2012). Using high-throughput sequencing, Xu et al. (2013a) identified 546 conserved miRNA families as well as 15 novel miRNAs from a radish root library. Despite the importance of the embryo, the major component of seed, for genetic breeding, little is known of the miRNA-modulated embryogenesis processes in radish. To elucidate the roles of miRNAs regulating radish embryogenesis, we constructed two libraries, one at 0 day after pollination (DAP) and 4-15 DAP library. We employed a next generation sequencing (NGS) platform (Solexa sequencing) to generate millions of short reads with a high level of accuracy. In the present study, known and novel miRNA families were isolated from the two ovules libraries of radish, some of which showed significant expression during radish embryogenesis. Additionally, potential target genes were predicted by KOBAS 2.0 and validated by qRT-PCR technology. Together, the large-scale identification of embryogenesis-specific miRNAs and their targets enhanced our understanding of gene expression patterns and provided new regulatory insights for embryogenesis of radish. Data obtained here will provide a solid foundation for further investigation of molecular mechanisms of plant embryogenesis and seed development.
Materials and Methods
Plant Materials
After vernalization treatment, a radish (R. sativus L.) advanced inbred line ‘NAU-DY13’ was grown in a growth chamber with a 16-h light at 26 °C/8-h dark at 20 °C cycle for 1 week. To examine the tissue-specific expression patterns of miRNAs, the petals, calyxes, anthers, and zygotic embryos were harvested separately after pollination and immediately frozen in liquid nitrogen and stored at −80 °C for further use.
Small RNA Library Construction and High-Throughput Sequencing
Total RNAs from ovules of radish were isolated using Trizol® Reagent (Invitrogen) following the manufacturer’s instructions. The two sRNA libraries [pre-pollinated (0_DAP library) and pollinated (4-15_DAP library) ovules] were prepared according to reported procedures (Hafner et al. 2008; Xu et al. 2013a, b). In brief, the small RNA fragments with a length of 18–30 nt were separated and purified by polyacrylamide gel electrophoresis, and subsequently ligated to Solexa adapters at each end. Assembled small RNAs were reverse transcribed to single-stranded cDNA using SuperScript II Reverse Transcriptase (Invitrogen). Finally, deep sequencing was carried out on the Illumina Genome Analyzer II (Illumina, San Diego, USA).
Analysis of Small RNA Sequencing Data and Identification of miRNA Precursors
Small RNA reads were obtained from Illumina HiSeq™ analysis. The clean reads were screened from raw data by filtering low quality reads, trimming sequences with 5′-primer contaminants or with poly-A tails, and removing reads smaller than 15 nt or longer than 30 nt. The unique RNAs were mapped to the radish reference sequences containing genomic survey sequences (GSS), EST sequences, and the mRNA transcriptome sequences (Li et al. 2009; Xu et al. 2013b). Sequences with a perfect match along their entire length were retained for subsequent analysis.
By performing a BLASTn search against known non-coding RNAs deposited in the GenBank and the Rfam (RNA family) database (Xu et al. 2013b), the sequences having an exact match with rRNAs, tRNAs, snRNAs, and snoRNAs were identified and excluded from further analysis (Wei et al. 2011; Xu et al. 2013a; Yang et al. 2013b). The remaining unique sequences were aligned with known miRNAs from miRBase 20.0 (http://www.mirbase.org/index.shtml) with a maximum of two mismatches. Unannotated reads were predicted for novel miRNAs using Mireap software (https://sourceforge.net/projects/mireap/). The criteria by Meyers et al. (2008) were used for screening the candidate novel miRNAs. Secondary structures of candidate miRNA precursors were confirmed by Mfold software (Xu et al. 2013b; Yang et al. 2013b).
Differential Expression Analysis of miRNAs During Embryogenesis in Radish
The expression abundance of miRNAs in two libraries was normalized to one million by total clean reads of miRNAs per sample (Gao et al. 2012). The differential expression of miRNAs between the two libraries was calculated as: Fold-change = log2 (miRNA normalized reads in 4-15_DAP/miRNA normalized reads in 0_DAP). The P value was calculated based on previously established methods (Li et al. 2009). The miRNAs with P ≤ 0.05 and relative ratios (4-15_DAP/0_DAP) greater than 2 or less than 0.5, along with, were considered up-regulated or down-regulated during embryogenesis, respectively.
Prediction and Annotation of Potential Targets by Bioinformatics Methods
The potential targets of radish miRNAs were predicted by the plant small RNA target analysis server as described by Dai and Zhao (2011) and Barvkar et al. (2013). To understand the potential functions of the miRNA-targeted genes, gene ontology (GO) annotations were assigned using the Blast2GO program (Galla et al. 2009; Zhai et al. 2013). The target sequences were allocated to the corresponding functional categories on the basis of the BLAST searches by GO annotation, using default parameters. GO enrichment analysis provided an effective method to predict candidate genes targeted by novel miRNAs. Functional relationships between predicted genes and novel miRNAs were determined using the following formula, where N represents the number of all successfully annotated genes; M represents the number of all genes mapped to a certain GO term; and n and m represent the number of target gene candidates in N and M, respectively.
in which, GO terms with corrected p-value ≤ 0.05 were defined as significantly enriched in candidate genes and the enrichment value of gene number from GO basic analysis was used to describe the enrichment level for each GO term. Additionally, KEGG Orthology Based Annotation System (KOBAS 2.0; http://kobas.cbi.pku.edu.cn/home.do/) was employed to predict the biological functions of genes (Xie et al. 2011).
qRT-PCR Validation of miRNAs and Their Potential Target Genes
Quantitative real-time PCR (qRT-PCR) was employed to experimentally validate the results by high-throughput sequencing of radish miRNAs and their targets. Total RNAs were obtained from five radish samples (0, 4, 8, 15, and 30 DAP) as described above using Trizol according to the manufacturer’s protocol. RNA samples were reverse-transcribed according to previous reports (Xu et al. 2013b; Yang et al. 2013b). All reactions were performed on an iCycler iQ real-time PCR detection system (BIO-RAD) with three replications, which were carried out in a total volume of 20 μl, including 0.2 μM primer pairs, 2 μl diluted cDNA, and 10 μl 2 × SYBR Green PCR Master Mix (TaKaRa Bio Inc., Dalian, China). The miRNA amplification reactions were incubated at 95 °C for 30 s, followed by 45 cycles of 95 °C for 5 s, 58 °C for 15 s, and 72 °C for 20 s. The primers for the miRNAs and their targets are shown in Supplementary Table S1. The 5.8S rRNA was used as the reference gene (Xu et al. 2013a).
Results
Global Analysis of Sequences from Small RNA Libraries
To characterize differences in zygotic embryogenesis at the small RNAome level and to identify known and novel miRNAs in radish embryos, we constructed small RNA libraries at 0_DAP and 4-15_DAP. In total, approximately 29.08 million raw reads representing 4.66 million unique sequences were generated by the Solexa sequencing technology (Supplementary Table S2). After filtering low quality reads and trimming adaptor/acceptor contaminations, 13,868,010 clean tags remained from the 0_DAP library and 15,209,357 remained from the 4-15_DAP library. Of these reads, 10.97 % were 0_DAP library-specific with 38.22 % unique sRNAs, 11.77 % were 4-15_DAP library-specific with 48.61 % unique sRNAs, and 77.26 % were present in both with 13.17 % unique sRNAs (Supplementary Fig. S1). In total, 935,713 (6.74 %) and 950,747 (6.25 %) sequences were found to be similar to known miRNAs (Supplementary Table S2).
Thereafter, sequences ranging from 15 to 30 nt in length were extracted to analyze the small RNA length distribution in radish (Fig. 1). The length distribution of the sRNAs was non-uniform in each library, and high abundance in 21–24 nt reads was observed, which accounted for 94.76 % and 93.76 % reads in the 0_DAP and 4-15_DAP libraries, respectively. In both libraries, the 24-nt reads were the abundant read length (70.16 % and 43.17 %, respectively). The small RNAs were further analyzed by BLASTn to filter rRNA, snRNA, snoRNA and tRNA, making up 648,272 reads (67,440 unique sequences, 4.68 %) in the 0_DAP library and 689,428 reads (74,656 unique sequences, 4.53 %) in the 4-15_DAP library (Table 1), respectively. By comparing these reads with all precursors and mature sequences of miRNAs released in miRBase 20.0 in plant, a total of 7,022 (0_DAP) and 11,164 (4-15_DAP) miRNA analogous sequences were screened out. The remaining 2,308,286 (0_DAP) and 2,773,122 (4-15_DAP) unannotated unique fragments were further screened for novel miRNAs.

Length distribution of small RNAs in radish ovule libraries at 0_DAP and 4-15_DAP
Identification of Known miRNA Families During Radish Embryogenesis
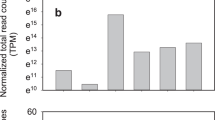
From the identified 7,022 (0_DAP) and 11,164 (4-15_DAP) known individual candidate miRNAs (Table 1), a BLASTn search against known miRNAs from various species resulted in identification of 178 sequences belonging to 23 conserved and 20 non-conserved families in both libraries (Table 2; Supplementary Table S3). Most of the identified miRNA families such as miR156/157, miR159, miR165/166, miR167, and miR169 are highly conserved in a variety of plant species (Lin and Lai 2013; Mao et al. 2012). In this study, they were also predicted in radish miRNA dataset containing 16, 12, 18, 9, and 16 members, respectively; whereas miR394 and miR397 had only one member in conserved miRNA families (Fig. 2a). Furthermore, different miRNA subsets marked as lowercase were in each conserved (Fig. 2b) and non-conserved miRNA family. In addition, as expected, some known but non-conserved miRNAs (miR477, 535, 827, 829, 2,111, and 2,118) were identified in our dataset, which have been previously reported in only a few plant species.

Sizes and abundance of identified known miRNA families from radish. a Distribution of known miRNA family size in radish. b Complexity of the conserved miRNA family members identified in radish. c Counts of each known miRNA family in radish (Raphanus sativus L.)
Next generation sequencing technology offers an effective way to estimate the expression abundance of miRNAs. The number of miRNA reads in the library is recognized as an index for estimating the relative abundance of miRNAs. In this study, the read number varied from one to 450,617, and the expression levels of several miRNA families (for instance, miR156/157, miR165/166, and miR167) were extremely high, with miR167 being the most abundant in both libraries (Fig. 2c). In contrast, a few miRNA families including miR394 and miR395 displayed very low levels of expression, with no more than 20 reads in conserved miRNA families (Table 2). Interestingly, different members in the same miRNA family also showed considerably variable expressional levels (Supplementary Table S3).
Identification of Novel Candidate miRNAs During Radish Embryogenesis
In addition to the identification of known miRNAs, 28 predicted novel radish miRNA families representing 38 unique miRNA sequences were also identified (Table 3, Supplementary Table S4). Only one member was identified in the majority of these novel miRNA families, whereas rsa-miRn3, rsa-miRn5, rsa-miRn6, rsa-miRn12, rsa-miRn13, rsa-miRn22, rsa-miRn23, and rsa-miRn28 (“rsa” represented R. sativus) generated more than one locus (Supplementary Table S4). Furthermore, compared with the conserved miRNAs, the relative expression abundance of novel miRNAs was much lower in both libraries.
Candidate miRNA precursors were predicted and identified, with minimum free energy (MFE) value ranging from −93.6 to −20 kcal mol−1 of secondary hairpin structures and an average length of 146 nt. According to the strict criteria for the annotation of novel miRNAs (Meyers et al. 2008; Li et al. 2011; Chen et al. 2012), 10 novel miRNAs were detected. Precursor structures for these miRNAs are shown in Supplementary Fig. S2. To investigate the homology of these 10 novel miRNAs in other plant species, the sequences were employed in BLASTn searches in National Center for Biotechnology Information (NCBI) databases. With the exception of rsa-miRn7, rsa-miRn16, and rsa-miRn18, no mismatches were found, indicating that these three novel miRNAs are all radish-specific.
Identification of Embryogenesis Related miRNAs in Radish
To gain deeper insight into the roles of miRNAs relevant to embryogenesis in radish, we performed differential expression pattern analysis of common and unique miRNAs. The miRNAs presented in Fig. 3 were considered up-regulated or down-regulated between the two libraries. As shown, a total of 46 known miRNAs and 10 potentially novel miRNAs were differentially expressed during embryogenesis (Supplementary Table S5). Among them, nearly two-thirds of the miRNAs related to embryogenesis showed down-regulated patterns contrasted to miRNAs in the 0_DAP library. In contrast, 16 known miRNAs and four predicted novel miRNAs (rsa-miRn16, rsa-miRn19, rsa-miRn20, and rsa-miRn26) showed contrary expression patterns (Fig.4a, b). Of these, eight miRNAs were differentially expressed at a ratio greater than 10-fold, including one predicted novel miRNA. A total of four miRNAs (miR159b, miR396d, miR825-5p, and miR845a-3p) were detected only in the 0_DAP library, whereas miR169r-3p, miR395k-3p, miR535b, and rsa-miRn19 were identified only in the 4-15_DAP library, indicating these miRNAs might be induced or repressed at a specific period of embryo formation. The two miRNAs showing the greatest change in expression levels were miR396d (17.31-fold) and miR169r-3p (14.58-fold), each expressed exclusively in only one of the two libraries, providing further evidence for the temporal role of miRNAs in radish embryogenesis (Supplementary Table S5).

Validation and comparative relative expression of differentially-expressed known (a) and novel (b) microRNAs between radish ovule libraries at 0_DAP and 4-15_DAP

GO enrichment multiple analysis in the classification of biological process
Prediction and Annotation of miRNA Targets in Radish
For a better understanding of the biological functions of these miRNAs, it is necessary to identify the putative target sites of the miRNA candidates. In this study, blast2go analysis predicted 3,746 targeted sequences for 169 known and novel miRNAs from the transcripts of R. sativus libraries, with a location number of 7,711 (Supplementary Table S6). According to the BLASTX results, sequences corresponding to the putative targets were successfully associated with GO terms (Supplementary Fig. S3). Additionally, GO enrichment analysis facilitated the reorganization of the main biological functions that target genes acted (Fig. 4). The enrichment of the genes participated in various metabolic and reproductive processes including embryonic meristem development (GO: 0048508), embryo development ending in seed dormancy (GO: 0009793) and embryo development (GO: 0009790).
In addition, the miRNA targeted genes during embryogenesis of radish were further investigated to better understand the functions of these target genes. As a result, 56 and 10 target genes were identified for 22 annotated known conserved miRNA families and six novel miRNAs, respectively (Table 4, Supplementary Table S7-1). The prediction details of non-conserved miRNAs are shown in Supplementary Table S7-2. The results indicated that the majority of targets are known transcription factors such as Squamosa promoter-binding protein (SPL, targeted by miR156/157), Myb domain protein 101 (MYB101, cleaved by miR159), Auxin response factor (ARF) family (regulated by miR 160/161, 167), Homeobox-leucine zipper protein (HD-ZIP protein, sliced by miR165), Agamous-like MADS-box protein 12 (AGL12, targeted by miR164), Argonaute (AGO, cleaved by miR403), APETALA2-like protein (AP2, targeted by miR172), as well as Laccase (LAC, regulated by miR397) family transcription factor (Supplementary Table S7-1). Moreover, a few transcripts were annotated to genes involved in response to biotic and abiotic stress. For instance, miR160/161 targeted aluminum-activated malate transporter 9 (ALMT9) and miR164 regulated ethylene-responsive transcription factor ERF073 (HRE1). In general, target annotation indicated that the putative genes were involved in a wide range of plant biological processes, such as regulation of metabolism, signal transduction, and phytohormone response.
Validation of miRNAs and Their Target Genes Through qRT-PCR
To validate the dynamic expression pattern of miRNAs by deep sequencing and their putative targets and to obtain further knowledge about the possible functions of miRNAs, a total of six known miRNAs and 11 corresponding targets were randomly examined during five developmental stages of embryos and in various floral tissues by qRT-PCR. The expression patterns of the six miRNAs in various tissues are shown in Fig. 5a. These include miR156c, miR167a, miR390b, and miR397a, which had a similarly high expression pattern in embryo. By comparison, the expression of miR159b was most abundant in the calyx, with expression levels gradually declining in the embryo, petal and anther, respectively. miR390a appeared to be primarily restricted to the anther apparently, but expressed weakly in the embryo.

qRT-PCR validation of known miRNAs and their putative targets. Small RNAs (<200 nt) were extracted from various radish floral tissues (petal, calyx, anther, and embryo) and ovules at different days after pollination (0, 4, 8, 15, and 30 DAP). Relative expression levels of miRNAs and targets were calculated using 5.8S rRNA and Actin gene as a control, respectively. Each bar shows the mean ± SE of triplicate assays
Among these miRNAs, miR390a and miR390b, both belonging to the same family, showed differential expression over five developmental stages. Moreover, the expression of three miRNAs, miR156c, 159b, and miR397a, reached a peak at 4 DAP; conversely, their target genes show the lowest expression level at this stage (Fig. 5b, c, d, and f). Similarly, miR167a expressed relatively high levels at 30 DAP (Fig. 5e), showing an inverse relation between miR167a and its corresponding target transcripts (ARF8, gi|332783910|gb|FY434046.1|FY434046; ARF8-1, Unigene24299_CKA; glycoside hydrolase family 47 protein, CL5433.Contig1_CKA), suggesting miRNA-mediated regulation of their potential targets. Overall, the results from comparisons of the expression levels in ovules pre- and post-pollinated at three stages of 4, 8, and 15 DAP corresponded closely with results from deep sequencing, implying that the miRNA expression levels detected by high-throughput RNA-seq technology are reliable. These observations indicated that most miRNAs in radish were expressed preferentially in one or two floral tissues and that some miRNAs might participate in the process of embryo morphogenesis and seed development.
Discussion
Embryogenesis represents a key phase in the life cycle in plants (Goldberg et al. 1994; Braybrook and Harada 2008; Palovaara et al. 2013). As regulators of gene expression, miRNAs are widely present in plants, and their diverse functions have been intensively studied in many plant species (Liang et al. 2010; Song et al. 2011). So far, however, no studies have been performed on the miRNA regulatory networks during radish embryogenesis. To reveal the molecular basis of miRNA-mediated gene regulation during embryogenesis in radish, two small RNA libraries from ovules before and after pollination were constructed by a high-throughput sequencing strategy. Some conserved and novel miRNAs involved in radish embryogenesis were identified. In addition, many known or putative potential target genes involved in embryogenesis of radish were predicted by bioinformatics approaches. To our current knowledge, this study is the first report on identification and characterization of miRNAs during embryogenesis in radish.
Characteristics of R. sativus miRNAs Related to Embryogenesis
Identification of a comprehensive set of miRNAs during embryogenesis is a critical step to facilitate our understanding of miRNA-mediated regulatory mechanisms of embryo formation and development. In this study, the length distribution showed that the 24 nt length class dominated in the dataset followed by 21 nt miRNAs, in accordance with the observation reported in some other species such as Arabidopsis, Oryza sativa, and Medicago truncatula (Voinnet 2009; Eyles et al. 2013; Ma et al. 2013). Recently, multiple studies have shown that the majority of known miRNAs in the plant kingdom are evolutionarily conserved (Chen et al. 2012; Barvkar et al. 2013), most of which are represented with moderately or highly expressed reads and some of which have conserved functions (Wei et al. 2013; Xu et al. 2013a). In the current study, the reads of several conserved miRNAs were sequenced more than 10,000 times in 0_ and/or 4-15_DAP libraries, such as miR156/157, miR158, and miR165/166 (Table 2), whereas the majority of non-conserved miRNAs showed relatively low reads with no more than 100, inferring that conserved miRNAs exhibited relatively higher expression abundance compared with non-conserved counterparts (Wang et al. 2012b). So far, only a few studies of small RNAs have been carried out in radish (Muvva et al. 2012; Xu et al. 2013a), and novel miRNAs involved in the process of embryogenesis have not been discovered.
An increasing number of studies have reported a number of conserved and non-conserved miRNAs regulated differentially during embryogenesis in Arabidopsis (Williams et al. 2005; Rodriguez et al. 2010; Ágyi and Havelda 2013). In Arabidopsis, Williams et al. (2005) demonstrated that miR166 with dynamic expression pattern is required for meristem formation during embryogenesis. Using genome-wide transcript profiling, Nodine and Bartel (2010) found that miR156 showed high activity throughout early embryogenesis, while the expression level was reduced at the late development stage. Subsequently, it was reported that Arabidopsis embryos mutant for the strong allele of DICER-LIKE1 matured much earlier, indicating that miRNAs are key regulators of the timing of the maturation program during early embryogenesis (Willmann et al. 2011). Moreover, Ágyi and Havelda (2013) employed in situ hybridization technique to assess the expression level and the mode of ten miRNAs (miR158, miR160, miR161, miR164, miR166, miR167, miR168, miR169, miR396, and miR472), and found that the majority of miRNAs showed uniform accumulation across the embryo. However miR167 showed a gradient-like expression profile. In the present investigations, by high-throughput sequencing technology, a total of 29 known miRNAs (Supplementary Table S5) were identified to be differentially expressed during radish embryogenesis. For instance, miR167, miR319, miR393, miR395, miR397, and miR408 were up-regulated, whereas miR164, miR171, and miR400 were down-regulated. As with the previous study in Arabidopsis, miR156, miR158, miR164, miR167, and miR169 showed high abundance during radish embryogenesis, suggesting these miRNAs were involved in the regulatory networks during embryogenesis. However, it was found that several reported miRNA families (miR160, miR162, miR166, miR172, and miR827) did not change significantly during embryogenesis in radish, although some of them were reported to control basic development in Arabidopsis (Williams et al. 2005; Wu et al. 2009). Furthermore, through comparative analysis of other investigated known miRNAs, it was noticed that miR169r, miR395k, miR396a, miR535b, and miR829 were specific to the 4-15_DAP library with a relatively high expression level. Some non-conserved miRNAs, such as miR829 and miR857, significantly increase in expression during radish embryogenesis, but were not previously identified during Arabidopsis embryogenesis, indicating that these miRNAs may be responsible for multiple embryonic patterning events in radish embryo morphogenesis and maturation. Nevertheless, further studies are still needed to clarify regulation mechanism associated with various miRNAs during radish embryogenesis.
Expression Patterns of miRNA Based on qRT-PCR
By analyzing the functions of the miRNAs and their target genes according to their temporal and spatial expression patterns, we were able to infer possible roles of miRNAs and their targets in plants (Gao et al. 2012; Mao et al. 2012). QRT-PCR based differential expression analysis of six miRNA families indicates potentially diverse regulatory roles in different developmental stages of embryogenesis and floral tissues in radish. Many miRNAs displayed temporal or tissue-specific expression patterns, which were consistent with some previous studies (Rubio-Somoza et al. 2009; Lan et al. 2012). It was reported that miR156, one of the largest miRNA families in plants, plays pivotal regulatory roles during the phase of juvenile-to-adult transition by targeting SPL genes in Arabidopsis (Wu et al. 2009; Li et al. 2012) and Brassica napus (Xu et al. 2012). Similarly, in the present study, miR156c expressed the highest level at 4 DAP, suggesting that miR156c was interrelated to early embryonic developmental stages. This result is consistent with several previous studies (Wu et al. 2011; Yang et al. 2013b). In addition, we found miR156c was predominantly expressed in the embryo compared with petal, calyx, and anther, implying that miR156c may be embryo specific in radish as reported in B. napus (Huang et al. 2013).
Recently, miR159, miR166, mir167, miR390, and miR397 were studied in some other species with slightly different roles during embryogenesis (Williams et al. 2005); Jung and Park 2007; Buxdorf et al. 2010; Zhang et al. 2012). As expected, miR159b, miR390b, and miR397a transcripts accumulated during early stages of radish zygotic embryo morphogenesis, and miR167a expressed at relatively high levels during middle and late embryo development, consistent with findings in Arabidopsis (Ágyi and Havelda 2013), orange and longan (Wu et al. 2011; Lin and Lai 2013). In larch or other gymnosperms, miR156, miR159, and miR390 play regulatory roles during cotyledonary embryo development rather than in the early phase of embryo formation, which were distinct from those of angiosperms (Oh et al. 2008; Zhang et al. 2012). Moreover, the majority of the miRNAs and their corresponding target genes showed negative correlation by qRT-PCR, indicating that these miRNAs likely regulate their corresponding predicted target genes (Yang et al. 2013a, b; Barvkar et al. 2013). Interestingly, in many cases, different targets of the same miRNA were up-regulated while others were expressed inversely, further suggesting that the regulation of the steady-state levels of miRNA targets is complex, as suggested previously (Willmann et al. 2011). The miRNA-target pair experiments further inferred that the miRNAs mentioned above may be crucial and major contributors to the network regulating embryogenesis and seed development in radish.
Functions of Predicted Target Genes During Radish Embryogenesis
The high-throughput sequencing technology and bioinformatics analysis provide us reliable and efficient approaches for massive data message of miRNAs and their potential targets with possible functions during embryogenesis in several plant species (Zhang et al. 2012; Lin and Lai 2013; Yang et al. 2013b). However, only a few target genes correlated to embryogenesis have been predicted and confirmed experimentally in vegetable crops (Braybrook and Harada 2008; Nodine and Bartel 2010).
A number of miRNA target genes were predicted in this study, and many of them encode members of large families of transcription factors including MYBs, SPLs, AP2, and NAC-domain proteins. Additionally, some miRNA targets encode enzymes and hormone receptors like ARFs (Table 4, Supplementary Table S7). All these target genes were highly conserved among plant species (Nonogaki 2010; Wang et al. 2012b; Eyles et al. 2013). In Arabidopsis, SPL transcription factors targeted by miR156 were involved in a broad range of developmental processes including flowering (Yu et al. 2012), shoot maturation (Nonogaki 2010), male fertilization (Xing et al. 2010) and the early stage of development (Nonogaki 2010). In addition, SPL genes perform significant functions in regulating embryogenesis and embryo development (Nodine and Bartel 2010; Willmann et al. 2011). In the present study, three miRNAs (miR156/157, miR159 and rsa-miRn12) targeted SPL3 and SPL13 transcription factors belonging to two different classes of the miR156-mediated SPL gene family, differing slightly from results observed in Arabidopsis (Nodine and Bartel 2010; Wu et al. 2009). As previously illustrated, the former was able to accelerate the transition from juvenile- to adult-phase and promote flowering (Nonogaki 2010; Li et al. 2012), whereas the SPL13 seemed to negatively regulate shoot apical meristem activity and leaf primordial development (Nonogaki 2010), suggesting that these miRNAs targeted different SPL-regulated genes and may control various morphological traits, such as embryogenesis in radish.
Moreover, another miRNA (miR172) targeted by AP2 could be activated by the upstream actor miR156 (targeted by SPLs) forming a miRNA gene regulation cascade and functioning to regulate the transitions between developmental stages and to specify floral organ identity governed by complex regulatory systems (Wang et al. 2009; Zhu and Helliwell 2011). In addition, miR397 target laccases were associated to lignifications and thickening of the cell wall in secondary cell growth (Zhang et al. 2012). Previous studies indicated abundant and widespread expression of miR159 in plants (Barvkar et al. 2013) and it targeted mostly the MYB transcription factors, among which MYB101 and MYB33 were demonstrated as positive regulators of ABA signaling during germination (Reyes and Chua 2007).
Plant growth regulators (PGRs), ARFs and Aux/IAAs in auxin particularly, are widely considered to be important during embryogenesis in plants (Rademacher et al. 2012; Yang et al. 2012). As illustrated in the present study, miR160/161 and miR167 participate in the auxin signal transduction by regulating the expression of ARF8, ARF16, and ARF17 in accordance with those in O. sativa (Lan et al. 2012), cotton (Wang et al. 2012d), M. truncatula (Eyles et al. 2013) and tomato (Lopez-Gomollon et al. 2012). In plants, ARF8 can modulate the transcription of certain GH3-like genes balancing the level of active 3-indole acetic acid (IAA). Moreover, the miR390, as a trigger of trans-acting short-interfering RNAs (tasiARFs), is also an auxin-sensitive miRNA controlling the growth of the newly formed primordia (e.g. lateral root initiation and embryogenesis) (Marin et al. 2010; Wan et al. 2012) and is involved in the auxin-signalling pathway in rice (Meng et al. 2009). In this study, miR390b was found to be highly expressed at 4 DAP, indicating that it functions during the early stages of radish embryogenesis. Although further analysis of the interrelation between miRNAs and targets are still required, the results could serve as the basis for further in-depth studies of plant miRNAs involved in auxin signaling during plant embryogenesis.
Conclusion
In conclusion, application of small RNA sequencing technology and bioinformatics provides for the first time an extensive perspective of miRNAs and target genes involved in radish embryogenesis. Sequence analyses and qRT-PCR validation revealed that miR156c, miR159b, miR390b and miR397a function at an early stage and are potentially significant to radish embryogenesis. Moreover, gene ontology categorization and enrichment analysis of the differentially expressed genes demonstrates that a number of transcription and regulatory factors are required for radish embryogenesis. This investigation represents a significant step towards illuminating the roles of miRNAs and advancing our understanding of target gene regulation during embryogenesis and seed development in radish.
References
Ágyi Á, Havelda Z (2013) Analysis of gradient-like expression of miR167 in Arabidopsis thaliana embryonic tissue. J Plant Biology 56:336–344
Barvkar VT, Pardeshi VC, Kale SM, Qiu S, Rollins M, Datla R, Gupta VS, Kadoo NY (2013) Genome-wide identification and characterization of microRNA genes and their targets in flax (Linum usitatissimum): Characterization of flax miRNA genes. Planta 237:1149–1161
Braybrook SA, Harada JJ (2008) LECs go crazy in embryo development. Trends Plant Sci 13:624–630
Buxdorf K, Hendelman A, Stav R, Lapidot M, Ori N, Arazi T (2010) Identification and characterization of a novel miR159 target not related to MYB in tomato. Planta 232:1009–1022
Carrington JC, Ambros V (2003) Role of microRNAs in plant and animal development. Science 301:336–338
Chen L, Ren Y, Zhang Y, Xu J, Sun F, Zhang Z, Wang Y (2012) Genome-wide identification and expression analysis of heat-responsive and novel microRNAs in Populus tomentosa. Gene 504:160–165
Chorostecki U, Crosa VA, Lodeyro AF, Bologna NG, Martin AP, Carrillo N, Schommer C, Palatnik JF (2012) Identification of new microRNA-regulated genes by conserved targeting in plant species. Nucleic Acids Res 40:8893–8904
Coudert Y, Perin C, Courtois B, Khong NG, Gantet P (2010) Genetic control of root development in rice, the model cereal. Trends Plant Sci 15:219–226
Dai X, Zhao PX (2011) psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39:W155–W159
Eyles RP, Williams PH, Ohms SJ, Weiller GF, Ogilvie HA, Djordjevic MA, Imin N (2013) microRNA profiling of root tissues and root forming explant cultures in Medicago truncatula. Planta 238:91–105
Galla G, Barcaccia G, Ramina A, Collani S, Alagna F, Baldoni L, Cultrera NG, Martinelli F, Sebastiani L, Tonutti P (2009) Computational annotation of genes differentially expressed along olive fruit development. BMC Plant Biology 9:128
Gao Z, Shi T, Luo X, Zhang Z, Zhuang W, Wang L (2012) High-throughput sequencing of small RNAs and analysis of differentially expressed microRNAs associated with pistil development in Japanese apricot. BMC Genomics 13:371
Goldberg RB, Gd P, Yadegari R (1994) Plant embryogenesis: zygote to seed science. Science 266:605–614
Hafner M, Landgraf P, Ludwig J, Rice A, Ojo T, Lin C, Holoch D, Lim C, Tuschl T (2008) Identification of microRNAs and other small regulatory RNAs using cDNA library sequencing. Methods 44:3–12
Huang D, Koh C, Feurtado JA, Tsang EW, Cutler AJ (2013) MicroRNAs and their putative targets in Brassica napus seed maturation. BMC Genomics 14:140
Jung JH, Park CM (2007) MIR166/165 genes exhibit dynamic expression patterns in regulating shoot apical meristem and floral development in Arabidopsis. Planta 225:1327–1338
Lan Y, Su N, Shen Y, Zhang R, Wu F, Cheng Z, Wang J, Zhang X, Guo X, Lei C, Wang J, Jiang L, Mao L, Wan J (2012) Identification of novel miRNAs and miRNA expression profiling during grain development in indica rice. BMC Genomics 13:264
Li H, Dong Y, Sun Y, Zhu E, Yang J, Liu X, Xue P, Xiao Y, Yang S, Wu J, Li X (2011) Investigation of the microRNAs in safflower seed, leaf, and petal by high-throughput sequencing. Planta 233:611–619
Li RQ, Yu C, Li YR, Lam TW, Yiu SM, Kristiansen K, Wang J (2009) SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25:1966–1967
Li S, Yang X, Wu F, He Y (2012) HYL1 controls the miR156-mediated juvenile phase of vegetative growth. J Exp Bot 63:2787–2798
Liang C, Zhang X, Zou J, Xu D, Su F, Ye N (2010) Identification of miRNA from Porphyra yezoensis by high-throughput sequencing and bioinformatics analysis. PloS One 5:e10698
Lin Y, Lai Z (2013) Comparative analysis reveals dynamic changes in miRNAs and their targets and expression during somatic embryogenesis in Longan (Dimocarpus longan Lour.). PloS One 8:e60337
Liu PP, Montgomery TA, Fahlgren N, Kasschau KD, Nonogaki H, Carrington JC (2007) Repression of AUXIN RESPONSE FACTOR10 by microRNA160 is critical for seed germination and post-germination stages. Plant J 52:133–146
Lopez-Gomollon S, Mohorianu I, Szittya G, Moulton V, Dalmay T (2012) Diverse correlation patterns between microRNAs and their targets during tomato fruit development indicates different modes of microRNA actions. Planta 236:1875–1887
Ma X, Shao C, Wang H, Jin Y, Meng Y (2013) Construction of small RNA-mediated gene regulatory networks in the roots of rice (Oryza sativa). BMC Genomics 14:510
Mao W, Li Z, Xia X, Li Y, Yu J (2012) A combined approach of high-throughput sequencing and degradome analysis reveals tissue specific expression of microRNAs and their targets in cucumber. PloS One 7:e33040
Marin E, Jouannet V, Herz A, Lokerse AS, Weijers D, Vaucheret H, Nussaume L, Crespi MD, Maizel A (2010) miR390, Arabidopsis TAS3 tasiRNAs, and their AUXIN RESPONSE FACTOR targets define an autoregulatory network quantitatively regulating lateral root growth. Plant Cell 22:1104–1117
Martin RC, Liu PP, Goloviznina NA, Nonogaki H (2010) microRNA, seeds, and darwin?: diverse function of miRNA in seed biology and plant responses to stress. J Exp Bot 61:2229–2234
Meng Y, Huang F, Shi Q, Cao J, Chen D, Zhang J, Ni J, Wu P, Chen M (2009) Genome-wide survey of rice microRNAs and microRNA-target pairs in the root of a novel auxin-resistant mutant. Planta 230:883–898
Meyers BC, Axtell MJ, Bartel B, Bartel DP, Baulcombe D, Bowman JL, Cao X, Carrington JC, Chen X, Green PJ, Griffiths-Jones S, Jacobsen SE, Mallory AC, Martienssen RA, Poethig RS, Qi Y, Vaucheret H, Voinnet O, Watanabe Y, Weigel D, Zhu JK (2008) Criteria for annotation of plant MicroRNAs. Plant Cell 20:3186–3190
Muvva C, Tewari L, Aruna K, Ranjit P, MD ZS, MD KAM, Veeramachaneni H (2012) In silico identification of miRNAs and their targets from the expressed sequence tags of Raphanus sativus. Bioinformation 8:98–103
Nodine MD, Bartel DP (2010) MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Genes Dev 24:2678–2692
Nonogaki H (2010) MicroRNA gene regulation cascades during early stages of plant development. Plant Cell Physiol 51:1840–1846
Oh TJ, Wartell RM, Cairney J, Pullman GS (2008) Evidence for stage-specific modulation of specific microRNAs (miRNAs) and miRNA processing components in zygotic embryo and female gametophyte of loblolly pine (Pinus taeda). New Phytol 179:67–80
Palovaara J, Saiga S, Weijers D (2013) Transcriptomics approaches in the early Arabidopsis embryo. Trends Plant Sci 18:514–521
Rademacher EH, Lokerse AS, Schlereth A, Llavata-Peris CI, Bayer M, Kientz M, Freire Rios A, Borst JW, Lukowitz W, Jurgens G, Weijers D (2012) Different auxin response machineries control distinct cell fates in the early plant embryo. Dev Cell 22:211–222
Reyes JL, Chua NH (2007) ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J 49:592–606
Rodriguez RE, Mecchia MA, Debernardi JM, Schommer C, Weigel D, Palatnik JF (2010) Control of cell proliferation in Arabidopsis thaliana by microRNA miR396. Development 137:103–112
Rubio-Somoza I, Cuperus JT, Weigel D, Carrington JC (2009) Regulation and functional specialization of small RNA-target nodes during plant development. Curr Opin Plant Biol 12:622–627
Song QX, Liu YF, Hu XY, Zhang WK, Ma B, Chen SY, Zhang JS (2011) Identification of miRNAs and their target genes in developing soybean seeds by deep sequencing. BMC Plant Biology 11:5
Sunkar R (2010) MicroRNAs with macro-effects on plant stress responses. Semin Cell Dev Biol 21:805–811
Sunkar R, Kapoor A, Zhu JK (2006) Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Plant Cell 18:2051–2065
Sunkar R, Zhu JK (2004) Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell 16:2001–2019
Voinnet O (2009) Origin, biogenesis, and activity of plant microRNAs. Cell 136:669–687
Wan LC, Wang F, Guo X, Lu S, Qiu Z, Zhao Y, Zhang H, Lin J (2012) Identification and characterization of small non-coding RNAs from Chinese fir by high throughput sequencing. BMC Plant Biology 12:146
Wang C, Han J, Liu C, Kibet KN, Kayesh E, Shangguan L, Li X, Fang J (2012a) Identification of microRNAs from Amur grape (Vitis amurensis Rupr.) by deep sequencing and analysis of microRNA variations with bioinformatics. BMC Genomics 13:122
Wang F, Li L, Liu L, Li H, Zhang Y, Yao Y, Ni Z, Gao J (2012b) High-throughput sequencing discovery of conserved and novel microRNAs in Chinese cabbage (Brassica rapa L. ssp. pekinensis). Mol Genet Genomics 287:555–563
Wang J, Czech B, Weigel D (2009) miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 138:738–749
Wang ZJ, Huang JQ, Huang YJ, Li Z, Zheng BS (2012c) Discovery and profiling of novel and conserved microRNAs during flower development in Carya cathayensis via deep sequencing. Planta 236:613–621
Wang ZM, Xue W, Dong CJ, Jin LG, Bian SM, Wang C, Wu XY, Liu JY (2012d) A comparative miRNAome analysis reveals seven fiber initiation-related and 36 novel miRNAs in developing cotton ovules. Mol Plant 5:889–900
Wei LQ, Yan LF, Wang T (2011) Deep sequencing on genome-wide scale reveals the unique composition and expression patterns of microRNAs in developing pollen of Oryza sativa. Genome Biol 12:R53
Wei M, Wei H, Wu M, Song M, Zhang J, Yu J, Fan S, Yu S (2013) Comparative expression profiling of miRNA during anther development in genetic male sterile and wild type cotton. BMC Plant Biology 13:66
Williams L, Grigg SP, Xie M, Christensen S, Fletcher JC (2005) Regulation of Arabidopsis shoot apical meristem and lateral organ formation by microRNA miR166g and its AtHD-ZIP target genes. Development 132:3657–3668
Willmann MR, Mehalick AJ, Packer RL, Jenik PD (2011) MicroRNAs regulate the timing of embryo maturation in Arabidopsis. Plant Physiol 155:1871–1884
Wu G, Park MY, Conway SR, Wang JW, Weigel D, Poethig RS (2009) The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 138:750–759
Wu XM, Liu MY, Ge XX, Xu Q, Guo WW (2011) Stage and tissue-specific modulation of ten conserved miRNAs and their targets during somatic embryogenesis of Valencia sweet orange. Planta 233:495–505
Xiang D, Venglat P, Tibiche C, Yang H, Risseeuw E, Cao Y, Babic V, Cloutier M, Keller W, Wang E, Selvaraj G, Datla R (2011) Genome-wide analysis reveals gene expression and metabolic network dynamics during embryo development in Arabidopsis. Plant Physiol 156:346–356
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong S, Kong L, Gao G, Li CY, Wei L (2011) KOBAS 2.0: a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res 39:W316–W322
Xing S, Salinas M, Hohmann S, Berndtgen R, Huijser P (2010) miR156-targeted and nontargeted SBP-box transcription factors act in concert to secure male fertility in Arabidopsis. Plant Cell 22:3935–3950
Xu L, Wang Y, Xu Y, Wang L, Zhai L, Zhu X, Gong Y, Ye S, Liu L (2013a) Identification and characterization of novel and conserved microRNAs in radish (Raphanus sativus L.) using high-throughput sequencing. Plant Sci 201–202:108–114
Xu L, Wang Y, Zhai L, Xu Y, Wang L, Zhu X, Gong Y, Yu R, Limera C, Liu L (2013b) Genome-wide identification and characterization of cadmium-responsive microRNAs and their target genes in radish (Raphanus sativus L.) roots. J Exp Bot 64:4271–4287
Xu MY, Dong Y, Zhang QX, Zhang L, Luo YZ, Sun J, Fan YL, Wang L (2012) Identification of miRNAs and their targets from Brassica napus by high-throughput sequencing and degradome analysis. BMC Genomics 13:421
Xue LJ, Zhang JJ, Xue HW (2009) Characterization and expression profiles of miRNAs in rice seeds. Nucleic Acids Res 37:916–930
Yan K, Liu P, Wu CA, Yang GD, Xu R, Guo QH, Huang JG, Zheng CC (2012) Stress-induced alternative splicing provides a mechanism for the regulation of microRNA processing in Arabidopsis thaliana. Mol Cell 48:521–531
Yang J, Liu X, Xu B, Zhao N, Yang X, Zhang M (2013a) Identification of miRNAs and their targets using high-throughput sequencing and degradome analysis in cytoplasmic male-sterile and its maintainer fertile lines of Brassica juncea. BMC Genomics 14:9
Yang X, Wang L, Yuan D, Lindsey K, Zhang X (2013b) Small RNA and degradome sequencing reveal complex miRNA regulation during cotton somatic embryogenesis. J Exp Bot 64:1521–1536
Yang X, Zhang X, Yuan D, Jin F, Zhang Y, Xu J (2012) Transcript profiling reveals complex auxin signalling pathway and transcription regulation involved in dedifferentiation and redifferentiation during somatic embryogenesis in cotton. BMC Plant Biology 12:110
Yazawa K, Takahata K, Kamada H (2004) Isolation of the gene encoding Carrot leafy cotyledon1 and expression analysis during somatic and zygotic embryogenesis. Plant Physiol Biochem 42:215–223
Yu S, Galvao VC, Zhang YC, Horrer D, Zhang TQ, Hao YH, Feng YQ, Wang S, Schmid M, Wang JW (2012) Gibberellin regulates the Arabidopsis floral transition through miR156-targeted SQUAMOSA promoter binding-like transcription factors. Plant Cell 24:3320–3332
Zhai L, Xu L, Wang Y, Cheng H, Chen Y, Gong Y, Liu L (2013) Novel and useful genic-SSR markers from de novo transcriptome sequencing of radish (Raphanus sativus L.). Mol Breed. doi:10.1007/s11032-013-9978-x
Zhang H, Ogas J (2009) An epigenetic perspective on developmental regulation of seed genes. Mol Plant 2:610–627
Zhang J, Zhang S, Han S, Wu T, Li X, Li W, Qi L (2012) Genome-wide identification of microRNAs in larch and stage-specific modulation of 11 conserved microRNAs and their targets during somatic embryogenesis. Planta 236:647–657
Zhang XN, Li X, Liu JH (2013) Identification of conserved and novel cold-responsive microRNA in trifoliate orange (Poncirus trifoliata (L.) Raf.) using high-throughput sequencing. Plant Mol Biol Rep. doi:10.1007/s11105-013-0649-1
Zhu QH, Helliwell CA (2011) Regulation of flowering time and floral patterning by miR172. J Exp Bot 62:487–495
Acknowledgements
This work was supported in part by grants from the Program for the National Natural Science Foundation of China (31171956, 31372064), the Key Technologies R & D Program of Jiangsu Province, China (BE2010328,BE2013429), the PAPD and JASTIF [CX(12)2006]. We thank Dr. Oliver R. E. from North Dakota State University, ND, USA for her critical review and helpful comments during the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhai, L., Xu, L., Wang, Y. et al. Genome-Wide Identification of Embryogenesis-Associated microRNAs in Radish (Raphanus sativus L.) by High-Throughput Sequencing. Plant Mol Biol Rep 32, 900–915 (2014). https://doi.org/10.1007/s11105-014-0700-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-014-0700-x